
Abstract
The marine bacteria of the Vibrionaceae family are significant from the point of view of their role in the marine geochemical cycle, as well as symbionts and opportunistic pathogens of aquatic animals and humans. The well-known pathogens of this group, Vibrio cholerae, V. parahaemolyticus, and V. vulnificus, are responsible for significant morbidity and mortality associated with a range of infections from gastroenteritis to bacteremia acquired through the consumption of raw or undercooked seafood and exposure to seawater containing these pathogens. Although generally regarded as susceptible to commonly employed antibiotics, the antimicrobial resistance of Vibrio spp. has been on the rise in the last two decades, which has raised concern about future infections by these bacteria becoming increasingly challenging to treat. Diverse mechanisms of antimicrobial resistance have been discovered in pathogenic vibrios, the most important being the membrane efflux pumps, which contribute to antimicrobial resistance and their virulence, environmental fitness, and persistence through biofilm formation and quorum sensing. In this review, we discuss the evolution of antimicrobial resistance in pathogenic vibrios and some of the well-characterized efflux pumps’ contributions to the physiology of antimicrobial resistance, host and environment survival, and their pathogenicity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Vibrionales of the phylum proteobacteria are represented by Gram-negative, curved bacteria that inhabit coastal marine environments and are either free-living or live in association with marine crustaceans (Ghenem et al. 2017; Baker-Austin et al. 2018). The cholera epidemic caused by Vibrio cholerae is as ancient as the human race. This bacterial microorganism caused seven known epidemics between 1816 and the present, killing millions (Sherman 2007; Deen et al. 2020). As it was known in ancient times, the blue death is associated with the massive cholera-associated diarrhea that turns an infected individual’s body into a blue tinge owing to extensive dehydration and consequential capillary rupture (Morris 2007; Wade 2022). The earliest documented cholera-like epidemic could be traced back to Greece in 400 BC (Kaper et al. 1995; Ramamurthy and Ghosh 2021). The true causes of pandemics that ravaged humanity from time immemorial were unknown until Louis Pasteur disproved the theory of abiogenesis in 1862 and unequivocally established the role of microbes in food spoilage and disease, and his counterpart Robert Koch in Germany discovered the causative agents of cholera, anthrax, diphtheria, and tuberculosis (Kaper et al. 1995; Blevins and Bronze 2010).
The causative agent of cholera, V. cholerae, is a non-halophilic member of the family Vibrionaceae with over 100 species, 12 of which are associated with human infections more often (Baker-Austin et al. 2018). The Vibrio species are oxidase- and catalase-positive, ferment glucose, produce indole from tryptophan, and are motile by polar flagella. The other important species that cause food-borne infections are V. parahaemolyticus and V. vulnificus. Vibrio spp. can cause opportunistic infections such as in wounds, ears, and eyes and septicemia. V. parahaemolyticus causes gastrointestinal infections associated with consuming raw or undercooked fish and shellfish. In contrast, V. vulnificus causes wound infections and septicemia. These Vibrio spp. are widely distributed in coastal-marine water, sediment, plankton, and aquatic animals (Colwell et al. 1977). Other Vibrio spp. that are involved in sporadic food-borne intestinal or extraintestinal infections include V. furnissi, V. mimicus, V. anguillarum, V. fluvialis, V. damsele, V. furnissi, and V. metschnikovii (Colwell et al. 1977; Morris 2003; Baker-Austin et al. 2018). V. fischeri is a well-known endosymbiont of light organs of squids, and V. harveyi is a pathogen of marine crustaceans, particularly shrimp. Other Vibrio species are also opportunistic pathogens of different life stages of fish and shellfish.
The vibrios, except V. cholerae and V. mimicus, are halophilic. Their numbers increase with increasing temperature in temperate waters, with the high numbers recorded during warm summer months when the temperature is > 25 °C, which also correlates with the higher rates of infections (Motes et al. 1998; Hounmanou et al. 2023). Infections are often associated with the consumption of raw seafood containing high (> 105 CFU/g) numbers of vibrios, particularly the filter-feeding bivalve mollusks, which accumulate bacteria to concentrations several folds higher than that in the surrounding water (Baker-Austin et al. 2018). Vibrios respond uniquely to unfavorable environmental conditions such as very low temperatures and high salinity by entering into a dormant state called “viable but non-culturable” (VBNC) state (Colwell 2000; Oliver 2010). In the VBNC state, bacteria are metabolically inactive. These bacteria do not grow on standard laboratory media but retain their ability to infect and cause disease (Huq and Colwell 1996).
Epidemiology of Vibrio spp.
Vibrio cholerae
V. cholerae remains the most devastating Vibrio spp., still responsible for water-borne infections and deaths in developing countries. About 3–5 million cholera cases with 100,000 deaths are reported annually (Deen et al. 2020; Saha and Ganguly 2021; CDC 2023). Cholera is endemic in 51 countries, particularly in Sub-Saharan Africa and Southeast Asia (Harris et al. 2012; Ali et al. 2015; Guillaume et al. 2018). Although over 200 serotypes of V. cholerae have been identified based on the lipopolysaccharide antigens (Morris 2003), the severe form of cholera known as “cholera gravis” is caused by V. cholerae O1, identified by its ability to agglutinate with the O1 antiserum. All other V. cholerae that do not agglutinate with O1 antiserum are termed “non-O1.” The pandemic strains of O1 are further grouped into “Classical” and “El Tor” biotypes (Kaper et al. 1995; Reidl and Klose 2002). Epidemiological studies suggest a succession of these biotypes in cholera pandemics. The Classical biotype caused the first six pandemics, while the ongoing pandemic involves the El Tor biotype (Mukhopadhyay et al. 2014). Based on the antigenic differences, these are further subdivided into Ogawa, Inaba, and Hikojima serotypes (Kaper et al. 1995). The severity of cholera caused by V. cholerae O1 is due to cholera toxin production (Kaper et al. 1995). The non-O1 strains do not produce cholera toxin; hence, they are non-pathogenic or cause mild gastrointestinal infections. However, certain strains are occasionally involved in severe cases of intestinal and extraintestinal infections (Bag et al. 2008; Mukherjee et al. 2014).
In 1992, a non-O1 serotype involved in a cholera outbreak similar to that caused by O1 V. cholerae was attributed to the serotype O139, which also produced the cholera toxin (Ramamurthy et al. 1993). This strain is the only non-O1 V. cholerae reported thus far to be capable of causing a severe form of cholera as the O1 serotype. Although V. cholerae O139 reportedly originated from the El Tor strain, they are quite distinct antigenically (Albert et al. 1993; Faruque et al. 2003b). Large cholera outbreaks occurred in India and Bangladesh due to the O139 serotype in 1992 (Albert et al. 1993; Nair et al. 1994). However, subsequently, this serotype has not been responsible for any new cholera cases, and the El Tor biotype has caused almost all cholera infections worldwide. Recently, Yemen has been the focal point of large outbreaks involving over 364,000 cases and 639 deaths in 2019 (Deen et al. 2020; UNHCR), while countries such as Afghanistan, DR Congo (Ingelbeen et al. 2019), Haiti (Rubin et al. 2022), Ethiopia, and Cameroon continue to report the most cases (Deen et al. 2020; WHO 2022; ECDC 2023).
The virulence of V. cholerae, by far, relies on its ability to elaborate cholera toxin, CT, an A-B type toxin with one A subunit and five B subunits, which exerts its lethal effects on the physiology of intestinal epithelial cells by NAD-ribosylation of the adenylate cyclase complex leading to uncontrolled production of cyclic AMP (cAMP) molecules (Kaper et al. 1995). This results in continuous loss of water and electrolytes from the cells, which manifests as the classical rice water stool of cholera. In addition to CT, the Zonula occludens toxin (Zot), the accessory cholera enterotoxin (Ace), and a core encoded pilin (Cep) are important for the virulence of V. cholerae. Other accessory virulence factors that contribute to the ability of V. cholerae successfully to colonize the intestinal epithelium include the toxin-coregulated pilus (TCP), regulatory proteins like ToxR, membrane porins, iron-regulated outer membrane proteins, the O-antigen of the lipopolysaccharide, and accessory colonization factors (Faruque et al. 2003a).
The non-O1, non-O139 V. cholerae (NOVC) serotypes are involved in intestinal and extraintestinal infections (Dutta et al. 2013; Deshayes et al. 2015; Vezzulli et al. 2020). Although NOVC lacks CT and TCP, they possess other putative virulence factors such as a heat-stable toxin (NAG-ST), a hemolysin (Hly), RTX toxins (Repeats in ToXin), hemagglutinin protease (HapA), toxin regulatory protein (ToxR), outer membrane proteins (Omp), and the type III (T3SS) and type VI (T6SS) secretion systems. However, these factors are not consistently present in all NOVC isolates (Bag et al. 2008; Ceccarelli et al. 2015; Neogi et al. 2019). Reported cases of NOVC bacteremia involve persons with predisposing conditions such as liver cirrhosis, diabetes, and malignancy, with seafood and water as major sources of infection (Chen et al. 2015; Deshayes et al. 2015). Most reported cases of NOVC involved acute gastroenteritis, biliary tract infection, and primary bacteremia. In contrast, occasionally, peritonitis, skin and soft tissue infection, and urinary tract infection have been reported (Chen et al. 2015).
In 1998, investigation of an unusual surge in the incidence of cholera-like infections involving non-O1 V. cholerae in Calcutta, India, revealed the involvement of serotypes such as O144, O11, O6, O8, O12, O19, O39, and O58 that exhibited distinct cytotoxic effect on CHO and HeLa cells, but uniformly lacked any virulence factor associated with O1/O139 V. cholerae (Sharma et al. 1998). Case studies have reported the involvement of non-O1 V. cholerae in pulmonary infection (Shannon and Kimbrough 2006), pneumonia (Marinello et al. 2017), septicemia with meningitis (Hao et al. 2015), necrotizing fasciitis (Ottaviani et al. 2011), and traumatic wound infection (Hirota et al. 2010).
Vibrio parahaemolyticus
V. parahaemolyticus is a Gram-negative, halophilic bacterium widespread in coastal waters and seafood. Fujino first discovered the microorganism following an outbreak of food poisoning in 1950 associated with consuming partially cooked sardine (shirasu) in Japan (Chakraborty et al. 1997). These microorganisms are natural inhabitants of inshore marine waters of temperatures above 15 °C, varying salinity and estuaries, and prevalent in warm summer (DePaola et al. 2003; Ndraha and Hsiao 2019; Fernández-Vélez et al. 2023).
V. parahaemolyticus is a major cause of seafood-borne illness worldwide, characterized by gastroenteritis and wound infections. The disease is distinguished by diarrhea, headache, vomiting, nausea, abdominal cramps, and low fever. Consumption of raw or partially cooked seafood, especially bivalves like oysters and clams, is frequently associated with food-borne infections caused by V. parahaemolyticus. The pathogenic strains of V. parahaemolyticus are distinguished by the presence of hemolysins, thermostable direct hemolysin (TDH) factor, and/ a TDH-related hemolysin (TRH) encoded by tdh and trh genes with about 70% nucleotide sequence similarity (Nishibuchi et al. 1985, 1989). A pandemic clone of O3:K6 harboring the tdh gene but not trh has been responsible for many outbreaks in Asia and the US since its first report from Kolkata, India, in the mid-nineties (Okuda et al. 1997).
Subsequently, other serotypes such as O1:KUT, O4:K68, and O1:K25 were also found to belong to the pandemic group of V. parahaemolyticus, presumably evolved from O3:K6 serotype since all these serovars were genetically indistinguishable based on their arbitrarily primed PCR, ribotyping and pulsed-field gel electrophoresis (PFGE) patterns (Okuda et al. 1997; Matsumoto et al. 2000; Okura et al. 2003; Chowdhury et al. 2004). The pandemic clones are characterized by specialized genetic traits that distinguish them from pre-pandemic strains of the same serovars, such as the presence of the tdh and the absence of the trh gene and a characteristic toxRS sequence (toxRS/new) with point mutations that can be used to detect the pandemic clones using a group-specific PCR (GS-PCR) (Matsumoto et al. 2000). Recently, a new type of V. parahaemolyticus, “O4:KUT-recAin,” with a new type of K antigen and a 25 kb genomic island inserted in the housekeeping gene recA has been reportedly a predominant cause of infections in China (Chen et al. 2020).
The tdh- and trh-positive V. parahaemolyticus are frequently encountered in marine fish and shellfish. Bivalve mollusks such as clams, oysters, scallops, etc., are important sources of human infections since these filter-feeding organisms accumulate V. parahaemolyticus in their muscle to levels much higher than in the surrounding water and sufficient to cause human infections (Deepanjali et al. 2005; HongYou et al. 2014; Mok et al. 2019; Hong To et al. 2020). Wild-caught and farmed shrimp are also important vehicles of pathogenic V. parahaemolyticus (Ayyappan et al. 2018; Changsen et al. 2023). V. parahaemolyticus has been recognized as the causative agent of acute hepatopancreatic necrosis disease in shrimp, further emphasizing this bacterium’s increasing importance as a zoonotic pathogen (Hong To et al. 2020).
Vibrio vulnificus
V. vulnificus can cause a range of infections, including gastroenteritis, sepsis, necrotizing fasciitis, and cellulitis, with a case fatality rate as high as 50%, particularly when susceptible individuals with underlying debilitations such as chronic liver disease and iron overloaded-condition (Kathleen et al. 2016; Heng et al. 2017). V. vulnificus is responsible for over 95% of the fatal cases of seafood-borne infections in the US (Kathleen et al. 2016). Nearly 25% of V. vulnificus infections are associated with the exposure of open wounds to seawater containing this pathogen (Bross et al. 2007).
Like all other Vibrio spp., V. vulnificus is widely distributed in coastal-marine water associated with the chitinous exoskeleton of marine crustaceans and gets concentrated to infectious levels in filter-feeding bivalve mollusks (Heng et al. 2017; Bonnin-Jusserand et al. 2019). While infections occur with raw or undercooked seafood consumption, wound infections leading to bacteremia can occur when exposed to seawater containing these bacteria or while handling seafood. The abundance of V. vulnificus increases with seawater warmer above 20 °C and at a moderate salinity of 15–25 ppt in temperate waters (Motes et al. 1998).
In contrast, in the tropics, V. vulnificus abundance is influenced more by salinity than the water temperature (Parvathi et al. 2004). At lower temperatures (< 13 °C), the bacterium enters into a viable but non-culturable (VBNC) state (Oliver 2010). Of three known biotypes of V. vulnificus, biotype 1 is predominantly responsible for human infections, biotype 2 is an eel pathogen, and biotype 3, a hybrid between biotypes 1 and 2 first reported in Israel, is a pathogen of freshwater fish such as tilapia and carps. However, the microorganism has been reported to cause human infections, such as severe soft tissue infections, with a relatively lower mortality rate than biotype 1 (Zaidenstein et al. 2008; Jones and Oliver 2009; Oliver et al. 2012). Climate change leading to an increase in seawater surface temperature is predicted to dramatically increase the cases of V. vulnificus infections in the future (Archer et al. 2023).
Unlike V. cholerae and V. parahaemolyticus, no specific virulence factor has been associated with the pathogenesis of V. vulnificus. Some of the important virulence factors include a cytotoxic pore-forming toxin (VvhA; V. vulnificus hemolysin), capsular polysaccharide (CPS), and a multifunctional autoprocessing repeats-in-toxin (MARTX) (Jones and Oliver 2009; Yuan et al. 2020; Li and Wang 2020). Other putative virulence factors include metalloproteases, collagenases and phospholipases, siderophores, fimbriae, and others (Gulig et al. 2005; Jones and Oliver 2009; Horseman and Surani 2011).
Antimicrobial resistance in Vibrio spp.
Antimicrobial agents are indispensable in human clinical medicine and have saved humankind from life-threatening microbial infections and continue to do so. Apart from being a therapeutic drug, these agents are also administered in sub-therapeutic doses among livestock, poultry, and aquaculture to prevent disease and promote growth (Lekshmi et al. 2017). Consequentially, imprudent use of these antimicrobial agents leads to the development and spread of bacterial antimicrobial resistance (Loo et al. 2020). While the use of antibiotics in aquaculture for the treatment of bacterial infections or as a prophylactic measure can promote the development of antibiotic resistance in Vibrio spp., the antibiotic residues in farm effluents can have a similar effect on the bacteria in the environment surrounding aquaculture farms. Although vibrios are usually sensitive to antimicrobials of veterinary and human importance (Kumar et al. 2017), there are incidence reports of multidrug-resistant vibrios with different antibiotic susceptibility profiles in clinical settings (Reyhanath and Ranjeet 2014; Tan et al. 2020). The antimicrobial resistance in vibrios in wild-caught and farmed fish and shellfish is a serious threat due to their zoonotic potential and the dissemination of such bacteria in the consumer community through the food chain (Martínez 2008).
The massive diarrheal condition observed in cholera patients causes dehydration and could be fatal if not treated early. Effective management of cholera involves oral rehydration, and antibiotic treatment is generally not warranted. However, antibiotics can drastically reduce the fecal shedding of V. cholerae and help prevent environmental contamination and transmission of cholera. V. cholerae is generally sensitive to frequently prescribed antimicrobials, including β-lactams, aminoglycosides, tetracycline, and quinolone antibiotics.
However, the incidences of antimicrobial resistance in cholera and non-cholera vibrios have increased (Costa et al. 2021). This condition can be attributed to their antibiotic exposure and the acquisition of antibiotic-resistance genes from other Gram-negative bacteria in the environment. Tetracycline and azithromycin have been the drugs of choice for treating cholera for close to four decades until the first emergence of tetracycline resistance in Bangladesh in 1979, in which the O1 isolates were also resistant to ampicillin, kanamycin, streptomycin, and trimethoprim-sulfamethoxazole (Glass et al. 1980).
On the other hand, several prior studies reported the isolation of antibiotic-resistant V. cholerae, such as biotype El Tor, from cholera cases (Kuwahara et al. 1967; Prescott et al. 1968). Rahal et al. (1973) reported V. cholerae resistant to tetracycline, chloramphenicol, sulphonamides, ampicillin, streptomycin, and kanamycin in Algeria (Rahal et al. 1973). Nearly 76% of V. cholerae El Tor strains isolated during the fourth epidemic outbreak in Tanzania were resistant to tetracycline, a phenomenon attributed to the extensive therapeutic and prophylactic use of tetracycline. (Towner et al. 1980). Following this, resistance to tetracycline and other commonly employed antibiotics in cholera therapy was reported from different geographical regions worldwide (Threlfall et al. 1980; Ichinose et al. 1986; Ouellette et al. 1988; Tabtieng et al. 1989; Saraswathi and Deodhar 1990; Dalsgaard et al. 2000).
V. cholerae O1 El Tor serotype Ogawa isolated in Honduras were resistant to tetracycline, trimethoprim-sulfamethoxazole, kanamycin, gentamicin, chloramphenicol, ampicillin, cephalothin, and doxycycline (Dubon et al. 1997). Over 75% of the V. cholerae O1 strains isolated in Kolkata, India, were resistant to tetracycline (Bhattacharya et al. 2011). Multidrug-resistant V. cholerae El Tor Biotype, Ogawa serotype resistant to tetracycline, trimethoprim, co-trimoxazole, nalidixic acid, polymyxin B, spectinomycin, streptomycin, and sulfamethoxazole was reported from a cholera outbreak from Odisha, Eastern India in 2010 (Jain et al. 2016). Similarly, V. cholerae O1 El Tor Ogawa isolated from 2012 to 2015 cholera outbreaks in Mozambique were 100% resistant to sulphamethoxazole-trimethoprim, while more than 90% of the isolates were resistant to ampicillin, nalidixic acid, chloramphenicol, and nitrofurantoin, and were susceptible to azithromycin and streptomycin (Dengo-Baloi et al. 2017). In recent times, frequent isolation of V. cholerae O1 and O139 strains resistant to all clinically employed drugs has been reported from different parts of the world (Verma et al. 2019; Chatterjee et al. 2020).
Antimicrobial resistance is being reported regularly among vibrios of pathogenic importance. The β-lactamase-associated resistance mechanisms were identified in V. harveyi, V. alginolyticus, and V. parahaemolyticus associated with seafood in Italy (Ottaviani et al. 2001). In Mexico, 70% of Vibrio isolates from penaeid shrimp were resistant to carbenicillin and ampicillin and harbored resistance plasmids (Molina-Aja et al. 2002). Multiple antimicrobial resistance was also reported in Vibrio strains from wastewater final effluents, indicating the spread of resistance genes in a sewage plant, posing a threat to communities dependent on such water bodies (Okoh and Igbinosa 2010).
An investigation on farmed fishes from South Korea during 2005–2007 reported drug resistance in V. parahaemolyticus and V. alginolyticus with maximum resistance against ampicillin (Oh et al. 2011). Multi-drug resistance was also observed in both strains, with a higher incidence in V. alginolyticus (Oh et al. 2011). Studies from across the globe have reported plasmid-mediated resistance against clinically relevant antibiotics in vibrios from wild-caught and farmed fish and shellfish (Manjusha and Sarita 2011; Reyhanath and Ranjeet 2014; Letchumanan et al. 2015; Loo et al. 2020). Antibiotic resistance with an average MAR index of 0.77 among the Vibrio isolates from crustaceans of retail markets has been reported from Egypt (Ahmed et al. 2018).
Several reports suggest that V. parahaemolyticus is becoming increasingly resistant to previously effective antibiotics such as tetracycline, aminoglycosides, cephalosporins, fluoroquinolones, and others (Elmahdi et al. 2016; Dutta et al. 2021; Grudlewska-Buda et al. 2023). Irrespective of the geographic region, resistance to ampicillin, penicillins, and tetracyclines is observed in V. parahaemolyticus worldwide (Elmahdi et al. 2016). V. parahaemolyticus isolated from seafood in Malaysia exhibits resistance to ampicillin, cefazolin, and penicillin. Resistance to more than one antibiotic was found in 90% of the isolates, with the MAR index ranging from 0.04 to 0.71 (Tan et al. 2020). As high as 24% of the seafood samples in Bulgaria were found contaminated with V. parahaemolyticus resistant to ampicillin, cefepime, and ceftazidime, with the MAR index ranging from 0.10 to 0.30. (Stratev et al. 2023). A study on the market samples of seafood sold in Vietnam reported 86% of V. parahaemolyticus isolates to be resistant to at least one antibiotic tested, with the highest resistance observed against ampicillin, followed by cefotaxime, ceftazidime, trimethoprim-sulfamethoxazole, and tetracycline (Vu et al. 2022).
Numerous studies have reported the isolation of V. parahaemolyticus resistant to antibiotics such as ampicillin and cephalothin (Silva et al. 2018), ampicillin, cefazolin, and penicillin (Tan et al. 2020), ampicillin, gentamicin, tetracycline, and fluoroquinolones (Lei et al. 2020), ampicillin, ampicillin/sulbactam, cefuroxime, cefoxitin, ceftazidime, cefepime, and colistin (Coutinho et al. 2019), cephalothin, cefoxitin and ceftazidime (da Silva et al. 2021), imipenem (Lee et al. 2018), amoxicillin/clavulanate, cefoxitin, cefotaxime, ceftazidime, cefuroxime, cefepime and trimethoprim/sulfamethoxazole in a New Delhi metallo-β-lactamase (blaNDM)-positive isolate (Briet et al. 2018), ampicillin, cefpodoxime, cefotaxime, ceftizoxime, tetracycline, ceftriaxone, ciprofloxacin and nalidixic acid (Parthasarathy et al. 2021).
V. vulnificus is susceptible to most antimicrobial agents such as β-lactams, third-generation cephalosporins, carbapenems, aminoglycosides, tetracyclines, fluoroquinolones, trimethoprim-sulfamethoxazole, and chloramphenicol. Nevertheless, the antimicrobial resistance in this pathogen is gradually increasing, with reports emerging on the isolation of strains resistant to clinically employed antibiotics such as ampicillin, doxycycline, tetracycline, aminoglycosides, macrolides, ciprofloxacin, amoxicillin, imipenem, aztreonam, and cephalosporins (Han et al. 2007; Baker-Austin et al. 2009; Elmahdi et al. 2016).
Mechanisms of antimicrobial resistance in Vibrio spp.
Early reports suggested the presence of a transmissible resistance (R) factor in V. cholerae (Prescott et al. 1968; Rahal et al. 1973; Hedges and Jacob 1975; Yokota and Kuwahara 1977). Subsequent studies on the antibiotic susceptibility of V. cholerae, a self-transmissible 62-kb, chromosomally integrating genetic element called the SXT element, which carried resistance genes to sulfamethoxazole, trimethoprim, and streptomycin was found in serotype O139 (Waldor et al. 1996). The genetic mechanisms of antimicrobial resistance identified in clinical and environmental isolates of V. cholerae include mobile genetic elements such as plasmids, transposons, integrons, and integrating conjugative elements (ICEs) (Verma et al. 2019; Das et al. 2020). V. cholerae O1 El Tor strains isolated from outbreaks in Siberia and the Russian Far East carried SXT/R391 ICEs and genes such as strA, strB (streptomycin), catB9/floR (chloramphenicol/ florfenicol), sul2 (sulfonamide), dfrA1/ dhfR (trimethoprim, and cotrimoxazole), and tet(A) (tetracycline) (Gladkikh et al. 2020). The multidrug-resistant (MDR) V. cholerae El Tor Biotype Ogawa serotype isolates from the 2010 outbreak in Odisha, India, carried elements like class I integron, SXT element, aadA2 (aminoglycoside resistance), and dfrA1 (trimethoprim resistance) genes (Jain et al. 2016). The whole genome sequence (WGS) of an MDR V. cholerae strain contained genetic elements coding for resistance to multiple antibiotics such as sulfonamide (sul2), trimethoprim (dfrA1), chloramphenicol (catB9), florfenicol (floR), and tetracycline [tet(34)] (Mevada et al. 2023). Novel genomic islands identified in V. cholerae non-O1/non-O139 carried mobile genetic elements such as Class 1 integrons and quinolone resistance determinants (Morita et al. 2020).
The β-lactam genes such as blaCTX-M, blaTEM, and blaSHV have been detected in Vibrio isolates recovered from bivalves sold in retail markets (Dahanayake et al. 2020). The ESBL (extended-spectrum β-lactamase) genes blaCTX-M (87.5%), blaTEM (40.6%), and blaSHV (21.8%) were reported in Vibrio isolates from raw mussels in Korea (Hossain et al. 2020; Dahanayake et al. 2020). In another study, the β-lactamase encoding blaSHV genes were detected in Vibrio isolates from water samples coast off North-East Sardina. At the same time, none of the Vibrio spp. harbored the blaTEM gene (Zanetti et al. 2001). In India, the blaTEM gene in Vibrio isolates from estuaries, shrimp farms, and seafood samples has been reported (Silvester et al. 2019). While the blaCTX-M was not detected in any isolates, the blaNDM gene was found in 13.3% of the strains. A recent study made the first report of plasmid-encoded colistin resistance mcr-1 in a virulent strain of V. parahaemolyticus isolated from a shrimp sample in Hong Kong, China (Lei et al. 2019). The isolate carried blaCARB17 and quinolone resistance gene qnrVC5. Ampicillin resistance is widely prevalent in clinical and environmental isolates of V. parahaemolyticus, which, in addition to the production of β-lactamases, has been attributed to decreased synthesis of penicillin-binding proteins (Meng et al. 2023).
The quinolone resistance-determining regions (QRDR) of gyrA and parC and the plasmid-mediated quinolone resistance (PMQR) genes have been reported from fluoroquinolone-resistant V. parahaemolyticus with Ser-83-Ile, Ser-83-Phe substitutions in GyrA, and a Ser-85-Leu substitution in ParC (Lei et al. 2020). A novel class of chloramphenicol acetyltransferase, type C CAT or CATC, encoded by the catC gene identified in V. parahaemolyticus, confers varying levels of intrinsic resistance to chloramphenicol (Zhang et al. 2019). Recent studies have reported the occurrence of the blaNDM-harboring V. parahaemolyticus and V. vulnificus strains from seafood and the environment (Briet et al. 2018; Oyelade et al. 2018). The blaTEM and blaCMY genes are also found in V. parahaemolyticus and V. vulnificus isolates (Oyelade et al. 2018). The WGS of NDM-positive V. parahaemolyticus revealed the presence of multiple antibiotic resistance genes such as sul1, sul2, dfrA16, strA, strB and aadA2, floR and tet(A) (Briet et al. 2018). A study showed the multiple mediators of tetracycline resistance in V. parahaemolyticus involving five different types of tet genes, viz. tet(34), tet(A), tet(B), tet(M), and tet(E) (Ye et al. 2023). Three different mutations (Q513K, S522L, and H526Y) in the rpoB gene of V. vulnificus have been attributed to high resistance to rifampicin (Cutugno et al. 2020). The ESBL-producing V. vulnificus isolated from imported seafood harbored a plasmid encoding ISEc9 upstream of the blaCTX-M-55 and qnrS2 genes (Nakayama et al. 2023).
Antimicrobial efflux pumps
The known species of Vibrio are resistant to multiple structurally distinct antimicrobial agents using a variety of disparate solute transporter systems. The transport of antimicrobial solutes across the membranes of living cells, including those of Vibrio spp., is conferred by integral membrane proteins, collectively called solute transporters (Stein 2012). Passive transport systems do not use biological energy to drive the passage of solutes and water across the biological membrane of the Vibrios (Sten-Knudsen 1978). Such transport mechanisms are driven by the energy of the solute or substrate gradients across the membrane. Active solute transporters, however, are driven by the energy stored in ATP released upon hydrolysis, as seen in primary active transport (Stein 1986), and the release of energy inherent in the membrane gradients of ions, as documented in secondary active transport, or co-transport, systems (West 1980; Stein 1986).
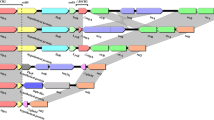
The efforts from the laboratory of Saier and colleagues led to the establishment and maintenance of the transporter classification database (TCD) https://www.tcdb.org/ (Saier et al. 2021). The TCD platform compiles transporters based on their functional properties and phylogenetic relationships between groups of homologous and related proteins. In general, transporters in living cells have been grouped into large superfamilies harboring sub-families with members that share similarities in sequence and structure, Fig. 1. Solute transporters are often evolutionarily conserved across all living taxa (Morita and Li 2016). The membrane transport proteins of Vibrio species fall into several large superfamilies (Table 1, Fig. 1). The ATP-binding cassette (ABC) superfamily encompasses the primary active transporters that utilize the hydrolysis of ATP to mediate the “uphill” transport of solutes across the membrane (Huda et al. 2003; Stephen et al. 2023). The efflux pumps of the secondary active transport type belong to the major facilitator superfamily (MFS), the resistance-nodulation-cell division (RND) superfamily, and the multidrug/oligosaccharidyl- lipid/polysaccharide (MOP) superfamily, the proteins of which function to extrude diverse solutes/substrates out of the cell and play critical roles in the antimicrobial resistance, virulence, and environmental persistence of Vibrio spp.

Efflux proteins belonging to different superfamilies/families in Vibrio spp. The ABC superfamily of efflux pumps are primary active transporters energized by hydrolysis of ATP. In contrast, all other groups of proteins belong to the secondary active transporters, which utilize an electropotential gradient of H+/Na+ (cations) across the membrane to power the movement of solutes
The multidrug/oligosaccharidyl-lipid/polysaccharide (MOP) superfamily of exporters includes members of the multidrug and toxic compound extrusion (MATE) (Kuroda and Tsuchiya 2009; Upadhyay et al. 2019; Claxton et al. 2021), the prokaryotic polysaccharide transporter (PST), the oligosaccharidyl-lipid flippase (OLF) from eukaryotes, and the mouse virulence factor (MVF) families of proteins (Hvorup et al. 2003). Likewise, the drug/metabolite transporter (DMT) superfamily contains related members of other transporter families, like the small multidrug resistance (SMR) family (Jack et al. 2001). The SMR family members QacEΔ1 and QacH have been studied in various V. cholerae non-O1 and V. parahaemolyticus isolates and have been shown to confer resistance to several quaternary ammonium compounds (Kazama et al. 1999; Ceccarelli et al. 2006). The SMR transport proteins have about 110 amino acids and four transmembrane domains, and they are postulated to oligomerize to form fully intact drug transporters (Jack et al. 2001).
The SMR and rhamnose transporter (RhaT) families from bacteria and the triose phosphate transporter (TPT) and the nucleotide-sugar transporter (NST) families from organelles of eukaryotic organisms and are discussed in detail elsewhere (Jack et al. 2001). More recently, the proteobacterial antimicrobial compound efflux (PACE) superfamily of proteins was discovered, harboring many multidrug efflux pumps energized by ion/substrate antiport, most uncharacterized (Hassan et al. 2021).
The major facilitator superfamily (MFS) consists of members that are passive and secondary active transporters from bacteria to humans (Henderson 1990, 1993; Griffith et al. 1992). The proteins of the resistance-nodulation-cell division (RND) class of secondary active transporters confer efflux of antimicrobial agents in bacteria, especially from the cells of the Vibrio genus of bacteria (Bina and Bina 2023). Together, the MFS and the RND transporters represent the most well-studied transporters of V. cholerae. This review article focuses on recent findings regarding the MFS, RND, and MATE transporters.
RND efflux pumps and their role in antimicrobial resistance and virulence
The RND efflux pumps are indispensable to the physiology of Vibrio spp. and play a critical role in their environmental persistence and pathogenesis (Stephen et al. 2022). V. cholerae, V. parahaemolyticus, and V. vulnificus genomes encode six, 12, and 11 RND efflux pumps, respectively (Taylor et al. 2012; Liu et al. 2022; Stephen et al. 2022). The unique structural architecture of the RND pumps consists of a tripartite protein assembly consisting of an outer membrane protein (OMP) and an inner membrane protein (IMP) connected by a periplasmic membrane fusion protein (MFP) (Fig. 2). The entire setup is organized into a continuous transmembrane channel, allowing the movement of substrates in an inward-outward fashion (protoplasm to the outside) without entering the periplasmic space (Nikaido 1994, 2011; Destoumieux-Garzón et al. 2014; Symmons et al. 2015). The antiport activity is vested with the inner membrane protein (IMP) energized by proton (H+) gradients. Although this membrane channel is expected to transport lipophilic substrates preferentially, the RND efflux pumps are generally non-specific and allow the extrusion of diverse substrates of all charge types (Nikaido 1998, 2011). As an exception to all known RND pumps, VexEF is energized by the electrochemical gradient of Na+ ions instead of H+ (Rahman et al. 2007).

Transport proteins from Vibrio bacteria. Depicted are known protein structures from species of the Vibrio genus, representative examples of which include a AcrAB-TolC of V. vulnificus (PDB 5V5S), b MsbA from V. cholerae (PBD 5TTP), c NorM (PDB 7PHP) from V. cholerae, d YddG, (PDB 5I20) a known DMT protein that is homologous to a putative permease from V. vulnificus, and e EmrD (PDB 2GFP), which is homologous to EmrD-3 of V. cholerae
The Vex group of the RND efflux pumps (VexAB, VexCD VexEF, VexGH, VexIJK, VexLM) from V. cholerae O1 are a group of bile-regulated proteins that confer resistance to bile and certain antimicrobial compounds. A whole-genome comparison of a non-O1 V. cholerae strain PS15 with V. cholerae O1 strain N16961 identified all of these efflux pumps except VexE (Mukherjee et al. 2014). The vexAB, vexCD, vexEF, vexGH, and vexIJK elements are on the large chromosome. At the same time, the vexLM gene is located on the small chromosome. All are organized into operons with an MFP-encoding gene upstream of the efflux pump (IMP) gene, and the operon does not contain the OMP-encoding gene (Bina and Bina 2023). Since bile acids are antimicrobial, bile resistance is necessary for successful intestinal colonization by a pathogenic bacterium such as V. cholerae. Early insights into the role of RND efflux pumps in bile resistance suggested the functional role of VexAB in intrinsic bile resistance in V. cholerae O1. At the same time, VexCD complemented this function of VexAB (Bina et al. 2006). The importance of RND efflux pumps in bile and antimicrobial resistance has been demonstrated using deletion mutants of V. cholerae. V. cholerae El Tor strain N16961 lacking all six RND efflux pumps was hypersensitive to bile, detergents, and antibiotics, produced significantly less cholera toxin and TCP protein, and was unable to colonize infant mouse intestines, suggesting the synergistic role of RND efflux pumps in the physiology and pathogenesis of V. cholerae (Bina et al. 2008).
Further, the absence of RND-mediated efflux resulted in the accumulation of metabolites in the periplasmic space, leading to the activation of periplasmic sensors involving two-component systems and the ToxR regulator as an adaptive response (Bina et al. 2018). This study conclusively showed the role of RND efflux pumps in regulating the expression of ToxR-mediated virulence factors, including TCP and the cholera toxin. However, this mechanism involves a complex network of proteins responding to stress in V. cholerae (Bina et al. 2018). Another study demonstrated the role of VexGH in Vibriobactin secretion in V. cholerae. Deleting the vexGH locus impaired homeostasis and reduced fitness under iron-limiting conditions (Kunkle et al. 2017; Du et al. 2018).
Without an identified OMP-encoding gene within the vex operon, researchers attempted to express V. cholerae Vex RND proteins in E. coli with the TolCvc outer membrane factor from V. cholerae non-O1 (Rahman et al. 2007). The VexAB and VexCD components could function using TolCvc and confer elevated MICs of cholates. VexAB-TolCvc also increased the MICs of erythromycin, novobiocin, rhodamine 6G, and TPPCl (Rahman et al. 2007). Similarly, VexEF-TolCvc was functional in an E. coli background and could efflux ethidium bromide in the presence of Na+. No definite antimicrobial substrate has been reported for the VexEF and VexLM pumps. However, VexEF showed efflux activity for bile, detergents, erythromycin, and novobiocin in an E. coli background (Rahman et al. 2007; Taylor et al. 2012; Bina and Bina 2023).
The efflux pumps homologous to VexAB, VexCD, and E. coli AcrAB (Fig. 2) encoded in the V. vulnificus genome have been shown to alter the bacterium’s antimicrobial susceptibilities. Increased susceptibility to erythromycin, ethidium bromide, acriflavine, and bile salts was observed in VexAB deletion mutants. Further, increased susceptibility was seen only to acriflavine in an AcrAB homolog mutant. No change in antimicrobial susceptibility was observed in a VexCD homolog deletion mutant, suggesting that these efflux pumps differ substantially in their preferred substrates and might contribute to other important physiological activities of the bacterium needing further investigation (Lee et al. 2015).
Twelve RND pumps of V. parahaemolyticus have been cloned in a hypersensitive E. coli background and characterized (Matsuo et al. 2007, 2013). Although VmeAB conferred significantly higher MICs of different antimicrobial agents in E. coli, the mutant strain of V. parahaemolyticus lacking VmeAB did not show the same resistance levels. However, a slight increase in the MICs was observed (Matsuo et al. 2007). In contrast, other efflux pumps, VmeCD, VmeEF, and VmeYZ, showed higher MICs in E. coli, and the effect was similar in deletion mutants of these efflux pump-encoding genes (Matsuo et al. 2013). Further, VmeAB, VmeCD, VmeEF, and VmeYZ co-expressed with VpoC, an outer membrane component in V. parahaemolyticus in E. coli, exhibited significantly higher MICs of various antimicrobials compared to the same efflux pumps functional with TolC of E. coli (Matsuo et al. 2013). VmeGHI, VmeJK, VmeLM, and VmeTUV were functional and could confer higher MICs only with VpoC. A null mutant lacking all 12 RND efflux pumps was highly sensitive to various antimicrobials. It showed a significantly lower fluid accumulation in rabbit ileal loops, suggesting that RND efflux pumps in V. parahaemolyticus contribute to its pathogenicity. These RND efflux pumps function to resist deoxycholates and other antimicrobials, are involved in the production or transport of siderophore vibrioferrin, and contribute to the pathogenicity of V. parahaemolyticus.
Proteomic profiling of the integral membrane proteins in V. parahaemolyticus revealed the involvement of the periplasmic adaptor subunit and the permease subunit of the RND efflux pump in virulence (Pérez-Acosta et al. 2018). The deletion of a membrane fusion protein, VmeL, belonging to the RND efflux pumps of V. parahaemolyticus resulted in lowered cytotoxicity towards HeLa cells, reduced virulence in a murine intraperitoneal infection assay, and an absence of lateral flagella hindering the motility (Liu et al. 2022). A ΔvmeL transcriptome analysis revealed a downregulation of genes related to the Type III Secretion 1 system (Liu et al. 2022), an important virulence factor involved in adhesion to the host cell (Wu et al. 2020; Liu et al. 2022). Similar studies in V. vulnificus involving the deletion of three RND efflux pumps VexBHZ showed increased cytotoxicity and biofilm formation (Lo et al. 2017). Analysis of whole genome sequences of Vibrio spp. can help identify new RND-type efflux pumps contributing to antimicrobial resistance (Lloyd et al. 2019).
MATE efflux pumps
The MATE group of ion/drug antiport systems is widely encountered in Vibrio spp. as well as in other Gram-negative and -positive bacteria. The first MATE-type transporter to be described was NorM from V. parahaemolyticus (Morita et al. 2000). Subsequently, proteins belonging to this group were reported from all domains of life performing essential tasks of detoxification and maintaining cellular homeostasis. These proteins actively extrude cationic substrates, including xenobiotics, drugs, toxic metabolites, hormones, and organic acids in plants (Kusakizako et al. 2020). The MATE efflux proteins fold into 12 transmembrane helices similar to the MFS transporters (Fig. 2). However, the topology of MATE proteins is distinctly different from the MFS family of efflux proteins, suggestive of different evolutionary origins of these two groups of membrane proteins (He et al. 2010; Kusakizako et al. 2020). Homologous MATE-type efflux pumps in other bacteria are NorM-VC, VcmA, and VcrM of V. cholerae (Huda et al. 2003), YdhE of Escherichia coli (Brown et al. 1999; Morita et al. 2000), BexA of Bacteroides thetaiotaomicron (Miyamae et al. 2001), PmpM from P. aeruginosa (He et al. 2004), NorM-NG of Neisseria gonorrhoeae (Rouquette-Loughlin et al. 2003), AbeM of Acinetobacter baumannii (Su et al. 2005), NorM of Erwinia amylovora (Burse et al. 2004), and KetM of K. pneumoniae (Ogawa et al. 2015). These transporters extrude cationic compounds such as DNA-intercalating dyes, detergents, aminoglycoside, and fluoroquinolone antibiotics. Although a majority of these are Na+-dependent, efflux pumps of this category either use H+ and Na+ simultaneously or H+ alone. For example, V. cholerae NorM can simultaneously couple with Na+ and H+, while AbeM of Acinetobacter baumannii is uniquely H+-dependent (Su et al. 2005; Jin et al. 2014). Similarly, VcmB, VcmD, and VcmH efflux pumps are Na+-dependent, while VcmN is Na+-independent (Begum et al. 2005). The homologous MATE efflux pumps are grouped into three clusters (Brown et al. 1999). Cluster 1 contains NorM of V. parahaemolyticus (NorM-VP), V. cholerae (NorM-VC), and YdhE of E. coli, while DinF of E. coli and its homologues such as VcmN of V. cholerae and PfMATE of Pyrococcus furiosus are in cluster 3 (Tanaka et al. 2013; Ogawa et al. 2015). Cluster 2 consists of eukaryotic MATE-type transporters. Mutational analyses have revealed that Asp-371 of NorM from V. cholerae is essential for substrate transport, and this residue is widely conserved across the Na+-dependent MATE transporters of cluster 1 (Otsuka et al. 2005; Ogawa et al. 2015). The cation-bound crystal structure of NorM-VC showed two amino acid residues, Glu-255 and Asp-371, located at the center of C-terminal lobes formed by TM7-12 constitute the substrate-binding pockets (He et al. 2010; Kusakizako et al. 2020). (Otsuka et al. 2005) showed that Asp-32, Glu-251, and Asp-367 in NorM-VP play important roles in the Na+-dependent drug transport process. Asp-367 in NorM-VP corresponds to Asp-371 of NorM-VC, and the replacement of Asp-367 with either alanine or asparagine severely impaired the drug transport activity of NorM-VP (Otsuka et al. 2005).
Lu and colleagues determined the crystal structure of Na+-coupled NorM-NG from Neisseria gonorrhoeae bound to three different substrates. They showed that Asp-41, Phe-265, Gln-284, Asp-355, and Asp-356 are essential for transport function, and these are supposedly conserved across prokaryotic and eukaryotic MATE-type transporters (Lu et al. 2013). Na+ and substrate bind distinct amino acids in NorM and bind simultaneously, in contrast to the general notion that Na+ and the substrate bind at two different stages corresponding to two different conformational statuses of the efflux protein (Lu et al. 2013). Asp-377 in NorA-NG and its corresponding residues Asp-367 and Asp-371 in NorA-VP and NorA-VC, respectively, have been shown to be critically important for substrate transport (Otsuka et al. 2005; Lu et al. 2013).
The crystal structure of another H+/drug antiport protein from V. cholerae, VcmN, and the subsequent molecular simulation studies have provided significant insights into the dynamics of ion coupling in this as well as other proteins of the DinF subfamily (Kusakizako et al. 2020; Castellano et al. 2021; Claxton et al. 2021). VcmN, originally proposed to be an H+/drug antiport with water bound to the N-lobe cavity, was later shown to exhibit Na+-dependent changes in conformation, suggesting that it could be Na+-driven like a majority of other MATE transporters (Kusakizako et al. 2020; Castellano et al. 2021; Claxton et al. 2021).
Two MATE efflux pumps of H- and D-type characterized from clinical isolates of V. fluvialis were dependent on Na+/K+ ions, and of these, the H-type pump extruded fluoroquinolones, ethidium bromide, and safranin, while the D-type extruded ethidium bromide only (Mohanty et al. 2012, 2021). Interestingly, unlike other MATE-type transporters, the H- and D-type pumps of V. fluvialis are made up of 10 and 11 TMS, respectively. However, molecular docking studies revealed a similar transport mechanism to NorM, with an aspartic acid residue in TM-1 acting as an ion binding site (Mohanty et al. 2021).
MFS efflux pumps
The multidrug efflux pumps of the MFS constitute a large group of related transporters from all known taxa (Pao et al. 1998; Saier et al. 1999). The MFS proteins share conserved sequences and structures, suggesting they share similar transport mechanisms across the membrane (Griffith et al. 1992; Varela and Griffith 1993). These transport systems confer resistance to antimicrobial and anticancer agents (Kumar et al. 2020; Hou et al. 2022). As such, these integral membrane proteins constitute suitable targets for modulation to restore the efficacy of chemotherapy for recalcitrant infections and cancer (Kumar et al. 2013b, 2016a; Stephen et al. 2022; Dhakne et al. 2023).
MFS efflux pumps of Vibrio spp.
According to the complete genome sequence of the V. cholerae O1 biovar El Tor toxigenic strain N16961, 18 genetic elements are thought to encode major facilitator superfamily transporters (Heidelberg et al. 2000). Several putative efflux pump-encoding genes from Vibrio spp. have been characterized physiologically (Stephen et al. 2022). The first MFS multidrug efflux pump from V. cholerae to be cloned, isolated, and studied for antimicrobial resistance is VceB, an integral membrane protein with 14 predicted transmembrane (TM) helices (Colmer et al. 1998). The vceB gene is encoded on the V. cholerae genome as part of the VceCRAB operon, where VceA and VceB are homologous to Escherichia coli proteins EmrA and EmrB, respectively (Lomovskaya and Lewis 1992). The VceCRAB locus encodes a multi-component membrane protein that effluxes multiple antimicrobial agents. VceC is considered an outer membrane protein by its functional similarity to OprM from Pseudomonas aeruginosa (Bai et al. 2010). VceR plays a functional role as a transcriptional regulator (Alatoom et al. 2007). VceA is a periplasmically-located membrane-fusion protein (Woolley et al. 2005). Therefore, the VceCRAB regulon encodes a tripartite antimicrobial efflux system consisting of VceC, VceA, and VceB (Woolley et al. 2005; Alatoom et al. 2007; Bai et al. 2010).
The second MFS multidrug efflux pump system discovered in V. cholerae is EmrD-3, physiologically studied in our laboratory (Smith et al. 2009). We found the emrD-3 gene on the chromosome of an O395 V. cholerae (Kumar et al. 2013a). The predicted structure of EmrD-3 encases another integral membrane protein, largely hydrophobic but with 12 TMs (Smith et al. 2009). We found that EmrD-3 confers transport of ethidium bromide across the membrane in a proton-driven manner and confers elevated resistance to multiple structurally distinct antimicrobial agents, such as linezolid, rifampin, erythromycin, and chloramphenicol, among others. Our laboratory performed a genomic comparative study of the emrD-3 gene from O395 with a toxigenic strain of V. cholerae N16961 and environmental isolate from Puget Sound, a strain denoted as PS15 (Kumar et al. 2013a; Mukherjee et al. 2014). We observed that emrD-3 was located on chromosome II of V. cholerae N16961 but absent from V. cholerae PS15 (Mukherjee et al. 2014). Due to the presence of EmrD-3 in toxigenic V. cholerae but missing in non-toxigenic counterparts, we anticipate that this multidrug efflux pump is a suitable target for antimicrobial resistance modulation to restore and enhance clinical treatment of severe cholera infections (Kumar et al. 2016a). Toward this, our laboratory discovered that both antimicrobial resistance levels and membrane drug transport activities by EmrD-3 are reduced by extracts of Allium sativum (garlic) and its pure bioactive component, allyl sulfide (Bruns et al. 2017). In the same study, we observed that A. sativum extract synergistically affects the V. cholerae growth with multiple antimicrobial agents.
From V. parahaemolyticus, the MFS transporter PvsC was discovered, showing the export of the iron siderophore vibrioferrin (Tanabe et al. 2003). This system is thought to enhance microbial virulence. The gene for PvsC is encoded on the pvsABCDE operon, and it is thought to play a role in the metabolism and efflux of Vibrio-specific siderophores (Tanabe et al. 2006).
Another set of MFS transporters from V. cholerae denoted as Mfs-1 through Mfs-5, five determinants totally, lose resistance to tetracycline and bile when the ORFs are deleted by mutation (Chen et al. 2013). The same study observed that expression programs of the mfs1-5 genes are controlled by a protein homologous to members of a family of well-characterized transcriptional regulators, namely LysR (Maddocks and Oyston 2008).
Role of efflux pumps in the physiology of Vibrio spp.
Using the deduced primary sequence (amino acid) of EmrD-3 as a query sequence in genomic databases, we found homologs in distantly related bacteria, like Proteus mirabilis and Bacillus cereus, but also in other species of the Vibrio genus, such as V. harveyi, V. vulnificus, and V. parahaemolyticus, indicating that these bacteria harbor conserved MFS multidrug resistance-conferring transporters as putative virulence factors (Smith et al. 2009; Mukherjee et al. 2014; Stephen et al. 2022).
Interestingly, the MFS VceB, EmrD-3, and Mfs1-5 transporters harbor elements of highly conserved sequence motifs, such as motif A, present on the loop between helices two and three of virtually all members of the MFS, and the antiporter motif, also called motif C, that resides in the fifth helix of MFS drug and multidrug efflux pumps (Henderson 1990; Varela et al. 1995; Colmer et al. 1998). The functional roles conferred by motifs A and C residues have been extensively studied in transporters of the MFS (Kumar et al. 2016b; Kakarla et al. 2017).
The signature sequence of motif A, “Gly-(X)3-Asp-Arg/Lys-X-Gly-Arg-Arg/Lys,” is highly conserved in virtually all members of the MFS, attesting to the functional importance of its residues in a variety of solute transporters from all living species including multidrug efflux pumps from known species of Vibrio bacteria (Griffith et al. 1992, 1994; Varela and Griffith 1993; Stephen et al. 2022). Thus, the homologous nature of the MFS transporters strongly suggests that molecular physiological studies of proteins from various species apply to the MFS transporters from the Vibrio genus. Structure–function studies of residues belonging to motif A have demonstrated critical roles in bacterial tetracycline efflux pumps, such as antimicrobial binding in TetA(C) of E. coli (McMurry et al. 1980), the substrate channel pathway (Yamaguchi et al. 1992), and elements of the transporter gate in TetA(B) (Someya et al. 2000). Residues of motif A were demonstrated to play important roles in stabilizing the transporter structure by salt-bridge formation in the YajR efflux pump of E. coli (Jiang et al. 2013). In various MFS transporters, motif A residues are proposed to mediate an essential role in conformational changes during substrate transport, such as in the LacY lactose symporter of E. coli (Varela and Wilson 1996). The LmrP drug transporter of Lactococcus lactis, with the protonation of acidic residues, influences a conformational switching system (Masureel et al. 2014). In the same study, the structure formed by residues of motif A appears to detect the energy status of the respiratory chain. This energy-sensing system should be relevant to the secondary active transporters of the MFS.
The laboratories of Skurray and Henderson identified a highly conserved consensus sequence in MFS efflux pumps that operate by an antiport process (Henderson 1990; Rouch et al. 1990). The signature sequence is “Gly-(X)8-Gly-(X)3-Gly-Pro-(X)2-Gly-Gly,” called the “antiporter motif” and later motif C (Varela and Griffith 1993; Varela et al. 1995; Ginn et al. 2000). Interestingly, when multiple amino acid sequence alignments were performed manually, the motif C appeared in symporters of the MFS, implicating a critical role for this conserved sequence in most proteins of the MFS (Yaffe et al. 2013). We showed by mutational analysis that the highly conserved glycine residue of the Gly-Pro dipeptide confers antimicrobial resistance in the TetA(C) tetracycline efflux pump encoded on the cloning plasmid pBR322 in host E. coli (Varela et al. 1995). The functional importance of the Gly-Pro dipeptide was demonstrated in TetA(L) and TetA(K) tetracycline efflux pumps from Staphylococcus aureus and Bacillus subtilis and shown to form a barrier to proton leakage, thus preventing extraneous ion-substrate energy coupling (Konishi et al. 1999; Jin and Krulwich 2002; De Jesus et al. 2005). Residues of motif C were shown to mediate conformational changes during the transport cycle of TetA(K) (Ginn et al. 2000), possibly influencing the direction of antimicrobial transport as predicted (Maiden et al. 1987; Pao et al. 1998; Saier et al. 1999). In the QacA multidrug efflux pump of S. aureus, residues of motif C bind antimicrobial substrates as they are translocated across the membrane (Hassan et al. 2006). An analysis of the structure–function relationship regarding motif C revealed its participation in forming a centrally-located substrate-binding cavity in the CaMdr1p drug transporter of antifungal agents from cells of Candida albicans, a eukaryote (Pasrija et al. 2007). Structurally speaking, in the mammalian vesicular monamine pump VMAT2, motif C forms an interface between the two large global bundles characteristic of the MFS transporters (Yaffe et al. 2013). Along these lines, residues of motif C form a molecular hinge structure that influences conformational changes during drug/ion antiport (Luo and Parsons 2010; Yaffe et al. 2013). Thus, the molecular structure formed by residues of motif C serves as a regulator of conformational switching during antimicrobial efflux in MFS transporters, a process relevant to virulence in cells of Vibrio species (Luo and Parsons 2010; Stephen et al. 2022).
Efflux pumps in biofilm formation and quorum sensing
The toxin-coregulated pilli (TCP) in V. cholerae is known to play a role in biofilm differentiation. The ΔTcpA (TCP pilin subunit) mutant in biofilms on the chitinous surface was undifferentiated, leading to high sensitivity to biocide like sodium dodecyl sulfate (SDS). Upon exposure to 0.2% SDS, the ΔTcpA mutant cells disassociated from the undifferentiated biofilm within 5 min, while the wildtype V. cholerae El Tor biofilm remained unaffected even after 30 min (Reguera and Kolter 2005). The RND efflux pump, like VexH in V. cholerae, is vital for the production of TCP (Taylor et al. 2012). Thus, the RND efflux pumps might also form biofilm on chitinous surfaces like copepods and zooplankton, which are associated with cholera outbreaks (Lipp et al. 2002). The biofilm formation capacity of V. parahaemolyticus was compromised upon deletion of VmeL, a membrane fusion protein belonging to the RND family of efflux pumps, conserved with more than 90% sequence similarity in V. diabolicus, V. harveyi, V. alginolyticus, V. chemaguriensis, and V. rotiferianus (Liu et al. 2022). The disruption of RND efflux pumps via deletion of the tolC-encoded component in V. cholerae leads to the activation of the family of LysR-type transcriptional regulator leuO (Weng et al. 2021); leuO is associated with biofilm formation in V. cholerae (Moorthy and Watnick 2005).
The relation between RND efflux pumps and quorum sensing (QS) has been investigated and proven in pathogens like Pseudomonas aeruginosa (Evans et al. 1998; Maseda et al. 2004), Escherichia coli (Rahmati et al. 2002), Salmonella enterica (Dawan et al. 2022), Acinetobacter baumannii (Abd El-Rahman et al. 2023). The QS molecules are known to act as a substrate for the efflux pumps, particularly those that cannot diffuse across the membrane passively (Rahmati et al. 2002; Piddock 2006). For V. cholerae, CqsA produces cholera autoinducer-1(CAI-1) (Wei et al. 2011), and LuxS produces autoinducer-2 (AI-2) (Bassler et al. 1993), known to play an essential role in virulence (Higgins et al. 2007) and biofilm formation (Hammer and Bassler 2003). A definitive part of RND efflux pumps in V. cholerae QS is yet to be established.
Conclusions
Cholera remains a serious healthcare concern on a global scale (Mandal et al. 2017). The causative agent of cholera, V. cholerae, and related species have acquired resistance to antimicrobial agents that are shared amongst them and unrelated microorganisms (Kitaoka et al. 2011). Of these resistance mechanisms, integral membrane transporters represent key multidrug resistance systems (Stephen et al. 2022). These bacterial multidrug efflux pumps make suitable targets for modulation to restore the clinical efficacy of antimicrobial agents in cases of severe infection (Kumar et al. 2016a; Stephen et al. 2022). Unfortunately, agents of resistance modulation are poorly understood, indicating that such studies are needed.
Future research programs can consider investigating new strategies for treating multidrug-resistant variants that cause severe disease (Varela and Kumar 2019). Thus, new modulators of antimicrobial efflux and other antimicrobial resistance mechanisms are needed. Putative modulators require translation to the clinical setting in regions with high cholera incidence and prevalence. Furthermore, studies of the physiological and molecular mechanisms of resistance modulation will provide new insights into the efficacy restoration of unrelated infections in which similar targets confound treatment.
Continued structure–function investigations are needed to fully understand the basic mechanisms of multidrug efflux in bacterial pathogens. For instance, the molecular mechanisms of energy transduction during primary and secondary active efflux pumps are not fully understood for antimicrobial efflux pumps in Vibrio species and related pathogens. Along these lines, a clear understanding is lacking of the relationships between the conformational changes that occur during transport and the mechanisms responsible for antimicrobial specificity. Interestingly, investigations of the highly conserved amino acid sequences shared by members of the various transporter superfamilies have provided insights into these mechanisms of solute translocation across the membrane. The MFS molecular hinge structure and its mode of operation during transport appear to provide a universal mechanism of solute transport. This characteristic may be beneficial not only for our basic understanding of transporter physiology but also as a means of addressing the problem of clinical resistance to antimicrobial-resistant bacterial pathogens.
Another field of study that is seemingly neglected involves our understanding of the transporter mechanisms that dictate substrate specificity. Some transporters are specific for a narrow range of substrates, like the MFS efflux pumps for the tetracycline class of antimicrobials (Nelson and Levy 2011). In contrast, many transporters are relatively more promiscuous, harboring many structurally dissimilar substrates (Alvarez-Ortega et al. 2013; Delmar et al. 2014; Ranaweera et al. 2015). It is poorly understood how these disparate transporter systems determine whether a given transporter possesses a single drug class of substrate specificity versus those of multiple drugs while simultaneously preventing leakage of ions or water that would dissipate the energetic driving forces of primary and secondary active transport.
Lastly, more work needs to be conducted regarding the nature of the pathway of substrates through the various transporters. How much multidrug transporters can accommodate the structurally distinctive antimicrobial agents is unclear. At the molecular physiological level, it remains fascinating how individual multidrug transporters appear to have a list of unique substrates for each. This issue of antimicrobial specificity remains relevant in the clinical setting and is a focus of future investigation.
References
Abd El-Rahman OA, Rasslan F, Hassan SS et al (2023) The RND efflux pump gene expression in the biofilm formation of Acinetobacter baumannii. Antibiotics 12:419. https://doi.org/10.3390/antibiotics12020419
Ahmed HA, El Bayomi RM, Hussein MA et al (2018) Molecular characterization, antibiotic resistance pattern and biofilm formation of Vibrio parahaemolyticus and V. cholerae isolated from crustaceans and humans. Int J Food Microbiol 274:31–37. https://doi.org/10.1016/j.ijfoodmicro.2018.03.013
Alatoom AA, Aburto R, Hamood AN, Colmer-Hamood JA (2007) VceR negatively regulates the vceCAB MDR efflux operon and positively regulates its own synthesis in Vibrio cholerae 569B. Can J Microbiol 53:888–900. https://doi.org/10.1139/W07-054
Albert MJ, Siddique AK, Islam MS et al (1993) Large outbreak of clinical cholera due to Vibrio cholerae non-O1 in Bangladesh. Lancet 341:704. https://doi.org/10.1016/0140-6736(93)90481-u
Ali M, Nelson AR, Lopez AL, Sack DA (2015) Updated global burden of cholera in Endemic Countries. PLoS Negl Trop Dis 9:e0003832. https://doi.org/10.1371/journal.pntd.0003832
Alvarez-Ortega C, Olivares J, Martínez JL (2013) RND multidrug efflux pumps: what are they good for? Front Microbiol 4:7. https://doi.org/10.3389/fmicb.2013.00007
Archer EJ, Baker-Austin C, Osborn TJ et al (2023) Climate warming and increasing Vibrio vulnificus infections in North America. Sci Rep 13:3893. https://doi.org/10.1038/s41598-023-28247-2
Ayyappan MV, Balange AK, Nayak BB, Kumar S (2018) Distribution of potentially pathogenic Vibrio parahaemolyticus in seafood and the aquatic environment of Mumbai, India. Fish Technol 55:205–211
Bag PK, Bhowmik P, Hajra TK et al (2008) Putative virulence traits and pathogenicity of Vibrio cholerae Non-O1, Non-O139 isolates from surface waters in Kolkata, India. Appl Environ Microbiol 74:5635–5644. https://doi.org/10.1128/AEM.00029-08
Bai J, Mosley L, Fralick JA (2010) Evidence that the C-terminus of OprM is involved in the assembly of the VceAB-OprM efflux pump. FEBS Lett 584:1493–1497. https://doi.org/10.1016/j.febslet.2010.02.066
Baker-Austin C, McArthur JV, Lindell AH et al (2009) Multi-site analysis reveals widespread antibiotic resistance in the marine pathogen Vibrio vulnificus. Microb Ecol 57:151–159. https://doi.org/10.1007/s00248-008-9413-8
Baker-Austin C, Oliver JD, Alam M et al (2018) Vibrio spp. infections. Nat Rev Dis Primers 4:8. https://doi.org/10.1038/s41572-018-0005-8
Bassler BL, Wright M, Showalter RE, Silverman MR (1993) Intercellular signalling in Vibrio harveyi: sequence and function of genes regulating expression of luminescence. Mol Microbiol 9:773–786. https://doi.org/10.1111/j.1365-2958.1993.tb01737.x
Begum A, Rahman MM, Ogawa W et al (2005) Gene cloning and characterization of four MATE family multidrug efflux pumps from Vibrio cholerae non-O1. Microbiol Immunol 49:949–957. https://doi.org/10.1111/j.1348-0421.2005.tb03690.x
Bhattacharya K, Kanungo S, Sur D et al (2011) Tetracycline-resistant Vibrio cholerae O1, Kolkata, India. Emerg Infect Dis 17:568–569. https://doi.org/10.3201/eid1703.101176
Bina XR, Bina JE (2023) Vibrio cholerae RND efflux systems: mediators of stress responses, colonization and pathogenesis. Front Cell Infect Microbiol 13:1203487. https://doi.org/10.3389/fcimb.2023.1203487
Bina JE, Provenzano D, Wang C et al (2006) Characterization of the Vibrio cholerae vexAB and vexCD efflux systems. Arch Microbiol 186:171–181. https://doi.org/10.1007/s00203-006-0133-5
Bina XR, Provenzano D, Nguyen N, Bina JE (2008) Vibrio cholerae RND family efflux systems are required for antimicrobial resistance, optimal virulence factor production, and colonization of the infant mouse small intestine. Infect Immun 76:3595–3605. https://doi.org/10.1128/IAI.01620-07
Bina XR, Howard MF, Taylor-Mulneix DL et al (2018) The Vibrio cholerae RND efflux systems impact virulence factor production and adaptive responses via periplasmic sensor proteins. PLoS Pathog 14:e1006804. https://doi.org/10.1371/journal.ppat.1006804
Blevins SM, Bronze MS (2010) Robert Koch and the ‘golden age’ of bacteriology. Int J Infect Dis 14:e744–e751. https://doi.org/10.1016/j.ijid.2009.12.003
Bonnin-Jusserand M, Copin S, Le Bris C et al (2019) Vibrio species involved in seafood-borne outbreaks (Vibrio cholerae, V. parahaemolyticus and V. vulnificus): review of microbiological versus recent molecular detection methods in seafood products. Crit Rev Food Sci Nutr 59:597–610. https://doi.org/10.1080/10408398.2017.1384715
Briet A, Helsens N, Delannoy S et al (2018) NDM-1-producing Vibrio parahaemolyticus isolated from imported seafood. J Antimicrob Chemother 73:2578–2579. https://doi.org/10.1093/jac/dky200
Bross MH, Soch K, Morales R, Mitchell RB (2007) Vibrio vulnificus infection: diagnosis and treatment. AFP 76:539–544
Brown MH, Paulsen IT, Skurray RA (1999) The multidrug efflux protein NorM is a prototype of a new family of transporters. Mol Microbiol 31:394–395. https://doi.org/10.1046/j.1365-2958.1999.01162.x
Bruns MM, Kakarla P, Floyd JT et al (2017) Modulation of the multidrug efflux pump EmrD-3 from Vibrio cholerae by Allium sativum extract and the bioactive agent allyl sulfide plus synergistic enhancement of antimicrobial susceptibility by A. sativum extract. Arch Microbiol 199:1103–1112. https://doi.org/10.1007/s00203-017-1378-x
Burse A, Weingart H, Ullrich MS (2004) NorM, an Erwinia amylovora multidrug efflux pump involved in in vitro competition with other epiphytic bacteria. Appl Environ Microbiol 70:693–703. https://doi.org/10.1128/AEM.70.2.693-703.2004
Castellano S, Claxton DP, Ficici E et al (2021) Conserved binding site in the N-lobe of prokaryotic MATE transporters suggests a role for Na+ in ion-coupled drug efflux. J Biol Chem 296:100262. https://doi.org/10.1016/j.jbc.2021.100262
CDC (2023) General Information, Cholera. https://www.cdc.gov/cholera/general/index.html. Accessed 13 Aug 2023
Ceccarelli D, Salvia AM, Sami J, Cappuccinelli P, Colombo MM (2006) New cluster of plasmid-located class 1 integrons in Vibrio cholerae O1 and a dfrA15 cassette-containing integron in Vibrio parahaemolyticus isolated in Angola. Antimicrob Agents Chemother 50(7):2493–2499
Ceccarelli D, Chen A, Hasan NA et al (2015) Non-O1/Non-O139 Vibrio cholerae carrying multiple virulence factors and V. cholerae O1 in the Chesapeake Bay Maryland. Appl Environ Microbiol 81:1909–1918. https://doi.org/10.1128/AEM.03540-14
Chakraborty S, Nair GB, Shinoda S (1997) Pathogenic vibrios in the natural aquatic environment. Rev Environ Health 12:63–80. https://doi.org/10.1515/REVEH.1997.12.2.63
Chang G (2003) Structure of MsbA from Vibrio cholera: a multidrug resistance ABC transporter homolog in a closed conformation. J Mol Biol 330:419–430. https://doi.org/10.1016/s0022-2836(03)00587-4
Changsen C, Likhitrattanapisal S, Lunha K et al (2023) Incidence, genetic diversity, and antimicrobial resistance profiles of Vibrio parahaemolyticus in seafood in Bangkok and eastern Thailand. PeerJ 11:e15283. https://doi.org/10.7717/peerj.15283
Chatterjee P, Kanungo S, Bhattacharya SK, Dutta S (2020) Mapping cholera outbreaks and antibiotic resistant Vibrio cholerae in India: an assessment of existing data and a scoping review of the literature. Vaccine 38:A93–A104. https://doi.org/10.1016/j.vaccine.2019.12.003
Chen S, Wang H, Katzianer DS et al (2013) LysR family activator-regulated major facilitator superfamily transporters are involved in Vibrio cholerae antimicrobial compound resistance and intestinal colonisation. Int J Antimicrob Agents 41:188–192. https://doi.org/10.1016/j.ijantimicag.2012.10.008
Chen Y-T, Tang H-J, Chao C-M, Lai C-C (2015) Clinical manifestations of non-O1 Vibrio cholerae infections. PLoS ONE 10:e0116904. https://doi.org/10.1371/journal.pone.0116904
Chen X, Li Y, Yao W et al (2020) A new emerging serotype of Vibrio parahaemolyticus in China is rapidly becoming the main epidemic strain. Clin Microbiol Infect 26:644.e1-644.e7. https://doi.org/10.1016/j.cmi.2019.09.024
Chowdhury NR, Stine OC, Morris JG, Nair GB (2004) Assessment of evolution of pandemic Vibrio parahaemolyticus by multilocus sequence typing. J Clin Microbiol 42:1280–1282. https://doi.org/10.1128/jcm.42.3.1280-1282.2004
Claxton DP, Jagessar KL, Mchaourab HS (2021) Principles of alternating access in multidrug and toxin extrusion (MATE) transporters. J Mol Biol 433:166959. https://doi.org/10.1016/j.jmb.2021.166959
Colmer JA, Fralick JA, Hamood AN (1998) Isolation and characterization of a putative multidrug resistance pump from Vibrio cholerae. Mol Microbiol 27:63–72. https://doi.org/10.1046/j.1365-2958.1998.00657.x
Colwell RR (2000) Viable but nonculturable bacteria: a survival strategy. J Infect Chemother 6:121–125. https://doi.org/10.1007/PL00012151
Colwell RR, Kaper J, Joseph SW (1977) Vibrio cholerae, Vibrio parahaemolyticus, and other vibrios: occurrence and distribution in Chesapeake Bay. Science 198:394–396
Costa WF, Giambiagi-deMarval M, Laport MS (2021) Antibiotic and heavy metal susceptibility of non-cholera Vibrio isolated from Marine Sponges and Sea Urchins: could they pose a potential risk to public health? Antibiotics (Basel) 10:1561. https://doi.org/10.3390/antibiotics10121561
Coutinho FH, Tschoeke DA, Clementino MM et al (2019) Genomic basis of antibiotic resistance in Vibrio parahaemolyticus strain JPA1. Mem Inst Oswaldo Cruz 114:e190053. https://doi.org/10.1590/0074-02760190053
Cutugno L, Mc Cafferty J, Pané-Farré J et al (2020) rpoB mutations conferring rifampicin-resistance affect growth, stress response and motility in Vibrio vulnificus. Microbiology (Reading) 166:1160–1170. https://doi.org/10.1099/mic.0.000991
da Silva LV, Ossai S, Chigbu P, Parveen S (2021) Antimicrobial and genetic profiles of Vibrio vulnificus and Vibrio parahaemolyticus isolated From the Maryland Coastal Bays. United States Front Microbiol 12:676249. https://doi.org/10.3389/fmicb.2021.676249
Dahanayake PS, Hossain S, Wickramanayake MVKS et al (2020) Manila clam (Ruditapes philippinarum) marketed in Korea as a source of vibrios harbouring virulence and β-lactam resistance genes. Lett Appl Microbiol 71:46–53. https://doi.org/10.1111/lam.13229
Dalsgaard A, Forslund A, Petersen A et al (2000) Class 1 integron-borne, multiple-antibiotic resistance encoded by a 150-kilobase conjugative plasmid in epidemic Vibrio cholerae O1 strains isolated in Guinea-Bissau. J Clin Microbiol 38:3774–3779. https://doi.org/10.1128/JCM.38.10.3774-3779.2000
Das B, Verma J, Kumar P et al (2020) Antibiotic resistance in Vibrio cholerae: understanding the ecology of resistance genes and mechanisms. Vaccine 38(Suppl 1):A83–A92. https://doi.org/10.1016/j.vaccine.2019.06.031
Dawan J, Li Y, Lu F et al (2022) Role of efflux pump-mediated antibiotic resistance in quorum sensing-regulated biofilm formation by Salmonella typhimurium. Pathogens 11:147. https://doi.org/10.3390/pathogens11020147
De Jesus M, Jin J, Guffanti AA, Krulwich TA (2005) Importance of the GP dipeptide of the antiporter motif and other membrane-embedded proline and glycine residues in tetracycline efflux protein Tet(L). Biochemistry 44:12896–12904. https://doi.org/10.1021/bi050762c
Deen J, Mengel MA, Clemens JD (2020) Epidemiology of cholera. Vaccine 38:A31–A40. https://doi.org/10.1016/j.vaccine.2019.07.078
Deepanjali A, Kumar HS, Karunasagar I (2005) Seasonal variation in abundance of total and pathogenic Vibrio parahaemolyticus bacteria in oysters along the southwest coast of India. Appl Environ Microbiol 71:3575–3580. https://doi.org/10.1128/AEM.71.7.3575-3580.2005
Delmar JA, Su CC, Yu EW (2014) Bacterial multidrug efflux transporters. Annu Rev Biophys 43:93–117. https://doi.org/10.1146/annurev-biophys-051013-022855
Dengo-Baloi LC, Semá-Baltazar CA, Manhique LV et al (2017) Antibiotics resistance in El Tor Vibrio cholerae 01 isolated during cholera outbreaks in Mozambique from 2012 to 2015. PLoS ONE 12:e0181496. https://doi.org/10.1371/journal.pone.0181496
DePaola A, Nordstrom JL, Bowers JC et al (2003) Seasonal abundance of total and pathogenic Vibrio parahaemolyticus in Alabama oysters. Appl Environ Microbiol 69:1521–1526
Deshayes S, Daurel C, Cattoir V et al (2015) Non-O1, non-O139 Vibrio cholerae bacteraemia: case report and literature review. Springerplus 4:575. https://doi.org/10.1186/s40064-015-1346-3
Destoumieux-Garzón D, Duperthuy M, Vanhove AS et al (2014) Resistance to antimicrobial peptides in vibrios. Antibiotics (basel) 3:540–563. https://doi.org/10.3390/antibiotics3040540
Dhakne P, Pillai M, Mishra S et al (2023) Refinement of safety and efficacy of anti-cancer chemotherapeutics by tailoring their site-specific intracellular bioavailability through transporter modulation. Biochim Biophys Acta 1878:188906. https://doi.org/10.1016/j.bbcan.2023.188906
Du D, Wang-Kan X, Neuberger A et al (2018) Multidrug efflux pumps: structure, function and regulation. Nat Rev Microbiol 16:523–539. https://doi.org/10.1038/s41579-018-0048-6
Dubon JM, Palmer CJ, Ager AL et al (1997) Emergence of multiple drug-resistant Vibrio cholerae O1 in San Pedro Sula. Honduras Lancet 349:924. https://doi.org/10.1016/s0140-6736(05)62699-2
Dutta D, Chowdhury G, Pazhani GP et al (2013) Vibrio cholerae non-O1, non-o139 serogroups and cholera-like diarrhea, Kolkata, India. Emerg Infect Dis 19:464–467. https://doi.org/10.3201/eid1903.121156
Dutta D, Kaushik A, Kumar D, Bag S (2021) Foodborne pathogenic vibrios: antimicrobial resistance. Front Microbiol 12:638331. https://doi.org/10.3389/fmicb.2021.638331
ECDC (2023) Cholera worldwide overview. https://www.ecdc.europa.eu/en/all-topics-z/cholera/surveillance-and-disease-data/cholera-monthly. Accessed 14 Aug 2023
Elmahdi S, DaSilva LV, Parveen S (2016) Antibiotic resistance of Vibrio parahaemolyticus and Vibrio vulnificus in various countries: a review. Food Microbiol 57:128–134. https://doi.org/10.1016/j.fm.2016.02.008
Evans K, Passador L, Srikumar R et al (1998) Influence of the MexAB-OprM multidrug efflux system on quorum sensing in Pseudomonas aeruginosa. J Bacteriol 180:5443–5447
Faruque SM, Kamruzzaman M, Meraj IM et al (2003a) Pathogenic potential of environmental Vibrio cholerae strains carrying genetic variants of the toxin-coregulated pilus pathogenicity Island. Infect Immun 71:1020–1025. https://doi.org/10.1128/IAI.71.2.1020-1025.2003
Faruque SM, Sack DA, Sack RB et al (2003b) Emergence and evolution of Vibrio cholerae O139. Proc Natl Acad Sci U S A 100:1304–1309. https://doi.org/10.1073/pnas.0337468100
Fernández-Vélez I, Bidegain G, Ben-Horin T (2023) Predicting the growth of Vibrio parahaemolyticus in oysters under varying ambient temperature. Microorganisms 11:1169. https://doi.org/10.3390/microorganisms11051169
Ghenem L, Elhadi N, Alzahrani F, Nishibuchi M (2017) Vibrio parahaemolyticus: a review on distribution, pathogenesis, virulence determinants and epidemiology. Saudi J Med Med Sci 5:93–103. https://doi.org/10.4103/sjmms.sjmms_30_17
Ginn SL, Brown MH, Skurray RA (2000) The TetA (K) tetracycline/H+ antiporter from Staphylococcus aureus: mutagenesis and functional analysis of motif C. J Bacteriol 182:1492–1498
Gladkikh AS, Feranchuk SI, Ponomareva AS et al (2020) Antibiotic resistance in Vibrio cholerae El Tor strains isolated during cholera complications in Siberia and the Far East of Russia. Infect Genet Evol 78:104096. https://doi.org/10.1016/j.meegid.2019.104096
Glass RI, Huq I, Alim AR, Yunus M (1980) Emergence of multiply antibiotic-resistant Vibrio cholerae in Bangladesh. J Infect Dis 142:939–942. https://doi.org/10.1093/infdis/142.6.939
Griffith JK, Baker ME, Rouch DA et al (1992) Membrane transport proteins: implications of sequence comparisons. Curr Opin Cell Biol 4:684–695. https://doi.org/10.1016/0955-0674(92)90090-y
Griffith JK, Cuellar DH, Fordyce CA et al (1994) Structure and function of the class C tetracycline/H+ antiporter: three independent groups of phenotypes are conferred by TetA (C). Mol Membr Biol 11:271–277. https://doi.org/10.3109/09687689409160437
Grudlewska-Buda K, Bauza-Kaszewska J, Wiktorczyk-Kapischke N et al (2023) Antibiotic resistance in selected emerging bacterial foodborne pathogens-an issue of concern? Antibiotics (basel) 12:880. https://doi.org/10.3390/antibiotics12050880
Guillaume Y, Ternier R, Vissieres K et al (2018) Responding to cholera in Haiti: implications for the national plan to eliminate cholera by 2022. J Infect Dis 218:S167–S170. https://doi.org/10.1093/infdis/jiy491
Gulig PA, Bourdage KL, Starks AM (2005) Molecular pathogenesis of Vibrio vulnificus. J Microbiol 43:118–131
Hammer BK, Bassler BL (2003) Quorum sensing controls biofilm formation in Vibrio cholerae. Mol Microbiol 50:101–104
Han F, Walker RD, Janes ME et al (2007) Antimicrobial susceptibilities of Vibrio parahaemolyticus and Vibrio vulnificus isolates from Louisiana Gulf and retail raw Oysters. Appl Environ Microbiol 73:7096–7098. https://doi.org/10.1128/AEM.01116-07
Hao Y, Wang Y, Bi Z et al (2015) A case of non-O1/non-O139 Vibrio cholerae septicemia and meningitis in a neonate. Int J Infect Dis 35:117–119. https://doi.org/10.1016/j.ijid.2015.05.004
Harris JB, LaRocque RC, Qadri F et al (2012) Cholera. Lancet 379:2466–2476. https://doi.org/10.1016/S0140-6736(12)60436-X
Hassan KA, Galea M, Wu J et al (2006) Functional effects of intramembranous proline substitutions in the staphylococcal multidrug transporter QacA. FEMS Microbiol Lett 263:76–85
Hassan KA, Maher C, Elbourne LD et al (2021) Increasing the PACE of characterising novel transporters by functional genomics. Curr Opin Microbiol 64:1–8. https://doi.org/10.1016/j.mib.2021.08.005
He G-X, Kuroda T, Mima T et al (2004) An H+-coupled multidrug efflux pump, PmpM, a member of the MATE family of transporters, from Pseudomonas aeruginosa. J Bacteriol 186:262–265. https://doi.org/10.1128/JB.186.1.262-265.2004
He X, Szewczyk P, Karyakin A et al (2010) Structure of a cation-bound multidrug and toxic compound extrusion transporter. Nature 467:991–994. https://doi.org/10.1038/nature09408
Hedges RW, Jacob AE (1975) A 98 megadalton R factor of compatibility group C in a Vibrio cholerae El Tor isolate from Southern U.S.S.R. Microbiology 89:383–386. https://doi.org/10.1099/00221287-89-2-383
Heidelberg JF, Eisen JA, Nelson WC et al (2000) DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406:477–483. https://doi.org/10.1038/35020000
Henderson PJ (1990) The homologous glucose transport proteins of prokaryotes and eukaryotes. Res Microbiol 141:316–328. https://doi.org/10.1016/0923-2508(90)90005-b
Henderson PJ (1993) The 12-transmembrane helix transporters. Curr Opin Cell Biol 5:708–721. https://doi.org/10.1016/0955-0674(93)90144-f
Heng S-P, Letchumanan V, Deng C-Y et al (2017) Vibrio vulnificus: an environmental and clinical burden. Front Microbiol. https://doi.org/10.3389/fmicb.2017.00997
Higgins DA, Pomianek ME, Kraml CM et al (2007) The major Vibrio cholerae autoinducer and its role in virulence factor production. Nature 450:883–886
Hirota D, Sachdev A, Thrupp L et al (2010) Traumatic arm wound infected with Vibrio cholerae in a non-immunocompromised Host. https://www.hmpgloballearningnetwork.com/site/wounds/case-report-and-brief-review/traumatic-arm-wound-infected-vibrio-cholerae-non. Accessed 9 Oct 2023
Hong To TT, Yanagawa H, Khanh Thuan N et al (2020) Prevalence of Vibrio parahaemolyticus causing acute hepatopancreatic necrosis disease of shrimp in shrimp, Molluscan shellfish and water samples in the Mekong Delta, Vietnam. Biology 9:312. https://doi.org/10.3390/biology9100312
HongYou C, LiHong T, Min C et al (2014) Investigation of diversity of Vibrio parahaemolyticus in molluscs. Dis Surveill 29:522–527
Horseman MA, Surani S (2011) A comprehensive review of Vibrio vulnificus: an important cause of severe sepsis and skin and soft-tissue infection. Int J Infect Dis 15:e157–e166. https://doi.org/10.1016/j.ijid.2010.11.003
Hossain S, Wickramanayake MVKS, Dahanayake PS, Heo GJ (2020) Occurrence of virulence and extended-spectrum β-lactamase determinants in Vibrio spp. isolated from marketed hard-shelled mussel (Mytilus coruscus). Microb Drug Resist 26:391–401. https://doi.org/10.1089/mdr.2019.0131
Hou Z, Gangjee A, Matherly LH (2022) The evolving biology of the proton-coupled folate transporter: new insights into regulation, structure, and mechanism. FASEB J 36:e22164. https://doi.org/10.1096/fj.202101704R
Hounmanou YMG, Engberg J, Bjerre KD, et al (2023) Correlation of high seawater temperature with Vibrio and Shewanella infections, Denmark, 2010–2018 - Volume 29, Number 3—March 2023 - Emerg Infect Dis J CDC. https://doi.org/10.3201/eid2903.221568
Huda MN, Chen J, Morita Y et al (2003) Gene cloning and characterization of VcrM, a Na+-coupled multidrug efflux pump, from Vibrio cholerae non-O1. Microbiol Immunol 47:419–427. https://doi.org/10.1111/j.1348-0421.2003.tb03379.x
Huq A, Colwell RR (1996) A microbiological paradox: viable but nonculturable bacteria with special reference to Vibrio cholerae. J Food Prot 59:96–101. https://doi.org/10.4315/0362-028X-59.1.96
Hvorup RN, Winnen B, Chang AB et al (2003) The multidrug/oligosaccharidyl-lipid/polysaccharide (MOP) exporter superfamily. Eur J Biochem 270:799–813
Ichinose Y, Ehara M, Watanabe S et al (1986) The characterization of Vibrio cholerae isolated in Kenya in 1983. J Trop Med Hyg 89:269–276
Ingelbeen B, Hendrickx D, Miwanda B et al (2019) Recurrent cholera outbreaks, Democratic Republic of the Congo, 2008–2017. Emerg Infect Dis 25:856
Jack DL, Yang NM, Saier MH Jr (2001) The drug/metabolite transporter superfamily. Eur J Biochem 268:3620–3639
Jain M, Kumar P, Goel AK (2016) Emergence of tetracycline resistant Vibrio cholerae O1 Biotype El Tor serotype Ogawa with classical ctxB gene from a cholera outbreak in Odisha, Eastern India. J Pathog 2016:1695410. https://doi.org/10.1155/2016/1695410
Jiang D, Zhao Y, Wang X et al (2013) Structure of the YajR transporter suggests a transport mechanism based on the conserved motif A. Proc Natl Acad Sci USA 110:14664–14669. https://doi.org/10.1073/pnas.1308127110
Jin J, Krulwich TA (2002) Site-directed mutagenesis studies of selected motif and charged residues and of cysteines of the multifunctional tetracycline efflux protein Tet(L). J Bacteriol 184:1796–1800. https://doi.org/10.1128/jb.184.6.1796-1800.2002
Jin Y, Nair A, van Veen HW (2014) Multidrug transport protein NorM from Vibrio cholerae simultaneously couples to sodium- and proton-motive force. J Biol Chem 289:14624–14632. https://doi.org/10.1074/jbc.M113.546770
Jones MK, Oliver JD (2009) Vibrio vulnificus: disease and pathogenesis. Infect Immun 77:1723–1733. https://doi.org/10.1128/IAI.01046-08
Kakarla P, Floyd J, Mukherjee M et al (2017) Inhibition of the multidrug efflux pump LmrS from Staphylococcus aureus by cumin spice Cuminum cyminum. Arch Microbiol 199:465–474. https://doi.org/10.1007/s00203-016-1314-5
Kaper JB, Morris JG, Levine MM (1995) Cholera. Clin Microbiol Rev 8:48–86
Kathleen MM, Samuel L, Felecia C, et al (2016) Antibiotic resistance of diverse bacteria from aquaculture in Borneo. Int J Microbiol 2016:2164761. https://www.hindawi.com/journals/ijmicro/2016/2164761/.
Kazama H, Hamashima H, Sasatsu M, Arai T (1999) Characterization of the antiseptic-resistance gene qacEΔ1 isolated from clinical and environmental isolates of Vibrio parahaemolyticus and Vibrio cholerae non-O1. FEMS Microbiol Lett 174(2):379–384
Kitaoka M, Miyata ST, Unterweger D, Pukatzki S (2011) Antibiotic resistance mechanisms of Vibrio cholerae. J Med Microbiol 60:397–407. https://doi.org/10.1099/jmm.0.023051-0
Konishi S, Iwaki S, Kimura-Someya T, Yamaguchi A (1999) Cysteine-scanning mutagenesis around transmembrane segment VI of Tn10-encoded metal-tetracycline/H(+) antiporter. FEBS Lett 461:315–318. https://doi.org/10.1016/s0014-5793(99)01490-8
Kumar S, He G, Kakarla P et al (2016a) Bacterial multidrug efflux pumps of the major facilitator superfamily as targets for modulation. Infect Disord Drug Targets 16:28–43. https://doi.org/10.2174/1871526516666160407113848
Kumar S, Ranjana K, Sanford LM et al (2016b) Structural and functional roles of two evolutionarily conserved amino acid sequence motifs within solute transporters of the major facilitator superfamily. Trends Cell Mol Biol 11:41–53
Kumar S, Lindquist IE, Sundararajan A et al (2013a) Genome sequence of non-O1 Vibrio cholerae PS15. Genome Announc. https://doi.org/10.1128/genomeA.00227-12
Kumar S, Mukherjee MM, Varela MF (2013b) Modulation of bacterial multidrug resistance efflux pumps of the major facilitator superfamily. Int J Bacteriol 2013:204141. https://doi.org/10.1155/2013/204141
Kumar S, Lekshmi M, Parvathi A et al (2017) Antibiotic resistance in seafood-borne pathogens. In: Foodborne pathogens and antibiotic resistance. Wiley, pp 397–415. https://doi.org/10.1002/9781119139188.ch17
Kumar S, Lekshmi M, Parvathi A et al (2020) Functional and structural roles of the major facilitator superfamily bacterial multidrug efflux pumps. Microorganisms 8:266. https://doi.org/10.3390/microorganisms8020266
Kunkle DE, Bina XR, Bina JE (2017) The Vibrio cholerae VexGH RND efflux system maintains cellular homeostasis by effluxing Vibriobactin. Mbio. https://doi.org/10.1128/mBio.00126-17
Kuroda T, Tsuchiya T (2009) Multidrug efflux transporters in the MATE family. Biochim Biophys Acta 1794(5):763–768. https://doi.org/10.1016/j.bbapap.2008.11.012
Kusakizako T, Miyauchi H, Ishitani R, Nureki O (2020) Structural biology of the multidrug and toxic compound extrusion superfamily transporters. Biochim Et Biophys Acta BBA Biomembr 1862:183154. https://doi.org/10.1016/j.bbamem.2019.183154
Kuwahara S, Goto S, Kimura M, Abe H (1967) Drug-sensitivity of El Tor vibrio strains isolated in the Philippines in 1964 and 1965. Bull World Health Organ 37:763–771
Lee S, Yeom J-H, Seo S et al (2015) Functional analysis of Vibrio vulnificus RND efflux pumps homologous to Vibrio cholerae VexAB and VexCD, and to Escherichia coli AcrAB. J Microbiol 53:256–261. https://doi.org/10.1007/s12275-015-5037-0
Lee L-H, Ab Mutalib N-S, Law JW-F et al (2018) Discovery on antibiotic resistance patterns of Vibrio parahaemolyticus in selangor reveals carbapenemase producing Vibrio parahaemolyticus in marine and freshwater fish. Front Microbiol 9:2513. https://doi.org/10.3389/fmicb.2018.02513
Lei T, Zhang J, Jiang F et al (2019) First detection of the plasmid-mediated colistin resistance gene mcr-1 in virulent Vibrio parahaemolyticus. Int J Food Microbiol 308:108290. https://doi.org/10.1016/j.ijfoodmicro.2019.108290
Lei T, Jiang F, He M et al (2020) Prevalence, virulence, antimicrobial resistance, and molecular characterization of fluoroquinolone resistance of Vibrio parahaemolyticus from different types of food samples in China. Int J Food Microbiol 317:108461. https://doi.org/10.1016/j.ijfoodmicro.2019.108461
Lekshmi M, Ammini P, Kumar S, Varela MF (2017) The food production environment and the development of antimicrobial resistance in human pathogens of animal origin. Microorganisms. https://doi.org/10.3390/microorganisms5010011
Letchumanan V, Yin W-F, Lee L-H, Chan K-G (2015) Prevalence and antimicrobial susceptibility of Vibrio parahaemolyticus isolated from retail shrimps in Malaysia. Front Microbiol 6:33. https://doi.org/10.3389/fmicb.2015.00033
Li G, Wang M-Y (2020) The role of Vibrio vulnificus virulence factors and regulators in its infection-induced sepsis. Folia Microbiol (praha) 65:265–274. https://doi.org/10.1007/s12223-019-00763-7
Lipp EK, Huq A, Colwell RR (2002) Effects of global climate on infectious disease: the cholera model. Clin Microbiol Rev 15:757–770. https://doi.org/10.1128/CMR.15.4.757-770.2002
Liu P, Zhang X, Wang Q et al (2022) Biological and transcriptional studies reveal VmeL is involved in motility, biofilm formation and virulence in Vibrio parahaemolyticus. Front Microbiol 13:976334
Lloyd NA, Nazaret S, Barkay T (2019) Genome-facilitated discovery of RND efflux pump-mediated resistance to cephalosporins in Vibrio spp. isolated from the mummichog fish gut. J Glob Antimicrob Resist 19:294–300
Lo C-C, Lin P-T, Chiang-Ni C et al (2017) Contribution of efflux systems to the detergent resistance, cytotoxicity, and biofilm formation of Vibrio vulnificus. Gene Rep 9:115–122. https://doi.org/10.1016/j.genrep.2017.09.004
Lomovskaya O, Lewis K (1992) Emr, an Escherichia coli locus for multidrug resistance. Proc Natl Acad Sci USA 89:8938–8942. https://doi.org/10.1073/pnas.89.19.8938
Loo KY, Letchumanan V, Law JWF et al (2020) Incidence of antibiotic resistance in Vibrio spp. Rev Aquac 12:2590–2608. https://doi.org/10.1111/raq.12460
Lu M, Symersky J, Radchenko M et al (2013) Structures of a Na+-coupled, substrate-bound MATE multidrug transporter. Proc Natl Acad Sci USA 110:2099–2104. https://doi.org/10.1073/pnas.1219901110
Lu W-J, Lin H-J, Janganan TK et al (2018) ATP-binding cassette transporter VcaM from Vibrio cholerae is dependent on the outer membrane factor family for its function. Int J Mol Sci 19:E1000. https://doi.org/10.3390/ijms19041000
Luo J, Parsons SM (2010) Conformational propensities of peptides mimicking transmembrane Helix 5 and Motif C in wild-type and mutant vesicular acetylcholine transporters. ACS Chem Neurosci 1:381–390. https://doi.org/10.1021/cn900033s
Maddocks SE, Oyston PCF (2008) Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology (reading) 154:3609–3623. https://doi.org/10.1099/mic.0.2008/022772-0
Maiden MC, Davis EO, Baldwin SA et al (1987) Mammalian and bacterial sugar transport proteins are homologous. Nature 325:641–643
Mandal A, Sengupta A, Kumar A et al (2017) Molecular epidemiology of extended-spectrum β-lactamase–producing Escherichia coli pathotypes in diarrheal children from low socioeconomic status communities in Bihar, India: emergence of the CTX-M Type. Infect Dis (auckl) 10:1178633617739018. https://doi.org/10.1177/1178633617739018
Manjusha S, Sarita GB (2011) Plasmid associated antibiotic resistance in Vibrios isolated from coastal waters of Kerala. Int Food Res J 18:1171–1181
Marinello S, Marini G, Parisi G et al (2017) Vibrio cholerae non-O1, non-O139 bacteraemia associated with pneumonia, Italy 2016. Infection 45:237–240. https://doi.org/10.1007/s15010-016-0961-4
Martínez JL (2008) Antibiotics and antibiotic resistance genes in natural environments. Science 321:365–367
Maseda H, Sawada I, Saito K et al (2004) Enhancement of the mexAB-oprM efflux pump expression by a quorum-sensing autoinducer and its cancellation by a regulator, MexT, of the mexEF-oprN efflux pump operon in Pseudomonas aeruginosa. Antimicrob Agents Chemother 48:1320–1328. https://doi.org/10.1128/aac.48.4.1320-1328.2004
Masureel M, Martens C, Stein RA et al (2014) Protonation drives the conformational switch in the multidrug transporter LmrP. Nat Chem Biol 10:149–155. https://doi.org/10.1038/nchembio.1408
Matsumoto C, Okuda J, Ishibashi M et al (2000) Pandemic spread of an O3:K6 clone of Vibrio parahaemolyticus and emergence of related strains evidenced by arbitrarily primed PCR and toxRS sequence analyses. J Clin Microbiol 38:578–585
Matsuo T, Hayashi K, Morita Y et al (2007) VmeAB, an RND-type multidrug efflux transporter in Vibrio parahaemolyticus. Microbiology 153:4129–4137. https://doi.org/10.1099/mic.0.2007/009597-0
Matsuo T, Nakamura K, Kodama T et al (2013) Characterization of all RND-type multidrug efflux transporters in Vibrio parahaemolyticus. MicrobiologyOpen 2:725–742. https://doi.org/10.1002/mbo3.100
McMurry L, Petrucci RE Jr, Levy BS (1980) Active efflux of tetracycline encoded by four genetically different tetracycline resistance determinants in Escherichia coli. Proc Natl Acad Sci 77:3974–3977
Meng X, Huang D, Zhou Q et al (2023) The influence of outer membrane protein on ampicillin resistance of Vibrio parahaemolyticus. Can J Infect Dis Med Microbiol 2023:8079091. https://doi.org/10.1155/2023/8079091
Mevada V, Patel R, Dudhagara P et al (2023) Whole genome sequencing and pan-genomic analysis of multidrug-resistant Vibrio cholerae VC01 isolated from a clinical sample. Microorganisms 11:2030. https://doi.org/10.3390/microorganisms11082030
Miyamae S, Ueda O, Yoshimura F, Hwang J, Tanaka Y, Nikaido H (2001) A MATE family multidrug efflux transporter pumps out fluoroquinolones in Bacteroides thetaiotaomicron. Antimicrob Agents Chemother 45(12):3341–3346. https://doi.org/10.1128/aac.45.12.3341-3346.2001
Mohanty P, Patel A, Bhardwaj AK (2012) Role of H- and D- MATE-type transporters from multidrug resistant clinical isolates of Vibrio fluvialis in conferring fluoroquinolone resistance. PLoS ONE 7:e35752. https://doi.org/10.1371/journal.pone.0035752
Mohanty P, Shah A, Bhardwaj AK (2021) Functional insights of two MATE transporters from Vibrio fluvialis. bioRxiv 49:949
Mok JS, Ryu A, Kwon JY et al (2019) Abundance, antimicrobial resistance, and virulence of pathogenic Vibrio strains from molluscan shellfish farms along the Korean coast. Mar Pollut Bull 149:110559. https://doi.org/10.1016/j.marpolbul.2019.110559
Molina-Aja A, García-Gasca A, Abreu-Grobois A et al (2002) Plasmid profiling and antibiotic resistance of Vibrio strains isolated from cultured penaeid shrimp. FEMS Microbiol Lett 213:7–12. https://doi.org/10.1016/S0378-1097(02)00791-7
Moorthy S, Watnick PI (2005) Identification of novel stage-specific genetic requirements through whole genome transcription profiling of Vibrio cholerae biofilm development. Mol Microbiol 57:1623–1635. https://doi.org/10.1111/j.1365-2958.2005.04797.x
Morita Y, Kataoka A, Shiota S et al (2000) NorM of vibrio parahaemolyticus is an Na(+)-driven multidrug efflux pump. J Bacteriol 182:6694–6697. https://doi.org/10.1128/JB.182.23.6694-6697.2000
Morita D, Takahashi E, Morita M et al (2020) Genomic characterization of antibiotic resistance-encoding genes in clinical isolates of Vibrio cholerae non-O1/non-O139 strains from Kolkata, India: generation of novel types of genomic islands containing plural antibiotic resistance genes. Microbiol Immunol 64:435–444. https://doi.org/10.1111/1348-0421.12790
Morita Y, Li X-Z (2016) Antimicrobial resistance and drug efflux pumps in Vibrio and Legionella. In: Li XZ, Elkins C, Zgurskaya H (eds) Efflux-mediated antimicrobial resistance in bacteria. Adis, Cham, p 307–328
Morris JG (2003) Cholera and other types of vibriosis: a story of human pandemics and oysters on the half shell. Clin Infect Dis 37:272–280. https://doi.org/10.1086/375600
Morris RD (2007) The blue death : disease, disaster, and the water we drink, 1st edn. HarperCollins, New York
Motes ML, DePaola A, Cook DW et al (1998) Influence of water temperature and salinity on Vibrio vulnificus in Northern Gulf and Atlantic Coast oysters (Crassostrea virginica). Appl Environ Microbiol 64:1459–1465. https://doi.org/10.1128/AEM.64.4.1459-1465.1998
Mukherjee M, Kakarla P, Kumar S et al (2014) Comparative genome analysis of non-toxigenic non-O1 versus toxigenic O1 Vibrio cholerae. Genom Discov 2:1–15. https://doi.org/10.7243/2052-7993-2-1
Mukhopadhyay AK, Takeda Y, Balakrish Nair G (2014) Cholera outbreaks in the El Tor Biotype era and the impact of the new El Tor variants. In: Nair GB, Takeda Y (eds) Cholera outbreaks. Springer, Berlin, pp 17–47
Nair GB, Ramamurthy T, Bhattacharya SK et al (1994) Spread of Vibrio cholerae O139 Bengal in India. J Infect Dis 169:1029–1034. https://doi.org/10.1093/infdis/169.5.1029
Nakayama T, Yamaguchi T, Jinnai M et al (2023) ESBL-producing Vibrio vulnificus and V. alginolyticus harbour a plasmid encoding ISEc9 upstream of blaCTX-M-55 and qnrS2 isolated from imported seafood. Arch Microbiol 205:241. https://doi.org/10.1007/s00203-023-03569-x
Ndraha N, Hsiao H-I (2019) The risk assessment of Vibrio parahaemolyticus in raw oysters in Taiwan under the seasonal variations, time horizons, and climate scenarios. Food Control 102:188–196. https://doi.org/10.1016/j.foodcont.2019.03.020
Nelson ML, Levy SB (2011) The history of the tetracyclines. Ann N Y Acad Sci 1241:17–32. https://doi.org/10.1111/j.1749-6632.2011.06354.x
Neogi SB, Chowdhury N, Awasthi SP et al (2019) Novel cholera toxin variant and ToxT regulon in environmental Vibrio mimicus isolates: potential resources for the evolution of Vibrio cholerae hybrid strains. Appl Environ Microbiol 85:e01977-e2018. https://doi.org/10.1128/AEM.01977-18
Nikaido H (1994) Prevention of drug access to bacterial targets: permeability barriers and active efflux. Science 264:382–388. https://doi.org/10.1126/science.8153625
Nikaido H (1998) Antibiotic resistance caused by gram-negative multidrug efflux pumps. Clin Infect Dis 27:S32–S41. https://doi.org/10.1086/514920
Nikaido H (2011) Structure and mechanism of RND-type multidrug efflux pumps. Adv Enzymol Relat Areas Mol Biol 77:1–60. https://doi.org/10.1002/9780470920541.ch1
Nishibuchi M, Ishibashi M, Takeda Y, Kaper JB (1985) Detection of the thermostable direct hemolysin gene and related DNA sequences in Vibrio parahaemolyticus and other vibrio species by the DNA colony hybridization test. Infect Immun 49:481–486. https://doi.org/10.1128/IAI.49.3.481-486.1985
Nishibuchi M, Taniguchi T, Misawa T et al (1989) Cloning and nucleotide sequence of the gene (trh) encoding the hemolysin related to the thermostable direct hemolysin of Vibrio parahaemolyticus. Infect Immun 57:2691–2697. https://doi.org/10.1128/IAI.57.9.2691-2697.1989
Ogawa W, Minato Y, Dodan H et al (2015) Characterization of MATE-type multidrug efflux pumps from Klebsiella pneumoniae MGH78578. PLoS ONE 10:e0121619. https://doi.org/10.1371/journal.pone.0121619
Oh EG, Son KT, Yu H et al (2011) Antimicrobial resistance of Vibrio parahaemolyticus and Vibrio alginolyticus strains isolated from farmed fish in Korea from 2005 through 2007. J Food Prot 74:380–386. https://doi.org/10.4315/0362-028X.JFP-10-307
Okoh AI, Igbinosa EO (2010) Antibiotic susceptibility profiles of some Vibrio strains isolated from wastewater final effluents in a rural community of the Eastern Cape Province of South Africa. BMC Microbiol 10:143. https://doi.org/10.1186/1471-2180-10-143
Okuda J, Ishibashi M, Hayakawa E et al (1997) Emergence of a unique O3:K6 clone of Vibrio parahaemolyticus in Calcutta, India, and isolation of strains from the same clonal group from Southeast Asian travelers arriving in Japan. J Clin Microbiol 35:3150–3155
Okura M, Osawa R, Iguchi A et al (2003) Genotypic analyses of Vibrio parahaemolyticus and development of a pandemic group-specific multiplex PCR assay. J Clin Microbiol 41:4676–4682
Oliver JD (2010) Recent findings on the viable but nonculturable state in pathogenic bacteria. FEMS Microbiol Rev 34:415–425. https://doi.org/10.1111/j.1574-6976.2009.00200.x
Oliver JD, Pruzzo C, Vezzulli L, Kaper JB (2012) Vibrio species. Food microbiology. John Wiley & Sons Ltd, pp 401–439
Otsuka M, Yasuda M, Morita Y et al (2005) Identification of essential amino acid residues of the NorM Na+/multidrug antiporter in Vibrio parahaemolyticus. J Bacteriol 187:1552–1558. https://doi.org/10.1128/JB.187.5.1552-1558.2005
Ottaviani D, Bacchiocchi I, Masini L et al (2001) Antimicrobial susceptibility of potentially pathogenic halophilic vibrios isolated from seafood. Int J Antimicrob Agents 18:135–140. https://doi.org/10.1016/S0924-8579(01)00358-2
Ottaviani D, Leoni F, Rocchegiani E et al (2011) Unusual case of necrotizing fasciitis caused by Vibrio cholerae O137. J Clin Microbiol 49:757–759. https://doi.org/10.1128/JCM.02257-10
Ouellette M, Gerbaud G, Courvalin P (1988) Genetic, biochemical and molecular characterization of strains of Vibrio cholerae multiresistant to antibiotics. Ann Inst Pasteur Microbiol 139:105–113
Oyelade AA, Adelowo OO, Fagade OE (2018) blaNDM-1-producing Vibrio parahaemolyticus and V. vulnificus isolated from recreational beaches in Lagos Nigeria. Environ Sci Pollut Res Int 25:33538–33547. https://doi.org/10.1007/s11356-018-3306-2
Pao SS, Paulsen IT, Saier MH Jr (1998) Major facilitator superfamily. Microbiol Mol Biol Rev 62:1–34
Parthasarathy S, Das SC, Kumar A et al (2021) Molecular characterization and antibiotic resistance of Vibrio parahaemolyticus from Indian oyster and their probable implication in food chain. World J Microbiol Biotechnol 37:145. https://doi.org/10.1007/s11274-021-03113-3
Parvathi A, Kumar HS, Karunasagar I, Karunasagar I (2004) Detection and enumeration of Vibrio vulnificus in oysters from two estuaries along the southwest coast of India, using molecular methods. Appl Environ Microbiol 70:6909–6913. https://doi.org/10.1128/AEM.70.11.6909-6913.2004
Pasrija R, Banerjee D, Prasad R (2007) Structure and function analysis of CaMdr1p, a major facilitator superfamily antifungal efflux transporter protein of Candida albicans: identification of amino acid residues critical for drug/H+ transport. Eukaryot Cell 6:443–453. https://doi.org/10.1128/EC.00315-06
Pérez-Acosta JA, Martínez-Porchas M, Elizalde-Contreras JM et al (2018) Proteomic profiling of integral membrane proteins associated to pathogenicity in Vibrio parahaemolyticus strains. Microbiol Immunol 62:14–23. https://doi.org/10.1111/1348-0421.12556
Piddock LJ (2006) Multidrug-resistance efflux pumps—not just for resistance. Nat Rev Microbiol 4:629–636
Prescott LM, Datta A, Datta GC (1968) R-factors in Calcutta strains of Vibrio cholerae and members of the enterobacteriaceae. Bull World Health Organ 39:971–973
Rahal K, Gerbaud GR, Chabbert YA (1973) Properties of a transferable resistance factor in Vibrio cholerae Biotype El Tor. Ann Microbiol (Paris) 124:283–294
Rahman MM, Matsuo T, Ogawa W et al (2007) Molecular cloning and characterization of all RND-type efflux transporters in Vibrio cholerae non-O1. Microbiol Immunol 51:1061–1070. https://doi.org/10.1111/j.1348-0421.2007.tb04001.x
Rahmati S, Yang S, Davidson AL, Zechiedrich EL (2002) Control of the AcrAB multidrug efflux pump by quorum-sensing regulator SdiA. Mol Microbiol 43:677–685
Ramamurthy T, Garg S, Sharma R et al (1993) Emergence of novel strain of Vibrio cholerae with epidemic potential in southern and eastern India. Lancet 341:703–704. https://doi.org/10.1016/0140-6736(93)90480-5
Ramamurthy T, Ghosh A (2021) A re-look at cholera pandemics from early times to now in the current era of epidemiology. J Disast Res 16:110–117. https://doi.org/10.20965/jdr.2021.p0110
Ranaweera I, Shrestha U, Ranjana K et al (2015) Structural comparison of bacterial multidrug efflux pumps of the major facilitator superfamily. Trends Cell Mol Biol 10:131
Reguera G, Kolter R (2005) Virulence and the environment: a novel role for Vibrio cholerae toxin-coregulated pili in biofilm formation on Chitin. J Bacteriol 187:3551–3555. https://doi.org/10.1128/jb.187.10.3551-3555.2005
Reidl J, Klose KE (2002) Vibrio cholerae and cholera: out of the water and into the host. FEMS Microbiol Rev 26:125–139. https://doi.org/10.1111/j.1574-6976.2002.tb00605.x
Reyhanath PV, Ranjeet K (2014) Incidence of multidrug resistant Vibrio parahaemolyticus isolated from Ponnani, South India. Iran J Microbiol 6:60–67
Rouch DA, Cram DS, DiBerardino D et al (1990) Efflux-mediated antiseptic resistance gene qacA from Staphylococcus aureus: common ancestry with tetracycline- and sugar-transport proteins. Mol Microbiol 4:2051–2062. https://doi.org/10.1111/j.1365-2958.1990.tb00565.x
Rouquette-Loughlin C, Dunham SA, Kuhn M et al (2003) The NorM efflux pump of Neisseria gonorrhoeae and Neisseria meningitidis recognizes antimicrobial cationic compounds. J Bacteriol 185:1101–1106. https://doi.org/10.1128/JB.185.3.1101-1106.2003
Rubin DHF, Zingl FG, Leitner DR et al (2022) Reemergence of cholera in Haiti. N Engl J Med 387:2387–2389. https://doi.org/10.1056/NEJMc2213908
Saha GK, Ganguly NK (2021) Spread and endemicity of cholera in india: factors beyond the numbers. J Infect Dis 224:S710–S716. https://doi.org/10.1093/infdis/jiab436
Saier MH Jr, Beatty JT, Goffeau A et al (1999) The major facilitator superfamily. J Mol Microbiol Biotechnol 1:257–279
Saier MH, Reddy VS, Moreno-Hagelsieb G et al (2021) The transporter classification database (TCDB): 2021 update. Nucleic Acids Res 49:D461–D467. https://doi.org/10.1093/nar/gkaa1004
Saraswathi K, Deodhar LP (1990) A study of V. cholerae strains isolated in Bombay. J Postgrad Med 36:128–130
Shannon JD, Kimbrough RC (2006) Pulmonary cholera due to infection with a non-O1 Vibrio cholerae strain. J Clin Microbiol 44:3459–3460. https://doi.org/10.1128/jcm.02343-05
Sharma C, Thungapathra M, Ghosh A et al (1998) Molecular analysis of non-O1, non-O139 Vibrio cholerae associated with an unusual upsurge in the incidence of cholera-like disease in Calcutta, India. J Clin Microbiol 36:756–763. https://doi.org/10.1128/jcm.36.3.756-763.1998
Sherman I (2007) Cholera. Twelve diseases that changed our world. John Wiley & Sons Ltd, pp 33–49
Silva IP, de Carneiro CS, Saraiva MAF et al (2018) Antimicrobial resistance and potential virulence of Vibrio parahaemolyticus isolated from water and bivalve mollusks from Bahia, Brazil. Mar Pollut Bull 131:757–762. https://doi.org/10.1016/j.marpolbul.2018.05.007
Silvester R, Pires J, Van Boeckel TP et al (2019) Occurrence of β-lactam resistance genes and plasmid-mediated resistance among vibrios isolated from Southwest Coast of India. Microb Drug Resist 25:1306–1315. https://doi.org/10.1089/mdr.2019.0031
Smith KP, Kumar S, Varela MF (2009) Identification, cloning, and functional characterization of EmrD-3, a putative multidrug efflux pump of the major facilitator superfamily from Vibrio cholerae O395. Arch Microbiol 191:903–911
Someya Y, Kimura-Someya T, Yamaguchi A (2000) Role of the charge interaction between Arg(70) and Asp(120) in the Tn10-encoded metal-tetracycline/H(+) antiporter of Escherichia coli. J Biol Chem 275:210–214. https://doi.org/10.1074/jbc.275.1.210
Stein WD (1986) CHAPTER 6 - primary active transport systems: chemiosmosis. In: Stein WD (ed) Transport and diffusion across cell membranes. Academic Press, pp 475–612
Stein W (2012) Transport and diffusion across cell membranes. Elsevier
Sten-Knudsen O (1978) Passive transport processes. In: Tosteson DC (ed) Concepts and models. Springer, Berlin, pp 5–113
Stephen J, Lekshmi M, Ammini P et al (2022) Membrane efflux pumps of pathogenic vibrio species: role in antimicrobial resistance and virulence. Microorganisms 10:382. https://doi.org/10.3390/microorganisms10020382
Stephen J, Salam F, Lekshmi M et al (2023) The major facilitator superfamily and antimicrobial resistance efflux pumps of the ESKAPEE pathogen Staphylococcus aureus. Antibiotics 12:343. https://doi.org/10.3390/antibiotics12020343
Stratev D, Fasulkova R, Krumova-Valcheva G (2023) Incidence, virulence genes and antimicrobial resistance of Vibrio parahaemolyticus isolated from seafood. Microb Pathog 177:106050. https://doi.org/10.1016/j.micpath.2023.106050
Su X-Z, Chen J, Mizushima T et al (2005) AbeM, an H+-coupled Acinetobacter baumannii multidrug efflux pump belonging to the MATE family of transporters. Antimicrob Agents Chemother 49:4362–4364. https://doi.org/10.1128/AAC.49.10.4362-4364.2005
Symmons MF, Marshall RL, Bavro VN (2015) Architecture and roles of periplasmic adaptor proteins in tripartite efflux assemblies. Front Microbiol 6:513. https://doi.org/10.3389/fmicb.2015.00513
Tabtieng R, Wattanasri S, Echeverria P et al (1989) An epidemic of Vibrio cholerae El Tor Inaba resistant to several antibiotics with a conjugative group C plasmid coding for type II dihydrofolate reductase in Thailand. Am J Trop Med Hyg 41:680–686. https://doi.org/10.4269/ajtmh.1989.41.680
Tan CW, Rukayadi Y, Hasan H et al (2020) Prevalence and antibiotic resistance patterns of Vibrio parahaemolyticus isolated from different types of seafood in Selangor, Malaysia. Saudi J Biol Sci 27:1602–1608. https://doi.org/10.1016/j.sjbs.2020.01.002
Tanabe T, Funahashi T, Nakao H et al (2003) Identification and characterization of genes required for biosynthesis and transport of the siderophore vibrioferrin in Vibrio parahaemolyticus. J Bacteriol 185:6938–6949. https://doi.org/10.1128/JB.185.23.6938-6949.2003
Tanabe T, Nakao H, Kuroda T et al (2006) Involvement of the Vibrio parahaemolyticus pvsC gene in export of the siderophore vibrioferrin. Microbiol Immunol 50:871–876
Tanaka Y, Hipolito CJ, Maturana AD et al (2013) Structural basis for the drug extrusion mechanism by a MATE multidrug transporter. Nature 496:247–251. https://doi.org/10.1038/nature12014
Taylor DL, Bina XR, Bina JE (2012) Vibrio cholerae vexH encodes a multiple drug efflux pump that contributes to the production of cholera toxin and the toxin co-regulated pilus. PLoS ONE 7:e38208
Threlfall EJ, Rowe B, Huq I (1980) Plasmid-encoded multiple antibiotic resistance in Vibrio cholerae El Tor from Bangladesh. Lancet 1:1247–1248. https://doi.org/10.1016/s0140-6736(80)91701-8
Towner KJ, Pearson NJ, Mhalu FS, O’Grady F (1980) Resistance to antimicrobial agents of Vibrio cholerae E1 Tor strains isolated during the fourth cholera epidemic in the United Republic of Tanzania. Bull World Health Organ 58:747–751
Tseng TT, Gratwick KS, Kollman J et al (1999) The rnd permease superfamily: an ancient, ubiquitous and diverse family that includes human disease and development proteins. J Mol Microbiol Biotechnol 1:107–125
Upadhyay N, Kar D, Deepak Mahajan B et al (2019) The multitasking abilities of MATE 1285 transporters in plants. J Exp Bot 70:4643–4656. https://doi.org/10.1093/jxb/erz246
Varela MF, Griffith JK (1993) Nucleotide and deduced protein sequences of the class D tetracycline resistance determinant: relationship to other antimicrobial transport proteins. Antimicrob Agents Chemother 37:1253–1258. https://doi.org/10.1128/aac.37.6.1253
Varela MF, Kumar S (2019) Strategies for discovery of new molecular targets for anti-infective drugs. Curr Opin Pharmacol 48:57–68. https://doi.org/10.1016/j.coph.2019.04.015
Varela MF, Wilson TH (1996) Molecular biology of the lactose carrier of Escherichia coli. Biochem Biophys Acta 1276:21–34. https://doi.org/10.1016/0005-2728(96)00030-8
Varela MF, Sansom CE, Griffith JK (1995) Mutational analysis and molecular modelling of an amino acid sequence motif conserved in antiporters but not symporters in a transporter superfamily. Mol Membr Biol 12:313–319. https://doi.org/10.3109/09687689509072433
Verma J, Bag S, Saha B et al (2019) Genomic plasticity associated with antimicrobial resistance in Vibrio cholerae. Proc Natl Acad Sci 116:6226–6231. https://doi.org/10.1073/pnas.1900141116
Vezzulli L, Baker-Austin C, Kirschner A et al (2020) Global emergence of environmental non-O1/O139 Vibrio cholerae infections linked with climate change: a neglected research field? Environ Microbiol 22:4342–4355. https://doi.org/10.1111/1462-2920.15040
Vu TTT, Hoang TTH, Fleischmann S et al (2022) Quantification and antimicrobial resistance of Vibrio parahaemolyticus in retail seafood in Hanoi, Vietnam. J Food Prot 85:786–791. https://doi.org/10.4315/JFP-21-444
Wade BM (2022) The blue death: cholera and reimagined community in nineteenth-century Havana. In: Venkatesan S, Chatterjee A, Lewis AD, Callender B (eds) Pandemics and epidemics in cultural representation. Springer Nature, Singapore, pp 81–103
Waldor MK, Tschäpe H, Mekalanos JJ (1996) A new type of conjugative transposon encodes resistance to sulfamethoxazole, trimethoprim, and streptomycin in Vibrio cholerae O139. J Bacteriol 178:4157–4165. https://doi.org/10.1128/jb.178.14.4157-4165.1996
Wei Y, Perez LJ, Ng W-L et al (2011) Mechanism of Vibrio cholerae autoinducer-1 biosynthesis. ACS Chem Biol 6:356–365
Weng Y, Fields EG, Bina TF et al (2021) Vibrio cholerae TolC is required for expression of the ToxR regulon. Infect Immunity. https://doi.org/10.1128/iai.00242-21
West IC (1980) Energy coupling in secondary active transport. Biochem Biophys Acta 604:91–126. https://doi.org/10.1016/0005-2736(80)90586-6
WHO (2022) WHO fact sheet. https://www.who.int/news-room/fact-sheets/detail/food-safety. Accessed 14 Jan 2023
Woolley RC, Vediyappan G, Anderson M et al (2005) Characterization of the Vibrio cholerae vceCAB multiple-drug resistance efflux operon in Escherichia coli. J Bacteriol 187:5500–5503. https://doi.org/10.1128/JB.187.15.5500-5503.2005
Wu C, Zhao Z, Liu Y et al (2020) Type III secretion 1 effector gene diversity among vibrio isolates from coastal areas in China. Front Cell Infect Microbiol. https://doi.org/10.3389/fcimb.2020.00301
Yaffe D, Radestock S, Shuster Y et al (2013) Identification of molecular hinge points mediating alternating access in the vesicular monoamine transporter VMAT2. Proc Natl Acad Sci 110:E1332–E1341
Yamaguchi A, Someya Y, Sawai T (1992) Metal-tetracycline/H+ antiporter of Escherichia coli encoded by transposon Tn10. The role of a conserved sequence motif, GXXXXRXGRR, in a putative cytoplasmic loop between helices 2 and 3. J Biol Chem 267:19155–19162
Ye L, Zheng Z, Xu Y et al (2023) Prevalence and genetic basis of tetracycline resistance in Vibrio parahaemolyticus isolates recovered from food products in Shenzhen, China during 2013 to 2021. Sci Total Environ 902:166026. https://doi.org/10.1016/j.scitotenv.2023.166026
UNHCR Yemen flooding escalates spread of cholera (2023). In: UNHCR Hong Kong. https://www.unhcr.org/hk/en/23296-yemen-flooding-escalates-spread-of-cholera.html. Accessed 8 Oct 2023
Yokota T, Kuwahara S (1977) Temperature-sensitive R plasmid obtained from naturally isolated drug-resistant Vibrio cholerae (Biotype El Tor). Antimicrob Agents Chemother 11:13–20. https://doi.org/10.1128/AAC.11.1.13
Yuan Y, Feng Z, Wang J (2020) Vibrio vulnificus Hemolysin: biological activity, regulation of vvhA expression, and role in pathogenesis. Front Immunol 11:599439. https://doi.org/10.3389/fimmu.2020.599439
Zaidenstein R, Sadik C, Lerner L et al (2008) Clinical characteristics and molecular subtyping of Vibrio vulnificus illnesses, Israel. Emerg Infect Dis 14:1875–1882. https://doi.org/10.3201/eid1412.080499
Zanetti S, Spanu T, Deriu A et al (2001) In vitro susceptibility of Vibrio spp. isolated from the environment. Int J Antimicrob Agents 17:407–409. https://doi.org/10.1016/S0924-8579(01)00307-7
Zhang G, Sun K, Ai G et al (2019) A novel family of intrinsic chloramphenicol acetyltransferase CATC in Vibrio parahaemolyticus: naturally occurring variants reveal diverse resistance levels against chloramphenicol. Int J Antimicrob Agents 54:75–79. https://doi.org/10.1016/j.ijantimicag.2019.03.012
Funding
The research studies reported from our laboratories and reflected in this publication were supported by Faculty Research and Instructional Development grants by ENMU and the National Institute of the General Medical Sciences (P20GM103451) awarded by the National Institutes of Health, plus the HSI-STEM program from the U.S. Department of Education (P031C110114).
Author information
Authors and Affiliations
Contributions
MFV and SK initiated and organized the project. AO, MA, ML, SK, JS, and MFV collected literature and wrote distinct sections of the manuscript. SK and MFV compiled, edited, and prepared the final form of the manuscript. All authors reviewed and agreed to the published form of the article.
Corresponding author
Ethics declarations
Conflict of interest
The authors here declare no conflict of interest.
Consent for publication
Not applicable.
Ethical approval
Not applicable.
Additional information
Communicated by Yusuf Akhter.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kumar, S., Lekshmi, M., Stephen, J. et al. Dynamics of efflux pumps in antimicrobial resistance, persistence, and community living of Vibrionaceae. Arch Microbiol 206, 7 (2024). https://doi.org/10.1007/s00203-023-03731-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00203-023-03731-5