
Abstract
Despite many years of research into chemical warfare agents, cytotoxic mechanisms induced by mustards are not well understood. Reactive oxygen and nitrogen species (ROS and RNS) are likely to be involved in chemical warfare agents induced toxicity. These species lead to lipid peroxidation, protein oxidation, and DNA injury, and trigger many pathophysiological processes that harm the organism. In this article, several steps of pathophysiological mechanisms and possible ways of protection against chemical warfare agents have been discussed. In summary, pathogenesis of mustard toxicity is explained by three steps: (1) mustard binds target cell surface receptor, (2) activates intracellular ROS and RNS leading to peroxynitrite (ONOO−) production, and (3) the increased ONOO− level damages organic molecules (lipids, proteins, and DNA) leading to poly(adenosine diphosphate-ribose) polymerase (PARP) activation. Therefore, protection against mustard toxicity could also be performed in these ways: (1) blocking of cell surface receptor, (2) inhibiting the ONOO− production or scavenging the ONOO− produced, and (3) inhibiting the PARP, activated by ONOO− and hydroxyl radical (OH•) induced DNA damage. As conclusion, to be really effective, treatment against mustards must take all molecular mechanisms of cytotoxicity into account. Combination of several individual potent agents, each blocking one of the toxic mechanisms induced by mustards, would be interesting. Therefore, variations of combination of cell membrane receptor blockers, antioxidants, nitric oxide synthase inhibitors, ONOO− scavengers, and PARP inhibitors should be investigated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction to chemical warfare agents
Chemical weapons (CW) were used for the first time on a large scale in World War I. The use of the vesicant sulfur mustard and the pulmonary agents phosgene and chlorine resulted in 1.3 million casualties. Since then, CW have been used in numerous incidents, e.g., sulfur mustard in the Iran–Iraq conflict and nerve agents against the Kurdish opposition in Iraq (Bismuth et al. 2004) and also in terroristic attacks by the Aum Shinrikyo section Japan. After the tragic events of September 11, 2001, the perception of the threat of using CW has increased (Goozner et al. 2002; Rosenbloom et al. 2002). As a result of this continuous threat, CW have been the subject of a considerable amount of toxicological research, with the ultimate goal of finding defensive measures against these agents. In addition, the use or alleged use of CW in war and terrorism has clearly established an urgent need for biological markers of exposure.
Sulfur mustard (SM; 2,2′-dichlorodiethyl sulfide) and nitrogen mustards (NM) are alkylating agents that have been used for many years as CW and therapeutic drugs, respectively. SM is a highly toxic CW and still remains a threat to both civilians and military personnel. Although some beneficial effects have been observed with some drugs in tissue culture systems, the antidote activity of the test compounds was always too weak to be used as protectants against SM (Kisby et al. 2000). NM were produced in the 1920s and 1930s as potential chemical warfare weapons. They are also known by their military designations as HN-1 [bis(2-chloroethyl)ethylamine], HN-2 [bis(2-chloroethyl)methylamine], and HN-3 [tris(2-chloroethyl)amine] (Lemire et al. 2004). HN-2, which has a similar molecular structure to SM, is commonly used as an anticancer drug and remains an important therapeutic agent in the treatment of early stage mycosis fungoides (Calvet et al. 1999). However, this agent is also extremely toxic and its use is accompanied by severe side effects (Smith et al. 1998).
Mustard gas causes injury via three major routes: (1) skin and eye damage after absorption through the integument and the ocular surface, respectively; (2) respiratory damage after inhalation; (3) systemic toxicity after ingestion or high exposures, manifested as gastrointestinal, circulatory, renal, and bone marrow toxicity. Inhalation of mustards primarily affects the laryngeal and tracheobronchial mucosa leading to tissue necrosis, airway inflammation, and lung oedema. At low-dose exposure, these chemicals act as lung irritants rather than acute toxicants, resulting in long-term airway diseases such as chronic bronchitis, lung fibrosis, and asthma (Karalliedde et al. 2000).
Despite many years of research into these agents, cytotoxic mechanisms induced by mustards and the initial events leading to cell death are still not fully understood. Papirmeister et al. (1985) have proposed a biochemical mechanism for mustard-induced toxicity involving the process of poly(adenosine diphosphate-ribose) polymerase (PARP) following DNA alkylation, resulting in rapid depletion of the NAD+/ATP metabolite leading to cell death (Meier et al. 2000).
Beside the alkylation of DNA, considered to be the most significant injury to cells from mustards, oxidative stress is likely involved in alkylating agents-acute toxicity (Giuliani et al. 1997; Rappenau et al. 1999). Indeed, alkylating agents are known to induce glutathione (GSH) depletion (Gross et al. 1993) which likely contributes to lipid peroxidation and cell death. Furthermore, there is abundance of evidence, that mustards cause nitric oxide (NO) production through nitric oxide synthase (NOS) induction leading to peroxynitrite formation (ONOO−) in the target cells.
In order to be really effective, a protective treatment against mustards must take all molecular mechanisms of cytotoxicity into account. It would be interesting to combine several individual potent agents, each blocking one of the toxic mechanisms induced by mustards. Therefore, several steps of target of antioxidants, potent NOS inhibitors, peroxynitrite scavengers, and PARP inhibitors have been discussed in this article.
Free oxygen radicals and antioxidant defense mechanism
Reactive oxygen species (ROS) are constantly generated under physiologic conditions as a consequence of aerobic metabolism. ROS include free radicals such as the superoxide (O −•2 ) anion, hydroxyl radicals (OH•), and the nonradical molecule hydrogen peroxide (H2O2). They are particularly transient species due to their high chemical reactivity and can react with DNA, proteins, carbohydrates, and lipids in a destructive manner. The cell is endowed with an extensive antioxidant defense system to combat against ROS, either directly by interception or indirectly through reversal of oxidative damage. When ROS overcome the defense systems of the cell and redox homeostasis is altered, the result is oxidative stress (Sies 1997) (Fig. 1). Oxidative stress is implicated in the pathogenesis of several diseases such as inflammatory, ischemic, neurodegenerative disorders, aging, diabetes, hypertension, and cancer (Erdelyi et al. 2005; Sies 1991).

Basic sources of ROS and principal defense mechanisms. Major sources of ROS production include respiration of mitochondria, oxidative burst of immune cells, and some environmental factors such as ultraviolet radiation, tobacco smoke, and exhaust gas. The generated superoxide is converted to H2O2 by superoxide dismutase (SOD). The two major defense systems against H2O2 are the GSH redox cycle and catalase (CAT)
Antioxidants defense mechanisms against ROS
Components of the antioxidant defense system function to prevent oxidative damage by intercepting ROS before they can damage intracellular targets. It consists mainly of three enzymes: superoxide dismutase (SOD), glutathione peroxidase (GSH-Px), and catalase (CAT) (Sies 1993, 1997). Three classes of SOD have been identified to date. These are mitochondrial Mn-SOD, cytosolic and extracellular Cu,Zn-SOD, and Ni-SOD (Faraci and Didion 2004; Miller 2004). All of these SOD isoforms destroy the free radical superoxide by converting it to H2O2. The primary defense mechanisms against H2O2 are CAT and GSH-Px. CAT is one of the most efficient enzymes known and cannot be saturated by H2O2 at any concentration. GSH-Px acts through the glutathione (GSH) redox cycle. The GSH system is probably the most important cellular defense mechanism that exists in the cell (Sies 1999) (Fig. 1).
Nitric oxide and nitric oxide synthase family
Nitric oxide is produced by a family of enzymes called NOS through enzymatic oxidation of the guanidino group of arginine. This occurs in two sequential monooxygenase reactions utilizing NADPH as cosubstrate and involving the utilization of molecular oxygen. Constitutive expression of two NOS isoforms is responsible for a low basal level of NO synthesis in neural cells (nNOS or NOS1) and in endothelial cells (eNOS or NOS3). Induction of the inducible isoform (iNOS or NOS2) by cytokines (e.g., tumor necrosis factor-α, interleukins), bacterial products (endotoxin/LPS), and chemical agents has been observed in virtually all cell types tested including macrophages, dendritic cells, fibroblasts, chondrocytes, osteoclasts, astrocytes, epithelial cells and results in the production of large amounts of NO (Moncada et al. 1991). The controversy arises from observations reporting both cytotoxic and cytoprotective effects of NO. In cases where NO was found cytotoxic, it was questioned whether NO directly or indirectly, through the formation of more reactive species such as peroxynitrite (ONOO−) exerted these effects (Szabo 1996).
The activated “devil triangle” in the target cell
As both excess NO or excess O −•2 decreases the bioavailability of ONOO−, equimolar concentrations of the radicals are ideal for ONOO− formation. ONOO− anion is in a pH-dependent protonation equilibrium with peroxynitrous acid (ONOOH). Homolysis of ONOOH gives rise to formation of the highly reactive hydroxyl radical (OH•) mediating molecular and tissue damage associated with ONOO− production (Radi et al. 2001).
ONOO− is formed when NO and O −•2 react in a near diffusion-limited reaction (Squatrito and Pryor 1998). The most powerful cellular antioxidant system protecting against the harmful effects of O −•2 is embodied by SOD (especially cytosolic Cu,Zn-SOD and mitochondrial Mn-SOD). However, it was shown that NO efficiently competes with SOD for superoxide (Beckman and Koppenol 1996) (Fig. 2). Beckman and Koppenol (1996) have therefore proposed that under conditions of increased NO production, NO can outcompete SOD for O −•2 resulting in ONOO− formation.

Formation of devil triangle; NO can be produced by nNOS, eNOS, and iNOS or in the mitochondria by mtNOS. Mustard can cause not only ROS production but also iNOS induction leading to NO overproduction. Under conditions of increased NO production, NO can outcompete SOD for superoxide anion resulting in peroxynitrite formation leading to PARP activation, lipid peroxidation, and protein oxidation
How is peroxynitrite harmful?
ONOO− is not a radical but is a stronger oxidant than its precursor radicals. It can directly react with target biomolecules via one- or two-electron oxidations (Alvarez and Radi 2003). Higher concentrations and the uncontrolled generation of ONOO− may result in unwanted oxidation and consecutive destruction of host cellular constituents. ONOO− may oxidize and covalently modify all major types of biomolecules, such as membrane lipids, thiols, proteins, and DNA. One of the most important mechanisms of cellular injury is a ONOO−-dependent increase in DNA strand breakage, which triggers the activation of PARP, a DNA repair enzyme. DNA damage causes PARP overactivation, resulting in the depletion of oxidized nicotinamide adenine dinucleotide (NAD) and adenosine triphosphate (ATP), and consequently in necrotic cell death (Virag and Szabo 2002).
DNA single-strand breakage is an obligatory trigger for the activation of PARP. ONOO−, as well as OH•, is the key pathophysiologically relevant triggers of DNA single-strand breakage (Schraufstatter et al. 1988). Moreover, nitroxyl anion, a reactive molecule derived from nitric oxide, is a potent activator of DNA single-strand breakage and PARP activation in vitro (Bai et al. 2001).
Approximately 15 years ago, it was generally assumed that triggers of DNA single-strand breakage are restricted to severe environmental toxic agents (e.g., genotoxic or cytotoxic drugs) or various forms of ionizing radiation (Gu et al. 1995; Lazebnik et al. 1994). The research into the potential role of PARP in pathophysiological processes gained a new momentum in the mid-1990s by studies linking the formation of NO synthases to DNA single-strand breakage and PARP activation, with subsequent energetic changes in the cell (Radons et al. 1994; Zhang et al. 1994). Subsequent studies clarified that the actual trigger of DNA single-strand breakage is ONOO−, rather than NO (Szabo et al. 1996). The identification of ONOO− as an important mediator of the cellular damage in various forms of inflammation stimulated significant interest into the role of the PARP related suicide pathway in various pathophysiological conditions. Endogenous production of ONOO− and other oxidants has been shown to lead to DNA single-strand breakage and PARP activation (Szabo 2003).
Possible mechanisms to block mustard toxicity
Current knowledge seems feasible that mustard toxicity comes from oxidative stress, iNOS induction, ONOO− production leading to lipid, protein, and DNA damage in the target cell. In order to cure such a high toxicity all molecular mechanisms should be considered. For example, we need an antidote to stop mustard get into the cell (e.g., mustard membrane receptor blocker), further, once mustard entered into the cell, then we need useful antioxidants, iNOS inhibitor, etc. If damage (e.g., lipid peroxidation, protein oxidation) occurred, repair mechanisms should be activated in order to cell survival. Steps for fight against mustard toxicity are summarized in Table 1.
Blocking of cell surface receptor of mustards
Sawyer (1998a, b, 1999) has shown that several arginine analogues have protective activity against the toxicity of SM in vitro and that these protective effects are not related to the potent NOS inhibiting activity of these drugs. l-thiocitrulline (l-TC) is an extremely potent protective agent that confers strictly prophylactic, but persistent protection against SM, while l-nitroarginine methyl ester (l-NAME) is effective when administered up to several hours after SM exposure and its effects are reversible upon removal of the drug (Sawyer 1998a, b). These different characteristics of protection strongly suggest that these drugs are acting at different sites in a cascade of events that is rapidly initiated by SM, but then progresses relatively slowly. This scenario predicts that, in combination, these drugs would result in additive or synergistic protection against SM.
Then the author has proposed a hypothesis on protective effects of l-TC. In this hypothesis, SM initiates its toxicity extremely rapidly through a cell surface-mediated event that can be blocked by l-TC. A signal is transduced into the cell that results in an additional event or lesion that manifests itself several hours later. This event/lesion progresses to cell death unless blocked reversibly by l-NAME. The supposition that the initial event in SM toxicity is a cell surface one is supported by the timing of l-TC protection against SM. The protection is strictly prophylactic, and l-TC is effective even when administered only 1 min prior to SM exposure of the cultures. Previous work has shown that l-TC does not chemically interact with SM, so this was not a scavenging effect. Although the lipophilic nature of SM would allow for toxic concentrations to quickly pass through the cell membrane to exert toxicity, it seems unlikely that the high millimolar l-TC levels required for protection would be able to penetrate into the cell within 1 min. It is more probable that l-TC blocks a cell surface receptor targeted by SM (Sawyer et al. 1996; Sawyer 1999).
Inhibiting the ONOO− production or scavenging the ONOO− produced
Three basic strategies serve the defense against ONOO− (Klotz and Sies 2003): the prevention of formation of the reactive species, interception with its damaging targets, and repair of damage done (Fig. 3).

Basic strategies serve the defense against peroxynitrite, the prevention of formation of the reactive species, interception with its damaging targets and repair of damage done. Prevention, NOS inhibitors (e.g., l-NAME, l-NOARG), in particular selective iNOS inhibitors (e.g., S-methylthiourea, aminoguanidine) may be useful to block the devil triangle. Antioxidants may also be effective on the same purpose. Interception, once peroxynitrite formed, scavengers of this may ameliorate the damage. Repair, repairing the damaged macromolecules depends on the cellular repairing mechanisms and some antioxidants such as vitamin E and C
Prevention
Prevention of the exposure of cells to ONOO− can simply be prevention of its formation. Generation of ONOO− can be prevented by inhibiting the formation of NO and/or of O −•2 by either inhibiting enzymatic systems responsible for the generation of these two radicals (NOS, xanthine oxidase, NADPH oxidase) or inhibiting their upregulation induced by inflammatory processes. Further, NO and O −•2 can be scavenged prior to their generating ONOO−.
Inhibiting the formation of superoxide
Beside DNA alkylation and the subsequent PARP activation mentioned above, in an earlier step, oxidative stress may contribute to NM-induced injury. It was shown that the antioxidant N-acetyl cysteine (NAC) was able to reduce the peroxide augmentation induced by NM in cell culture. The antioxidant property of NAC may intervene in this effect since it was observed that NAC prevents peroxide augmentation following tert-butylhydroperoxide (TBHP) treatment (Ochi and Miyaura 1989). Moreover, reduction of sulfhydryl alkylation, in particular GSH, using NAC, must also be strongly implicated in oxidative stress induced by NM. Among the other antioxidant molecules tested, only the lipid peroxidation inhibitor, α-tocopherol, slightly reduced the effect of NM on cell proliferation, in a dose-dependent manner. Furthermore, the lipid peroxidation inhibitors, silymarin (Morazzoni and Bombardelli 1995) and butylated hydroxytoluene (BHT), and the iron chelators desferroxamine (DFO) and 1,10-phenanthroline (OP), which have shown protective effects against NM toxicity in hepatocyte and skin models (Khan et al. 1992; Wormser and Nyska 1991), although were all ineffective on cell culture. On the other hand, all the tested antioxidants were effective at preventing the oxidative stress induced by the lipoperoxide TBHP. The oxidative stress induced by NM does not appear as fundamental in the NM-induced injury on cell culture, as metabolic injury is not prevented by several antioxidants. The failure of antioxidants, when used lonely, against mustard toxicity may be explained by the fact that, ROS, especially O −•2 , cannot be scavenged enough if enormous NO is present in the cell or tissue. Like SOD, antioxidants cannot defeat O −•2 if NO production is not blocked simultaneously (Fig. 2).
Inhibiting the formation of nitric oxide
Several different arginine analogue NOS inhibitors such as l-NAME have been shown to have protective activity against the toxicity of SM in primary cultures of chick embryo neurons (Sawyer 1998a, b). Sawyer (1998a, b) examined the possibility that the toxicity of SM was due to the induction or activation of NOS, thus liberating increased and toxic quantities of the reactive chemical species, NO. This seemed reasonable since many of the toxic effects of SM seemed to be parallel with the effects of NO overproduction. In these studies the author reasoned that if NO was indeed involved, then its inhibition by the well-characterized arginine analogue NOS inhibitor l-NAME should confer protection by preventing the SM-induced overproduction of NO. This approach was successful and l-NAME was found to confer protection against the toxicity of SM to an extent that was in excess of those obtained by any previously published protective drug regimens against the toxicity of SM. But there was a problem to reveal the mechanism; if the toxicity of SM was indeed due to its effects on NOS then SM toxicity, as well as the protection conferred by l-NAME pretreatment, should be dependent on the l-arginine concentration in the medium (Dawson et al. 1991, 1993); SM toxicity and l-NAME protection were independent of l-arginine concentration. NOS is a stereospecific enzyme requiring l-arginine as a substrate and inhibition of NOS is likewise stereospecific for the l-analogues of arginine (Dawson et al. 1991, 1993; Kerwin and Heller 1994; Rees et al. 1989); d-NAME was as efficacious as l-NAME in conferring protection.
In addition, Nyska et al. (2001) reported beneficial effects against SM skin toxicity when iNOS expression was reduced indirectly through epidermal inflammation reduction. Other anti-inflammatory methods depending on TNF-α inhibition were also suggested to be useful in SM-induced toxicity (Wormser et al. 2005).
However, since iNOS is responsible for the abundant NO synthesis and mainly responsible for the ONOO− production, future research is needed using highly selective iNOS inhibitors such as aminoguanidine, S-methylthiourea, and 1400W against mustard toxicity.
Interception
Low-molecular weight compounds such as carbon dioxide, thiols, ascorbate, and selenocompounds have been shown to react with ONOO− (Arteel and Sies 1999; Koppenol 1998). One of the most promising results came from ebselen (2-phenyl-1,2-benzisoselenazol-3[2H]-one), a lipid soluble selenoorganic compound able to detoxify ONOO−. We have previously shown that ebselen is a potent ONOO− scavenger (Korkmaz et al. 2005). This selenoorganic compound has also protective effect against NM-induced lung toxicity (unpublished data). Further research is needed to clarify the whole mechanism.
Repair
Repair of damage resulting from the reaction of ONOO− escaping the regulatory mechanisms serves the regeneration of normal cellular conditions. Correspondingly, the cellular repair systems available for damaged DNA, lipids, and proteins are employed. It is part of the restitution process for the cells to actively degrade mildly oxidized or nitrated proteins (Grune et al. 1998, 2001). One of the most important target molecules is DNA and DNA injury causes PARP activation which may lead to necrosis or apoptosis.
Inhibiting the PARP activated by ONOO− and OH• induced DNA damage
Numerous hypotheses have been proposed over the last 75 years to explain the toxicity of SM, including the involvement of inflammatory mediators, GSH depletion, energy depletion, or calcium perturbations (Dannenberg et al. 1985; Higuchi et al. 1988; Vindevoghel et al. 1994). None of these hypotheses have withstood experimental scrutiny with notable success. However, Papirmeister et al. (1985) proposed a scheme that incorporated many facets of these theories, as well as other experimental observations, in a unified hypothesis. They suggested that SM toxicity was initiated by DNA alkylation. Upon cellular recognition of genomic damage, normal DNA repair mechanisms would come into play to remove these lesions and the activity of PARP would become markedly elevated in order to affect repair. This increased activity would result in energy depletion (decrease of NAD/ATP) and the resultant inhibition of glycolysis would activate the hexose monophosphate shunt, causing protease release: activation and the subsequent pathology observed due to SM intoxication. Although elegant in concept, aspects of this hypothesis also seem to be failing the test of experimental evidence. While PARP inhibitors have been shown to have efficacy with respect to protecting against NAD depletion in a number of different systems, the correlation between NAD depletion and cytotoxicity is not clear, and PARP inhibitors do not confer significant protection against SM cytotoxicity in many of the systems thus far examined. Nevertheless, this theory has stimulated a tremendous amount of work that has shed more light on the toxicity of SM; although not enough to elucidate its mechanism of action. The PARP hypothesis has also been, in part, responsible for our current interest in NOS inhibitors.
Indeed, when examined closely, it seemed feasible that NOS induction or activation would explain the toxic effects of SM, since many of its toxic effects seem to have parallels with the biological effects and functions of NO including DNA damage, NAD depletion, cell and tissue toxicity, inflammation and shock. In earlier publications, Gross et al. (1985) have shown that SM lowers NAD concentrations in human skin grafted to athymic nude mice. Moreover, Yourick et al. (1991) indicated that NAD pretreatment reduces microvesicle formation in hairless guinea pigs cutaneously exposed to SM.
Pathogenesis of mustard toxicity
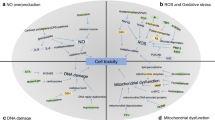
In the light of current data, pathogenesis of mustard toxicity can be summarized as following steps. First, mustard binds target cell surface receptor. Second, activates intracellular ROS and NO production leading to ONOO− production. Third, the increased ONOO− level damages lipids (lipid peroxidation), proteins (protein oxidation), and DNA (strand breaks) leading to PARP activation (Fig. 4). Antioxidants such as α-tocopherol, vitamin C, and intracellular mechanisms may repair the damaged lipid and proteins. Activated PARP may act in an extremely different way called the Yin and Yang of PARP activation. The death-promoting and the cytoprotective effects of poly(ADP-ribosylation) represent two seemingly opposing faces (the Yin and Yang) of PARP. Oxidative and nitrosative stress induced DNA breakage causes a high level of PARP activation, leading to the depletion of NAD and ATP and consequently to necrotic cell death. On the other hand, poly(ADP-ribosylation) facilitates DNA repair in cells subjected to treatment with alkylation agents or ionizing radiation.

The overall mechanism of mustard induced cell toxicity. Mustard can easily enter into the cell and causes both superoxide and nitric oxide overproduction. These two precursors may form peroxynitrite. Peroxynitrite is known as a strong nitrosative agent and causes lipid peroxidation, protein oxidation, and DNA damage. DNA damage leads to PARP activation and affects the cellular energetic levels. If the damage is severe enough, PARP overactivates and causes depletion of cellular NAD and ATP levels leading to cellular necrosis
As pointed out above, both sides of DNA damage-dependent PARP activation have recently been in the focus of therapeutic approaches. On the other hand, in particular cancer therapy, inhibition of PARP activity is an attractive option for suppressing repair and achieving more extensive cell killing, but this time via the apoptotic pathway. Therefore, the outcome of inhibition of PARP activity depends on the tissue background and additional pharmacologic treatments. Research is moving on very fast in this newly emerged field of therapy. But there are two caveats: first, long-lasting inhibition may have deleterious effects. Therefore, interference with this part of the repair machinery may give rise to mutations in cells outside the target of mustards. Second, as there are several PARP family members with unknown functions, nonspecific inhibition of poly(ADP-ribosylation) in general may have unexpected and severe side effects (Beneke et al. 2004).
Conclusion and future directions
In order to be really effective, a protective treatment against mustards must take all molecular mechanisms of cytotoxicity into account. Therefore, it would be interesting to combine several individual potent agents, each blocking one of the toxic mechanisms induced by mustards. Thus, a combination of cell membrane receptor blockers, antioxidants, NOS inhibitors, peroxynitrite scavengers, and PARP inhibitors must be investigated.
References
Alvarez B, Radi R (2003) Peroxynitrite reactivity with amino acids and proteins. Amino Acids 25:295–311
Arteel GE, Sies H (1999) Protection against peroxynitrite by cocoa polyphenol oligomers. FEBS Lett 462:167–170
Bai P, Bakondi E, Szabo E, Gergely P, Szabo C, Virag L (2001) Partial protection by poly(ADP-ribose) polymerase inhibitors from nitroxyl-induced cytotoxity in thymocytes. Free Radic Biol Med 31:1616–1623
Beckman JS, Koppenol WH (1996) Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol 271:C1424–C1437
Beneke S, Diefenbach J, Burkle A (2004) Poly(ADP-ribosyl)ation inhibitors: promising drug candidates for a wide variety of pathophysiologic conditions. Int J Cancer 111:813–818
Bismuth C, Borron SW, Baud FJ, Barriot P (2004) Chemical weapons: documented use and compounds on the horizon. Toxicol Lett 149:11–18
Calvet JH, Feurmann M, Llorente L, Loison F, Harf A, Marano F (1999) Comparative toxicity of sulfur mustard and nitrogen mustard on tracheal epithelial cells in primary culture. Toxicol In Vitro 13:859–866
Dannenberg AM, Pula PJ, Liu LH, Harada S, Tanaka F, Vogt RF, Kajiki A, Higuchi K (1985) Inflammatory mediators and modulators released in organ culture from rabbit skin lesions produced in vivo by sulfur mustard. I. Quantitative histopathology; PMN, basophil, and mononuclear cell survival; and unbound (serum) protein content. Am J Pathol 121:15–27
Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH (1991) Nitric oxide mediates glutamate neurotoxicity in primary cortical culture. Proc Natl Acad Sci (USA) 88:6368–6371
Dawson VL, Dawson TM, Bartley DA, Uhl GR, Snyder SH (1993) Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. J Neurosci 13:2651–2661
Erdelyi K, Bakondi E, Gergely P, Szabo C, Virag L (2005) Pathophysiologic role of oxidative stress-induced poly(ADP-ribose) polymerase-1 activation: focus on cell death and transcriptional regulation. Cell Mol Life Sci 62:751–759
Faraci FM, Didion SP (2004). Vascular protection: superoxide dismutase isoforms in the vessel wall. Arterioscler Thromb Vasc Biol 24:1367–1373
Giuliani I, Baeza-Squiban A, Marano F (1997) Early cytotoxic effects of mechlorethamine, a nitrogen mustard, on mammalian airway epithelium. Toxicol In Vitro 11:695–702
Goozner B, Lutwick LI, Bourke E (2002) Chemical terrorism: a primer for 2002. J Assoc Acad Minor Phys 13:14–18
Gross CL, Meier HL, Papirmeister B, Brinkley FB, Johnson JB (1985) Sulfur mustard lowers nicotinamide adenine dinucleotide concentrations in human skin grafted to athymic nude mice. Toxicol Appl Pharmacol 81:85–90
Gross CL, Innace JK, Hovatter RC, Meier HL, Smith WJ (1993) Biochemical manipulation of intracellular glutathione levels influences cytotoxicity to isolated human lymphocytes by sulphur mustard. Cell Biol Toxicol 9:259–268
Grune T, Blasig IE, Sitte N, Roloff B, Haseloff R, Davies KJA (1998) Peroxynitrite increases the degradation of aconitase and other cellular proteins by proteasome. J Biol Chem 273:10857–10862
Grune T, Klotz LO, Gieche J, Rudeck M, Sies H (2001) Protein oxidation and proteolysis by the nonradical oxidants singlet oxygen or peroxynitrite. Free Radic Biol Med 30:1243–1253
Gu Y, Sarnecki C, Aldape RA, Livingston DJ, Su MS (1995) Cleavage of poly(ADP-ribose) polymerase by interleukin-1 beta converting enzyme and its homologs TX and Nedd-2. J Biol Chem 270:18715–18718
Higuchi K, Kajiki A, Nakamura M, Harada S, Pula PJ, Scott AL, Dannenberg AM (1988) Proteases released in organ culture by acute dermal inflammatory lesions produced in vivo in rabbit skin by sulfur mustard: hydrolysis of synthetic peptide substrates for trypsin-like and chymotrypsin-like enzymes. Inflammation 12:311–334
Karalliedde L, Wheeler H, Maclehose R, Murray V (2000) Possible immediate and long-term health effects following exposure to chemical warfare agents. Public Health 114:238–248
Kerwin JF Jr, Heller M (1994) The arginine–nitric oxide pathway: a target for new drugs. Med Res Rev 14:23–74
Khan S, Ramwani JJ, O’Brien PJ (1992) Hepatocyte toxicity of mechlorethamine and other alkylating anticancer drugs. Biochem Pharmacol 43:1963–1967
Kisby GE, Springer N, Spencer PS (2000) In vitro neurotoxic and DNA-damaging properties of nitrogen mustard. J Appl Toxicol 20:S35–S41
Klotz LO, Sies H (2003) Defenses against peroxynitrite: selenocompounds and flavonoids. Toxicol Lett 140–141:125–132
Koppenol WH (1998) The basic chemistry of nitrogen monoxide and peroxynitrite. Free Radic Biol Med 25:385–391
Korkmaz A, Oter S, Sadir S, Coskun O, Topal T, Ozler M, Bilgiç H (2005) Peroxynitrite may be involved in bladder damage caused by cyclophosphamide in rats. J Urol 173:1793–1796
Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC (1994) Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature (London) 371:346–347
Lemire SW, Barr JR, Ashley DL, Olson CT, Hayes TL (2004) Quantitation of biomarkers of exposure to nitrogen mustards in urine from rats dosed with nitrogen mustards and from an unexposed human population. J Anal Toxicol 28:320–326
Meier HL, Millard C, Moser J (2000) Poly(ADP-ribose) polymerase inhibitors regulate the mechanism of sulfur mustard-initiated cell death in human lymphocytes. J Appl Toxicol 20:S93–S100
Miller AF (2004) Superoxide dismutases: active sites that save, but a protein that kills. Curr Opin Chem Biol 8:162–168
Moncada S, Palmer RMJ, Higgs EA (1991) Nitric oxide: physiology, pathophysiology, and pharmacology. Pharm Rev 43:109–142
Morazzoni P, Bombardelli E (1995) Silybum marianum (Carduus marianus). Fitoterapia 66:3–39
Nyska A, Lomnitski L, Maronpot R, Moomaw C, Broodsky B, Sintow A, Wormser U (2001) Effects of iodine on inducible nitric oxide synthase and cyclooxygenase-2 expression in sulfur mustard-induced skin. Arch Toxicol 74:768–774
Ochi T, Miyaura S (1989) Cytotoxicity of an organic hydroperoxide and cellular antioxidant defense system against hydroperoxides in cultured mammalian cells. Toxicology 55:69–82
Papirmeister B, Gross CL, Meier HL, Petrali JP, Johnson JB (1985) Molecular basis of mustard-induced vesication. Fundam Appl Toxicol 5:S134–S149
Radi R, Peluffo G, Alvarez MN, Naviliat M, Cayota A (2001) Unraveling peroxynitrite formation in biological systems. Free Radic Biol Med 30:463–488
Radons J, Heller B, Burkle A, Hartmann B, Rodriguez ML, Kroncke KD, Burkart V, Kolb H (1994) Nitric oxide toxicity in islet cells involves poly(ADP-ribose) polymerase activation and concomitant NAD depletion. Biochem Biophys Res Commun 199:1270–1277
Rappenau S, Baeza-Squbian A, Braut-Boucher F, Aubery M, Gendorm MC, Marano F (1999) Use of fluorescent probes to assess the early sulfhydryl depletion and oxidative stress induced by mechlorethamine on airway epithelium. Toxicol In Vitro 13:765–771
Rees DD, Palmer RMJ, Hodson HF, Moncada S (1989) A specific inhibitor of nitric oxide formation from l-arginine attenuates endothelium-dependent relaxation. Br J Pharmacol 96:418–424
Rosenbloom M, Leikin JB, Vogel SN, Chaudry ZA (2002) Biological and chemical agents: a brief synopsis. Am J Ther 9:5–14
Sawyer TW (1998a) Characterization of the protective effects of l-nitroarginine methyl ester (l-NAME) against the toxicity of sulphur mustard in vitro. Toxicology 131:21–32
Sawyer TW (1998b) Modulation of sulphur mustard toxicity by arginine analogues and related nitric oxide synthase inhibitors in vitro. Toxicol Sci 46:112–123
Sawyer TW (1999) Synergistic protective effects of selected arginine analogues against sulphur mustard toxicity in neuron culture. Toxicol Appl Pharmacol 155:169–176
Sawyer TW, Lundy PM, Weiss MT (1996) Protective effect of an inhibitor of nitric oxide synthase on sulphur mustard toxicity in vitro. Toxicol Appl Pharmacol 141:138–144
Schraufstatter I, Hyslop PA, Jackson JH, Cochrane CG (1988) Oxidant-induced DNA damage of target cells. J Clin Invest 82:1040–1050
Sies H (1991) Oxidative stress: from basic research to clinical application. Am J Med 91(3C):31S–38S
Sies H (1993) Strategies of antioxidant defense. Eur J Biochem 215:213–219
Sies H (1997) Oxidative stress: oxidants and antioxidants. Exp Physiol 82:291–295
Sies H (1999) Glutathione and its role in cellular functions. Free Radic Biol Med 27:916–921
Smith KJ, Smith WJ, Hamilton T, Skelton HG, Graham JS, Okerberg C, Moeller R, Hackley BE (1998) Histopathological and immunohistochemical features in human skin after exposure to nitrogen and sulfur mustard. Am J Dermopathol 20:22–28
Squatrito GL, Pryor WA (1998) Oxidative chemistry of nitric oxide: the roles of superoxide, peroxynitrite and carbon dioxide. Free Radic Biol Med 25:392–403
Szabo C (1996) The pathophysiological role of peroxynitrite in shock, inflammation, and ischemia-reperfusion injury. Shock 6:79–88
Szabo C (2003) Multiple pathways of peroxynitrite cytotoxicity. Toxicol Lett 140–141:105–112
Szabo C, Zingarelli B, O’Connor M, Salzman AL (1996) DNA strand breakage, activation of poly(ADP-ribose) synthetase and cellular energy depletion are involved in the cytotoxicity of macrophages and smooth muscle cells exposed to peroxynitrite. Proc Natl Acad Sci USA 93:1753–1758
Vindevoghel L, Capdevila C, Binder P, Adolphe M (1994) Cytotoxicity of sulphur mustard on a human keratinocyte cell line: direct effects compared to conditioned-medium effects. Toxicol Lett 71:227–234
Virag L, Szabo C (2002) The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev 54:375–479
Wormser U, Nyska A (1991) Protective effect of O-phenanthroline against mechlorethamine toxicity in the rat liver-slice system and in the guinea pig skin. Arch Toxicol 65:666–670
Wormser U, Brodsky B, Proscura E, Foley JF, Jones T, Nyska A (2005) Involvement of tumor necrosis factor-alpha in sulfur mustard-induced skin lesion; effect of topical iodine. Arch Toxicol 79:660–670
Yourick JJ, Clark CR, Mitcheltree LW (1991) Niacinamide pretreatment reduces microvesicle formation in hairless guinea pigs cutaneously exposed to sulfur mustard. Fundam Appl Toxicol 17:533–542
Zhang J, Dawson VL, Dawson TM, Snyder SH (1994) Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science (Washington DC) 263:687–689
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Korkmaz, A., Yaren, H., Topal, T. et al. Molecular targets against mustard toxicity: implication of cell surface receptors, peroxynitrite production, and PARP activation. Arch Toxicol 80, 662–670 (2006). https://doi.org/10.1007/s00204-006-0089-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-006-0089-x