
Abstract
In the Tat protein export pathway of Gram-negative bacteria, TatA and TatB are homologous proteins that carry out distinct and essential functions in separate sub-complexes. In contrast, Gram-positive Tat systems usually lack TatB and the TatA protein is bifunctional. We have used a mutagenesis approach to delineate TatA/B-type domains in the bifunctional TatAd protein from Bacillus subtilis. This involved expression of mutated TatAd variants in Escherichia coli and tests to determine whether the variants could function as TatA or TatB by complementing E. coli tatA and/or tatB mutants. We show that mutations in the C-terminal half of the transmembrane span and the subsequent FGP ‘hinge’ motif are critical for TatAd function with its partner TatCd subunit, and the same determinants are required for complementation of either tatA or tatB mutants in Escherichia coli. This is thus a critical domain in both TatA and TatB proteins. In contrast, substitution of a series of residues at the N-terminus specifically blocks the ability of TatAd to substitute for E. coli TatB. The results point to the presence of a universally conserved domain in the TatA/B-family, together with a separate N-terminal domain that is linked to the TatB-type function in Gram-negative bacteria.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Twin-Arginine Translocation (Tat) pathway is responsible for the export of a subset of periplasmic proteins across the bacterial plasma membrane. A notable attribute is its ability to transport proteins in a prefolded state, and this appears to be its primary role in bacteria (reviewed by Müller 2005; Robinson and Bolhuis 2004). Some Tat substrates are cofactor-containing redox enzymes involved in anaerobic respiratory pathways; these proteins require a co-factor to be inserted in the cytoplasm before translocation. Other cofactor-less proteins may fold either too rapidly or too tightly to be compatible with the alternative Sec-dependent protein export pathway (Müller 2005; Berks 1996; Santini et al. 1998; Weiner et al. 1998; Sargent et al. 1998).
Most of our information on the Tat pathway has been obtained from studies on the Gram-negative bacterium Escherichia coli. E. coli contains three genes that are essential for Tat function: tatA, tatB and tatC. A homologue of the tatA gene, tatE, is expressed elsewhere in the genome and is thought to be a cryptic gene duplication of tatA. The E. coli tatA gene encodes a membrane protein of 9.6 kDa that forms homo-oligomeric complexes that vary in size from less than 100 kDa to over 500 kDa (Gohlke et al. 2005; Oates et al. 2005). A small amount of TatA is also found in a membrane-localised 370 kDa complex with TatB (~18 kDa) and TatC (~28 kDa); this 370 kDa TatABC complex is thought to act as the initial receptor for Tat substrates (Alami et al. 2003; Bolhuis et al. 2001). Upon binding of substrate, it is thought that TatA complexes are recruited to form the translocation pore; it has been suggested that the TatA size variation in E. coli may be linked to the need to translocate proteins of different sizes without compromising the integrity of the membrane (Gohlke et al. 2005; Oates et al. 2005).
The E. coli TatA and TatB proteins share considerable homology and a similar structural arrangement, with a short N-terminal periplasmic domain (only a few residues), a single transmembrane (TM) span and a predicted cytoplasmic amphipathic helix that may align along the face of the membrane. This helix is followed by a largely unstructured C-terminal tail, with TatB having a longer extension than TatA. Several mutagenesis studies have been carried out to identify the regions of the TatA and TatB proteins that are important for translocation activity. Truncation analysis has shown that large sections of the C-terminal tails of these two proteins are not required for function (Lee et al. 2002). With both TatA and TatB proteins, the critical regions have been found to be the TM span, the cytoplasmic amphipathic helix and the small hinge region between the two helices (Barrett and Robinson 2005; Greene et al. 2007; Ize et al. 2002; Lee et al. 2006). Single amino acid substitutions within the amphipathic helices of TatA and TatB can have a drastic effect on translocation activity, but perhaps surprisingly, single substitutions within the TM spans can be tolerated and relatively little difference in translocation activity is observed in E. coli.
The Tat pathways of Gram-positive bacteria display some important differences, the main difference being the absence of a TatB component in almost all Gram-positive species (Jongbloed et al. 2004). The absence of TatB and the close similarity between the TatA and TatB proteins of Gram-negative bacteria suggested that the TatA protein in these systems might be bi-functional, fulfilling the roles of both TatA and TatB (Jongbloed et al. 2006). In support of this idea, we found that the TatAd protein of Bacillus subtilis complemented both E. coli ΔtatAE and ΔtatB mutant strains (Barnett et al. 2008). The TatAd protein forms a ~230 kDa complex with TatCd in the plasma membrane, together with a ~160 kDa homo-oligomeric TatAd complex (Barnett et al. 2008). These complexes may well be somewhat different to their E. coli counterparts in terms of complex size and composition, with the TatAd complex appearing to lack the heterogeneity observed with the E. coli TatA complex.
While the ability of TatAdCd to replace the endogenous TatABC system in E. coli suggests that Gram-positive TatAC-type systems use a broadly similar mechanism to their Gram-negative counterparts, it has also been suggested that Gram-positive Tat systems use a fundamentally different operating mechanism. In this alternative model, substrates bind to a cytoplasmic form of TatA, after which the complex docks onto membrane-bound TatC (Schreiber et al. 2006). Further studies on TatAC-type systems are clearly merited in order to resolve this important point.
In this report, we have identified residues that are important for the functioning of TatAd together with its cognate partner, TatCd and we show that several of the variant TatAd proteins have significant effects on TatAdCd complex assembly or stability. We have used a novel method to delineate the residues and domains that contribute to TatA/B-type functions by testing the ability of TatAd variants to specifically substitute for the TatA or TatB proteins in E. coli tat mutants. The results demonstrate the presence of an N-terminal TatB-type domain within TatAd, together with a separate domain that appears to be universally conserved among TatA/TatB proteins.
Experimental
Bacterial strains, plasmids and growth conditions
All of the strains and plasmids used are listed in Table 1. E. coli MC4100 (Casadaban and Cohen 1980) was used as the parental strain; ΔtatABCDE, ΔtatAE and ΔtatB (Sargent et al. 1999; Wexler et al. 2000) mutant versions have been described previously, arabinose resistant derivatives were used. All of the variant Tat proteins were expressed from the arabinose inducible pBAD24 vector.
E. coli cultures were grown aerobically in Luria broth or anaerobically in minimal TMAO/Glycerol media at 37°C (Barrett et al. 2005). tat genes were expressed from the arabinose inducible pBAD24 vector. Media was supplemented with ampicillin (100 μM) and arabinose (200 μM) where cells carried the pBAD24 plasmid.
Mutagenesis of TatAdCd and TatAd
Mutations were introduced into plasmids pBAdCd and pBAd-his (Barnett et al. 2008) that have been described previously. All mutations were introduced by site-directed mutagenesis using the Quick-Change™ system (Stratagene, USA). A full list of the primers used is available from the author upon request. All mutated genes were sequenced in full by the University of Warwick molecular biology service using Applied Biosystems Big Dye v3.1 and an ABI 3100 genetic analyserTM. The following sequencing primer was used: BADSEQ (5′ tatttgcacggcgtcaca 3′). Chromas software was used to analyse DNA sequences. All mutant pBAdCd plasmids were expressed in the E. coli ΔtatABCDE strain and all mutant pBAd-his plasmids were expressed in E. coli ΔtatAE and ΔtatB strains.
Production and stability of TatAd/Cd and TatAd mutants
An overnight culture (0.5 ml) was used to inoculate 9.5 ml of fresh LB, and cultures were grown for 3 h with induction of plasmids as described above. The OD600 of each of the cultures was measured and adjusted to 0.1. 200 μl of the diluted culture was pelleted before being resuspended in 30 ul of gel-loading buffer. Samples were applied directly onto 17.5% SDS–polyacrylamide gels and electrophoresis was performed using the C.B.S (USA) vertical gel system. After electrophoresis, proteins were transferred from the gels onto PVDF membrane (Amersham, UK) by Western blotting. Membranes were used for immunoblotting with anti-strep HRP conjugated antibodies (IBA, GmbH), for detection of TatCd and using specific anti-TatAd antibodies (Barnett et al. 2008) with subsequent detection using anti-rabbit HRP conjugate (Promega, USA) and ECL (Amersham, UK) detection reagents.
To test the expression levels of E. coli TatC in the ∆tatAE and ∆tatB strains, equal numbers of whole cells were subjected to SDS-gel electrophoresis and subsequent immunoblotting using anti-TatC antibodies (provided by Matthias Muller, Freiberg), with detection as described above.
Affinity purification of TatAdCd complexes
∆tatABCDE cells were grown aerobically to mid-exponential growth phase with induction of tatAdCd from plasmid pBAdCd using 0.5 mM arabinose. After 3 h of induction, cells were harvested and plasma membranes isolated as described previously (Barnett et al. 2008) and solubilised in 1% Dodecyl maltoside (DDM). Solubilised membranes were incubated with 5 μg/ml avidin to block any biotinylated proteins before application to an equilibrated 2 ml Streptactin affinity column (IBA, GmbH). The column was washed with five column volumes of equilibration buffer containing 20 mM Tris–HCl-pH 8.0, 150 mM NaCl and 0.02% DDM. Bound protein was eluted from the column in five 1.5 ml fractions using the same buffer as above but containing 2.5 mM desthiobiotin (Sigma, UK).
Circular dichroism spectroscopy of TatAdCd complexes
Purified TatAdCd complexes were prepared as described above. The protein concentration was determined using the Bradford assay and protein concentration diluted to 100 μg/ml in elution buffer. CD spectra were acquired using a Jasco J-815 spectrophotometer over 200–260 nm at a constant temperature of 4°C. For each sample, the average from 16 consecutive scans was obtained.
Translocation assays
Briefly, 0.5 ml of an overnight culture was used to inoculate 9.5 ml of fresh growth medium and cultures were grown for 3 h with induction of plasmids as described above. After 3 h, the OD600 of each culture was taken and cultures were diluted down to an OD600 of 0.4. Equal volumes of cell suspensions were harvested by centrifugation and cells were fractionated into periplasmic, cytoplasmic and membrane fractions using the previously described lysozyme/cold osmotic shock method (Randall and Hardy 1986). Following fractionation, equal volumes of samples were run on 10% native polyacrylamide gels that were subsequently stained for TorA activity using a well established gel based activity assay (Barnett et al. 2008; Silvestro et al. 1989).
Results
Expression of mutated TatAd variants together with TatCd in E. coli
The E. coli TatA and TatB proteins are related, yet they carry out completely different functions; TatB is tightly associated with TatC in the substrate-binding complex whereas TatA forms separate complexes of varying size. In this study, we have used the TatAd protein from B. subtilis as a tool to study the locations and importance of individual domains within the TatA and TatB subunits. The TatAdCd system is active when produced in E. coli and TatAd is bifunctional, able to complement both the ΔtatAE and ΔtatB mutants (Barnett et al. 2008). We reasoned that mutagenesis studies on TatAd would (1) enable us to study residues that are important for TatAd function in general and (2) permit the identification of regions that are specifically important for either a TatA or TatB function.
Figure 1 shows an alignment of the N-terminal sequence of TatAd with those of several other Gram-positive TatA proteins and the E. coli TatA and TatB subunits. Conserved features are restricted to the N-terminal half of the protein and residues within this section were randomly selected and substituted by Ala. The proteins were produced in E. coli ΔtatABCDE cells together with TatCd using the pBAdCd plasmid. In this way, we could test the effects of individual substitutions on ‘overall’ TatAd function in terms of its ability to support translocation together with TatCd. Several of the same TatAd variants were then produced (this time without TatCd) in E. coli ΔtatAE and ΔtatB mutant strains following expression from the pBAd-his plasmid. This allowed us to test the ability of the variant TatAd proteins to replace TatA and TatB individually.

Sequence alignment and topology of TatA proteins. The amino acid sequences of the TatA proteins of several Gram-positive bacteria, together with those of E. coli TatA and TatB, were aligned using the ClustalW programme. The first 50–60 amino acids are shown. Conserved residues are indicated (*) as are conservative substitutions. (:) The predicted transmembrane spanning domain (TM) and amphipathic helix (AMP) is indicated above the alignment
First, the expression and stability of the variant forms of the TatAd protein were tested when produced alongside TatCd using the pBAdCd plasmid in E. coli ΔtatABCDE cells. Following induction of the plasmids for 3 h as described in Experimental, E. coli cells were analysed by immunoblotting with antibodies to TatAd, and to the Strep-II tag present on TatCd (Fig. 2a). Overall we found that both the TatAd and TatCd proteins were present at broadly similar levels and the Figure shows a representative sample of the data, including all of the variants that were found to be defective in translocation. This confirms that any differences observed between the variant proteins are unlikely to be due to the presence of markedly different levels of TatAd or TatCd proteins, at least when TatAd and TatCd are produced together.

Expression and stability of TatAd mutants. a Extracts of E. coli ΔtatABCDE cells expressing wild type and mutated versions of the pBAdCds plasmid were analysed using SDS–polyacrylamide gels that were subsequently immunoblotted for detection of TatCd (using antibodies to the Strep-II tag), and TatAd (using specific anti-TatAd antibodies). b Wild-type TatAd was produced together with TatCd following expression from the pBAdCd plasmid in E. coli ΔtatABCDE cells (AdCd ∆tat panel), or in the absence of TatCd from the pBAdh plasmid in ΔtatAE and ΔtatB cells. As controls, strains lacking plasmid were analysed in parallel. The cells were fractionated into membrane and cytoplasmic samples (M, C). c Three of the TatAd variants were produced, either together with TatCd in E. coli ΔtatABCDE cells (AdCd ∆tat) or in the absence of TatCd from the pBAdh plasmid in ΔtatAE and ΔtatB cells as indicated. Cells were again fractionated into membrane and cytoplasm samples, run on SDS–PAGE gels and subsequently immunoblotted using specific anti-TatAd antibodies
We next compared the levels of TatAd when produced together with TatCd in ΔtatABCDE cells (as above) with those produced alone in ΔtatAE and ΔtatB strains. Under these conditions, the TatAd would be interacting with wild-type levels of native E. coli TatC and either TatAE or TatB accordingly. We also fractionated the cells into membrane and cytoplasm (M, C) samples to test whether the variant TatAd forms are inserted into the plasma membrane. This was done partly to test whether the mutations affect the ability of the proteins to insert into the membrane at all and partly to test whether the ratio of cytoplasmic: membrane-bound TatAd was changed, which may have relevance to the ‘cytoplasmic TatAd receptor’ translocation model described above. Figure 2b shows that production of wild-type TatAdCd in ΔtatABCDE cells (AdCd ∆tat panel) leads to the presence of membrane-bound TatAd plus a significant amount of cytoplasmic TatAd (approximately one-third of total TatAd), as found in previous studies (Barnett et al. 2008). The adjacent ‘∆tat’ control panel confirms that no TatAd signal is detected in the absence of TatAdCd production. TatAd is present in lower amounts when produced in ΔtatAE or ΔtatB strains, and the majority is again membrane-bound. Again, controls show that no TatAd is detected in ΔtatAE or ΔtatB strains lacking plasmid, as expected. When TatAd is expressed in the absence of TatCd, a C-terminal hexa-histidine tag is present. One possible explanation for the lower TatAd expression levels from these plasmids is that the his-tag is destabilising the protein complexes. Alternatively, the tag may reduce the avidity of the TatAd antibodies for the epitope leading to a lower estimate of protein expression.
Similar tests were carried out using three of the TatAd variants (S3A, N4A, I5A) as shown in Fig. 2c. A similar pattern is observed in each case: the bulk of the protein is membrane-bound, and TatAd is present at higher levels when produced together with TatCd from the pBAdCds plasmid in E. coli ΔtatABCDE cells. In other tests (data not shown), we observed similar behaviour for many of the variants, including G6A, P8A, G9A, L10A, I11A, L12A, F14A, V15A, I16A, L18A, F21A, G22A, P23A and G30A. Two of the variants (I19A and P27A) were present at extremely low levels in the ΔtatB strain (but at normal levels in ΔtatAE cells), suggesting that the alanine substitutions affect the stability of the proteins when functioning as a TatB-type protein. Overall, these data show that all of the TatAd variants are produced and inserted into the plasma membrane. However, the data do appear to demonstrate differences in TatAd stability depending on whether the protein is functioning as a bifunctional TatAd protein together with TatCd, or specifically substituting for E. coli TatA or TatB (when TatAd levels are invariably lower).
The N-terminal half of TatAd is critical for TatAdCd translocation activity
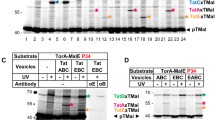
Tat-dependent export assays with these variant TatAd forms are shown in Fig. 3. E. coli ΔtatABCDE cells producing the TatAd variants together with wild-type TatCd were fractionated into periplasm (P), cytoplasm (C) and membrane (M) samples, and samples were loaded onto native polyacrylamide gels. Following electrophoresis, gels were assayed for the presence of active TMAO reductase (TorA), a known Tat substrate. Previous studies have shown that its presence in the periplasm is strictly Tat-dependent and the TatAdCd system is able to transport this protein with high efficiency (Barnett et al. 2008). In this assay, the native gel is stained dark blue with reduced methyl viologen. Transfer of the stained gel to buffer containing TMAO (the TorA substrate) reveals the presence of active TorA as the enzyme reduces the TMAO causing the oxidation of the methyl viologen. This leads to a clearing of the gel turbidity.

Identification of residues critical for TatAdCd-mediated export of TorA. Single amino acid substitutions were introduced into TatAd on plasmid pBAdCds using site-directed mutagenesis. Substituted versions were produced in E. coli ΔtatABCDE cells and cells fractionated into Periplasm (P), Cytoplasm (C) and Membrane (M) fractions. Cell fractions were run on native polyacrylamide gels that were assayed for the presence of active TorA. The figure shows a substitutions within the periplasmic domain, b substitutions within the transmembrane spanning domain (TM) and c substitutions within the hinge region and amphipathic helix
Figure 3a shows that alanine substitutions in the extreme N-terminal periplasmic region of TatAd (S3A, N4A, I5A and G6A) had no detectable effect on translocation activity by TatAdCd as active TorA is present in the periplasmic lane of each mutant. This strongly suggests that the identity of these N-terminal periplasmic residues is not critical in Tat function. This is not unexpected, as a sequence alignment of TatA proteins from Gram-positive bacteria and E. coli TatA and TatB shows that this N-terminal portion of the protein is not highly conserved (Fig. 1).
Pro8 is thought to mark the beginning of the TM helix according to a recently published solution NMR structure (Hu et al. 2010), and substitution of this residue by Ala (P8A variant) blocks export of TorA, with no periplasmic TorA activity observed (Fig. 3a). All TorA activity in this case is found in the cytosolic fraction. The sequence alignment in Fig. 1 shows that while E. coli TatA and TatB do not contain Pro at this position, all of the Gram-positive TatA proteins have Pro at either this position or an adjacent one. This appears to be an important difference between the TatA proteins of Gram-negative and Gram-positive bacteria.
The next four TatAd variants (G9A, L10A, I11A and L12A in the N-terminal region of the TM span) show efficient export of TorA to the periplasm, indicating that these residues are not critical (Fig. 3b). However, substitution of Phe14 (F14A) resulted in a complete loss of translocation activity with TorA activity found exclusively in the cytoplasmic and membrane fractions. This was surprising as this residue is not conserved among the TatA proteins of Gram-positive bacteria. However, a recent report describing the NMR solution structure of the TatAd protein has suggested a role for Phe14 in stabilising the hinge region of the protein via interactions of its aromatic ring with the hydrophobic side chains of Leu18 and Leu26 (Hu et al. 2010). Substitution of the following Val15 residue had no obvious effect on export. Summarising the above data, the N-terminal periplasmic domain and first half of the TM span are rather tolerant of alanine substitutions, with the notable exceptions of Pro8 and Phe14, which clearly play important roles in structure or activity.
The next three substitutions towards the C-terminal end of the TM span (I16A, I18A and I19A) have more dramatic effects and each almost blocks TorA transport, with only very weak TorA activity detected in the periplasm samples (Fig. 3b). Ile and Leu residues are rather conserved at these positions in Gram-positive TatA proteins (Fig. 1) and may be important for helix-helix interactions. As mentioned above, I18A has been found to stabilise the hinge region of TatAd via interactions with Phe14 suggesting an important structural or functional role (Hu et al. 2010).
The highly conserved hinge regions between the TM span and the amphipathic helices of E. coli TatA and TatB have been shown previously to be important for Tat function (Greene et al. 2007; Lee et al. 2006). E. coli TatA has a highly conserved FG motif while TatB shares the Gly residue with TatA but this is followed by a conserved Pro. The TatA proteins of Gram-positive bacteria generally contain all three residues in a highly conserved FGP motif (Fig. 1). Figure 3c shows that substitutions at all three positions (F21A, G22A and P23A) block translocation of TorA; no activity is detectable in the periplasmic fractions. This underlines the importance of this hinge region for function. It is also important to note that while these residues have been shown to be important for translocation activity in E. coli, substitution of these residues did not result in such a drastic reduction in translocation activity (Greene et al. 2007; Lee et al. 2006).
Finally, we made substitutions at four positions at the N-terminal end of the amphipathic helix. The K25A variant both supported only a low level of translocation activity, with a weak periplasmic TorA bands visible and L26A was blocked in translocation activity (Fig. 3c), while the P27A and G30A substitutions had no obvious effect on export of TorA. Interestingly, both Leu26 and Phe23 have been found to be important in maintaining the conformation of the TatAd hinge region (Hu et al. 2010). It should be noted that, while the TorA assays are not quantitative, they allow for any significant differences in translocation activity to be visualised. All translocation assays were conducted several times and reproducible results were obtained. In some of the gel panels, an additional second band is seen, particularly in the cytoplasmic lanes, but the significance of this band is unclear. Taken together, the results demonstrate critical roles for the C-terminal part of the TM span and the subsequent hinge region of TatAd when TatAdCd is produced in the E. coli tatABCDE background. These results are summarised in the left hand diagram in Fig. 4; filled circles represent residues where substitution leads to a complete, or near-complete, loss of activity and these clearly cluster in the C-terminus of the TM span and the hinge region.

Membrane topology of TatAd and residues important for TatAd function. A schematic representation of the topology of the TatAd protein in the plasma membrane. Substitutions that have a significant effect on translocation activity when TatAd and TatCd are produced together in E. coli ΔtatABCDE cells (left hand diagram), or when TatAd is produced alone in E. coli ΔtatAE or ΔtatB mutant strains, are shaded in black. The Figure highlights one domain that is important for TatAd function under all conditions tested (i.e. when produced in ΔtatABCDE, ΔtatAE or ΔtatB mutant strains) and a second domain that is critical for the ability of TatAd to function in place of E. coli TatB in the ∆tatB strain
Most of the translocation-defective TatAd mutants assemble into intact TatAdCd complexes. The above data show that a number of alanine substitutions in TatAd block translocation activity, but the data do not indicate whether the substitutions specifically affect the assembly or translocation activity of the Tat complexes. TatAd assembles with TatCd to form a ~300 kDa TatAdCd complex, and we carried out a study to determine whether the alanine substitutions that affect translocation activity had any gross affect on the formation of this complex. TatCd bears a C-terminal Strep-II tag, and we expressed the mutated tatAdCd operons in tatABCDE cells and affinity-purified the TatAdCd complexes using Streptactin affinity chromatography. Wild-type TatAdCd was purified by this method in a previous study (Barnett et al. 2008). Figure 5a shows silver-stained SDS–PAGE gels of the eluates; the TatCd protein is detected in each case and the TatAd protein is also observed to co-elute from the Streptactin affinity column along with TatCd (the identities of the TatCd and TatAd bands were confirmed by immunoblotting with anti-strep-II tag antibodies—data not shown). The TatAd bands stain rather weakly with silver as has been observed before (Barnett et al. 2008).

Purification and Circular dichroism of TatAdCd complexes containing mutated TatAd variants. a Purified wild-type TatAdCd complexes and several of the variant TatAd versions were analysed by SDS–PAGE with silver staining. The migration of TatCd and TatAd is indicated. b Circular dichroism was performed on purified wild-type and variant TatAdCd complexes. Purified protein (100 μg/ml) was used for analysis and data were acquired over a range of 200–260 nm. Averages were taken from 16 consecutive scans. The wild-type TatAdCd CD profile in shown in bold while variant proteins exhibiting different spectra are indicated
The purified TatAdCd complexes and the variant versions were analysed by Circular Dichroism spectroscopy to examine any changes to the overall secondary structure of the complexes arising from the alanine substitution. The results are shown in Fig. 5b. The spectrum from the wild-type complex is shown in bold and is typical of a structure rich in α-helical content, with minima at 208 and 222 nm and a maximum at 190 nm. All of the variant complexes tested show a closely matching spectrum with only slight differences observed with the I16A, I19A and F14A variants. I16A and F14A show more pronounced minima at 208 nm, although the spectra have the same overall shape which suggests that differing levels of protein (or impurity) may be responsible for this result. I19A, on the other hand, shows a more pronounced difference which suggests that its structure may differ from that of wild-type complex. Overall, the data suggest that most of the TatAd variants, with the possible exception of I19A, assemble into TatAdCd complexes with apparently correct overall secondary structure, at least as judged using this method.
The C-terminal end of the TM span and the hinge region of TatAd are critical for its ability to substitute for E. coli TatA
We next examined the effects of the same substitutions on the bifunctional nature of TatAd. When produced in E. coli, TatAd can complement both the ΔtatAE and ΔtatB mutant strains (Barnett et al. 2008), and we produced the TatAd variants (without TatCd) from the pBAd-his construct in either the E. coli ΔtatAE or ΔtatB mutant strains. The aim was to pinpoint residues that are specifically important for either the TatA or TatB roles. Figure 6 illustrates the export of TorA when the TatAd variants are produced in the E. coli ΔtatAE strain. In the positive controls, we found that wild-type TatAd was able to support efficient translocation of TorA when produced in E. coli ΔtatAE cells (‘Ad ∆AE’ samples). This is clearly indicated by the presence of a clear band representing active TorA in the periplasm sample. As negative controls, we ran samples from E. coli ΔtatAE cells (∆AE samples) and no export is detected.

Identification of residues in the bifunctional TatAd protein that is critical for its TatA function. Single amino acid substitutions were introduced into the TatAd protein on plasmid pBAdh using site-directed mutagenesis. Substituted versions were produced in E. coli ΔtatAE cells and cells fractionated into Periplasm (P), Cytoplasm (C) and Membrane (M) fractions. Cell fractions were run on native polyacrylamide gels that were assayed for the presence of active TorA. The figure shows data from a substitutions within the periplasmic domain, b substitutions within the transmembrane spanning domain (TM) and c substitutions within the hinge region and amphipathic helix
Substitutions at the extreme N-terminus of TatAd (S3A, N4A and I5A) have no apparent effect on TatAd function in ΔtatAE cells and full complementation of the E. coli ΔtatAE TorA export defect is observed. This is perhaps not surprising as the same substitution had no effect on translocation of TorA by TatAdCd. In contrast, the G6A variant is completely blocked in translocation (a more surprising finding since the same substitution has no detectable effect on export by TatAdCd; Fig. 3a). P8A exhibits efficient export of TorA (Fig. 6a). Since this substitution blocked export by TatAdCd, this result suggests that Pro8 is important for the TatB function of TatAd but only in combination with the E. coli TatAC subunits.
TatAd variants L10A, I11A, L12A, V15A, I16A and I18A (where the alanine substitutions are localised to the TM span), all permit export of TorA in ΔtatAE cells (Fig. 6b), although the I18A variant supports only a rather low level of translocation. After this point, substitutions in the C-terminal end of the TM span, hinge region and start of the amphipathic helix have a much more severe effect on export activity, and essentially no export is observed with I19A, F21A, G22A, P23A, K25A, P27A and G30A when produced in ΔtatAE cells (Fig. 6c). Of these, I19A, F21A, G22A, P23A, K25A similarly blocked export of TorA by TatAdCd (Fig. 3). In contrast, P27A and G30A supported efficient export of TorA by TatAdCd (Fig. 3) indicating that these mutations block the TatA function of TatAd when expressed in ΔtatAE cells. Again, all translocation assays were performed at least twice and reproducible results were obtained. The effects of these substitutions are summarised in Fig. 4b; the data demonstrate that the C-terminal half of the TM span and the hinge region are critical for the TatA-type function of TatAd when expressed in E. coli.
The extreme N-terminus and N-terminal half of TatAd are critical for a TatB function
The same TatAd variants were analysed in the E. coli ΔtatB strain to identify residues and regions important for the TatB-type activity of TatAd. In the control test, TatAd restores TorA export when produced in ΔtatB cells (‘Ad ΔB’ panel). Among the substitutions made in the N-terminal periplasmic domain of TatAd, S3A, N4A and I5A all cause a complete block in export of TorA in ΔtatB cells (Fig. 7a). These substitutions had no detectable effect when the protein was produced in ΔtatAE cells, strongly suggesting that, while this region of the protein is not important for a TatA role, it is essential for the TatB role but only in combination with E. coli TatAC, since the same substitutions do not affect function when TatAd is expressed alongside TatCd. Figure 7a also shows that the G6A variant (inactive when produced in the ΔtatAE cells) is also inactive when produced in the ΔtatB strain; we observe no TorA band on the gel in the periplasmic lane. The same substitution did not block TatAdCd-mediated export (Fig. 3) suggesting that it may inadvertently prevent TatAd from functionally interacting with E. coli Tat components.

Identification of residues in the bifunctional TatAd protein that is critical for its TatB function. Single amino acid substitutions were introduced into the TatAd protein on plasmid pBAdh using site-directed mutagenesis. Substituted versions were produced in E. coli ΔtatB cells and cells fractionated into Periplasm (P), Cytoplasm (C) and Membrane (M) fractions. Cell fractions were run on native polyacrylamide gels that were assayed for the presence of active TorA. The figure shows results obtained with proteins containing a substitutions within the periplasmic domain, b substitutions within the transmembrane spanning domain (TM) and c substitutions within the hinge region and amphipathic helix. d The production of E. coli TatC in the ∆tatAE and ∆tatB strains was determined by running whole cells on SDS–PAGE gels and immunoblotting with anti-TatC antibodies
Figure 7b shows that the N-terminal part of the TM span is also particularly important for the TatB function. Although the L10A variant translocates TorA efficiently in the ΔtatB strain, the L12A and I18A variants support less efficient activity, and the G9A, I11A, V15A and I16A variants are all blocked in export. Substitutions in the C-terminal end of the TM span and the hinge region also have severe effects on transport activity. Weak export is observed with I19A and F21A but the remainder (G22A and P23A in the hinge region, plus K25A, P27A and G34A in the amphipathic region) are inactive (Fig. 7c). The results are summarised in Fig. 4; the overall picture is one in which the C-terminal TM span and hinge regions are important under all three sets of conditions (‘total’ TatAd function, and when functioning in place of E. coli TatA or TatB), whereas substitutions in the N-terminal regions specifically affect the ability of TatAd to function as a TatB protein.
We considered it important to check that any differences observed in the ability of the TatAd variants to translocate TorA in the ∆tatAE and ∆tatB strains were not due to any downstream effects of these deletions on production and stability of E. coli TatC. ∆tatAE, ∆tatB and ∆tatABCDE strains were grown overnight and equal numbers of whole cells were subjected to SDS-gel electrophoresis and immunoblotting using specific anti-TatC antibodies. The data (Fig. 7d) show that in both the ∆tatAE and ∆tatB strains, the TatC protein is present at broadly similar levels and is therefore unlikely to contribute to the differences in TatAd function observed between the two strains. In the control lane, no TatC is detectable in the ∆tatABCDE strain as expected.
Discussion
Studies on the Tat pathways of the Gram-positive bacterium Bacillus subtilis have identified interesting differences between its TatAC-type pathways and the TatABC systems of Gram-negative bacteria. The absence of a TatB component in most Gram-positive bacteria led to the idea that the TatA proteins may be bifunctional, fulfilling both TatA and TatB roles (Jongbloed et al. 2006). In support of this idea, E. coli TatA proteins carrying single substitutions at the extreme N-terminus were found to support translocation of a sensitive TorA-MalE reporter protein (Blaudeck et al. 2005) and the native TorA protein (Barrett et al. 2007) in the absence of TatB. Although TatB is normally essential for Tat translocation in E. coli, these studies confirmed that TatB is, in many respects, similar to TatA in structural terms. Another study found that Colicin V could be translocated by the E. coli Tat pathway in the absence of TatB (Ize et al. 2002). Using a direct approach, we showed that the B. subtilis TatAd protein is able to complement both the E. coli ΔtatAE and ΔtatB mutant strains (Barnett et al. 2008). This makes TatAd a useful tool for identifying residues important for both TatA- and TatB-type functions, and two main points have emerged from this study.
First, the data highlight an essential domain that encompasses the C-terminal part of the TM span, the ‘hinge region’ (FGP motif) and initial part of the amphipathic helix. An entire series of alanine substitutions in these areas results in dramatic reductions in translocation activity by TatAdCd when produced in the E. coli ∆tatABCDE strain. Circular dichroism studies do not show any indication that the TatAd variants differ significantly in overall secondary structure compared to wild-type TatAd or that the alanine substitutions disrupt interactions with the TatCd partner subunit. Of those substitutions that block export by TatAdCd, the majority also block the ability of TatAd to substitute for either E. coli TatA or TatB in the ∆tatAE or ∆tatB strains. Analysis of Fig. 4 highlights the point that this stretch of amino acids plays a key role in the functioning of TatAd in all cases. Studies on the E. coli TatA and TatB proteins have shown that the same regions are important in those proteins (Barrett and Robinson 2005; Greene et al. 2007; Ize et al. 2002; Lee et al. 2006) but it is notable that many of the TatAd substitutions lead to an absolute block in translocation, while most of the above substitutions in the E. coli TatA/TatB proteins led to a drop in activity rather than an absolute block. In this sense, the TatAd variants give unusually clear-cut results (possible reasons are discussed below).
When the TatAd mutagenesis data are considered together with previous studies on the E. coli TatA/B proteins, the clear conclusion is that the C-terminal half of the TM span and ‘FGP hinge’ region are of paramount importance for the entire TatA/B-family of proteins, from both Gram-positive and Gram-negative bacteria. The actual roles of these domains in the overall translocation mechanism have yet to be determined with confidence, but we consider it likely that these domains may be involved in inter-subunit interactions between these family members. Given that TatB is closely associated with TatC in E. coli (Bolhuis et al. 2001) this region of TatB may form the site for communication with the TatA complex when the TatA complex coalesces with TatBC. In B. subtilis, the TatAd protein may interchangeably associate with TatC in the TatAC complex or with other TatAd subunits in a separate complex, but this region may again be the key site for association of the two complexes.
The second main conclusion is that other amino acid substitutions have very different effects on the ability of TatAd to complement the ∆tatB or ∆tatAE mutant strains. In particular, the extreme N-terminal part of the TatAd protein is critical for performing a TatB role in E. coli. A long series of substitutions (including most of the variants from S3A to I16A) lead to a total block in TorA export when the variant forms are produced in ∆tatB cells. These substitutions encompass the short periplasmic domain and the N-terminal half of the TM span; clearly, these regions contain determinants that are essential for the ability of TatAd to carry out a TatB function in E. coli. Of this long stretch of residues, only the G6A variant was found to prevent it functioning as TatA in E. coli (by blocking export activity in ∆tatAE cells expressing TatAd), again emphasising that this region has a special importance for the TatB-type role. These results are consistent with a previous study in which amino acid substitutions at the extreme N-terminus (at positions 2–6) of E. coli TatA enabled the protein to act as TatB (Blaudeck et al. 2005). Although the TatAd proteins are present at a slightly lower level when produced in ∆tatB cells compared with ∆tatAE cells, detectable levels of protein on Western blots is sufficient for translocation activity to be observed.
While these N-terminal substitutions block TorA export in ∆tatB cells producing TatAd, the same substitutions do not lead to a block in activity when TatAd is produced together with TatCd (i.e. when TatAdCd is produced in a tat null mutant). We believe that the results reflect the different copy numbers of the Tat systems under consideration. The TatAdCd subunits are co-ordinately produced after expression from a multicopy plasmid that has been shown to lead to the synthesis of Tat components at levels that are ca. 50-fold higher than those of the wild-type Tat system in E. coli (Bolhuis et al. 2001). In contrast, the ∆tatAE and ∆tatB complementation tests involve production of TatAd alone, which therefore interacts with 50-fold lower wild-type levels of the E. coli TatA and TatC subunits. These will be in limiting quantities and translocation defects will be more much readily exposed since the levels of complete translocase will be far lower. It is thus highly likely that the same N-terminal TatAd residues are important for functioning with TatCd, but that the much higher Tat copy number masks the reductions in translocation efficiency when TatAdCd are co-produced. In direct support of this idea, previous studies have shown that the vast majority of amino acid substitutions in E. coli TatB are functionally invisible when the variant forms are produced at relatively high levels using a multicopy plasmid (Barrett et al. 2005). Very few tat mutations lead to a genuinely complete block in export.
Finally, our data reveal one key determinant within the TatAd protein that may be absent from the TatA and TatB proteins of Gram-negative bacteria. Substitution of Pro8 leads to a complete block in export, and it is notable that this residue is effectively conserved in all Gram-positive TatA proteins. This suggests a core role for the residue in these proteins. The residue is also conserved in Streptomyces species and, since these Gram-positive organisms do contain TatABC-type systems, it would appear that this Pro residue is not correlated with a bifunctional activity for the TatA proteins.
References
Alami M, Luke I, Deitermann S, Eisner G, Koch HG, Brunner J, Müller M (2003) Differential interactions between a twin-arginine signal peptide and its translocase in Escherichia coli. Mol Cell 12:937–946
Barnett JP, Eijlander RT, Kuipers OP, Robinson C (2008) A minimal Tat system from a Gram-positive organism: a bifunctional TatA subunit participates in discrete TatAC and TatA complexes. J Biol Chem 283:2534–2542
Barrett CM, Robinson C (2005) Evidence for interactions between domains of TatA and TatB from mutagenesis of the TatABC subunits of the twin-arginine translocase. Febs J 272:2261–2275
Barrett CM, Mangels D, Robinson C (2005) Mutations in subunits of the Escherichia coli twin-arginine translocase block function via differing effects on translocation activity or tat complex structure. J Mol Biol 347:453–463
Barrett CM, Freudl R, Robinson C (2007) Twin arginine translocation (Tat)-dependent export in the apparent absence of TatABC or TatA complexes using modified Escherichia coli TatA subunits that substitute for TatB. J Biol Chem 282:36206–36213
Berks BC (1996) A common export pathway for proteins binding complex redox factors? Mol Microbiol 22:393–404
Blaudeck N, Kreutzenbeck P, Muller M, Sprenger GA, Freudl R (2005) Isolation and characterisation of bifunctional Escherichia coli TatA mutant proteins that allow efficient tat-dependent protein translocation in the absence of TatB. J Biol Chem 280:3426–3432
Bolhuis A, Mathers JE, Thomas JD, Barret CM, Robinson C (2001) TatB and TatC form a functional and structural unit of the twin-arginine translocase from Escherichia coli. J Biol Chem 276:20213–20219
Casadaban MJ, Cohen SN (1980) Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J Mol Biol 138:179–207
Gohlke U, Pullan L, McDevitt CA, Porcelli I, de Leeuw E, Palmer T, Saibil HR, Berks BC (2005) The TatA component of the twin-arginine protein transport system forms channel complexes of variable diameter. Proc Natl Acad Sci USA 102:10482–10486
Greene NP, Porcelli I, Buchanan G, Hicks MG, Schermann SM, Palmer T, Berks BC (2007) Cysteine scanning mutagenesis and disulfide mapping studies of the TatA component of the bacterial twin arginine translocase. J Biol Chem 282:23937–23945
Hu Y, Zhao E, Li H, Xia B, Jin C (2010) Solution NMR structure of the TatA component of the twin-arginine protein transport system from gram-positive bacterium Bacillus subtilis. J Am Chem Soc 45:15942–15944
Ize B, Gerard F, Zhang M, Chanal A, Voulhoux R, Palmer T, Filloux A, Wu LF (2002) In vivo dissection of the Tat translocation pathway in Escherichia coli. J Mol Biol 317:327–335
Jongbloed JD, Grieger U, Antelmann H, Hecker M, Nijland R, Bron S, van Dijl JM (2004) Two minimal Tat translocases in Bacillus. Mol Microbiol 54:1319–1325
Jongbloed JD, van der Ploeg R, van Dijl JM (2006) Bifunctional TatA subunits in minimal Tat protein translocases. Trends Microbiol 14:2–4
Lee PA, Buchanan G, Stanley NR, Berks BC, Palmer T (2002) Truncation analysis of TatA and TatB defines the minimal functional units required for protein translocation. J Bacteriol 184:5871–5879
Lee PA, Orriss GL, Buchanan G, Greene NP, Bond PJ, Punginelli C, Jack RL, Sansom MS, Berks BC, Palmer T (2006) Cysteine-scanning mutagenesis and disulfide mapping studies of the conserved domain of the twin-arginine translocase TatB component. J Biol Chem 281:34072–34085
Müller M (2005) Twin-arginine-specific protein export in Escherichia coli. Res Microbiol 156:131–136
Oates J, Barrett CM, Barnett JP, Byrne KG, Bolhuis A, Robinson C (2005) The Escherichia coli twin-arginine translocation apparatus incorporates a distinct form of TatABC complex, spectrum of modular TatA complexes and minor TatAB complex. J Mol Biol 346:295–305
Randall LL, Hardy SJ (1986) Correlation of competence for export with lack of tertiary structure of the mature species: a study in vivo of maltose-binding protein in E. coli. Cell 46:921–928
Robinson C, Bolhuis A (2004) Tat-dependent protein targeting in prokaryotes and chloroplasts. Biochim Biophys Acta 1694:135–147
Santini CL, Ize B, Chanal A, Muller M, Giordano G, Wu LF (1998) A novel sec-independent periplasmic protein translocation pathway in Escherichia coli. EMBO J 17:101–112
Sargent F, Bogsch EG, Stanley NR, Wexler M, Robinson C, Berks BC, Palmer T (1998) Overlapping functions of components of a bacterial Sec-independent protein export pathway. EMBO J 17:3640–3650
Sargent F, Stanley NR, Berks BC, Palmer T (1999) Sec-independent protein translocation in Escherichia coli. A distinct and pivotal role for the TatB protein. J Biol Chem 274:36073–36082
Schreiber S, Stenge R, Westermann M, Volkmer-Engert R, Pop O, Muller PM (2006) Affinity of TatCd for TatAd elucidates its receptor function in the Bacillus subtilis Twin-srginine translocation (Tat) translocase system. J Biol Chem 281:19977–19984
Silvestro A, Pommier J, Pascal MC, Giordano G (1989) The inducible trimethylamine N-oxide reductase of Escherichia coli K12: its localization and inducers. Biochim Biophys Acta 999:208–216
Weiner JH, Bilous PT, Shaw GM, Lubitz SP, Frost L, Thomas GH, Cole JA, Turner RJ (1998) A novel and ubiquitous system for membrane targeting and secretion of cofactor-containing proteins. Cell 93:93–101
Wexler M, Sargent F, Jack RL, Stanley NR, Bogsch EG, Robinson C, Berks BC, Palmer T (2000) TatD is a cytoplasmic protein with DNase activity. No requirement for TatD family proteins in sec-independent protein export. J Biol Chem 275:16717–16722
Acknowledgments
We greatly appreciate the gift of plasmids pBAdCd and pBAd-his from Prof. Oscar P. Kuipers (Groningen Biomolecular sciences and biotechnology institute, Haren, the Netherlands). We also thank Prof. Matthias Muller for the gift of TatC antibodies and J. Muller for anti-TatAd antibodies. This work was funded by a Biotechnology and Biological Sciences Research Council (BBSRC) grant to CR and SM and a BBSRC studentship to JPB.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Wolfgang Buckel.
Rights and permissions
About this article
Cite this article
Barnett, J.P., Lawrence, J., Mendel, S. et al. Expression of the bifunctional Bacillus subtilis TatAd protein in Escherichia coli reveals distinct TatA/B-family and TatB-specific domains. Arch Microbiol 193, 583–594 (2011). https://doi.org/10.1007/s00203-011-0699-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-011-0699-4