
Abstract
Gene probe hybridization was used to determine distribution and expression of co-metabolic genes at a contaminated site as it underwent in situ methanotrophic bioremediation of trichloroethylene (TCE). The bioremediation strategies tested included a series of air, air:methane, and air:methane:nutrient pulses of the test plot using horizontal injection wells. During the test period, the levels of TCE reduced drastically in almost all test samples. Sediment core samples (n = 367) taken from 0 m (surface)–43 m depth were probed for gene coding for methanotrophic soluble methane monooxygenase (sMMO) and heterotrophic toluene dioxygenase (TOD), which are known to co-metabolize TCE. The same sediment samples were also probed for genes coding for methanol dehydrogenase (MDH) (catalyzing the oxidation of methanol to formaldehyde) to assess specifically changes in methylotrophic bacterial populations in the site. Gene hybridization results showed that the frequency of detection of sMMO genes were stimulated approximately 250% following 1% methane:air (v/v) injection. Subsequent injection of 4% methane:air (v/v) resulted in an 85% decline probably due to nutrient limitations, since addition of nutrients (gaseous nitrogen and phosphorus) thereafter caused an increase in the frequency of detection of sMMO genes. Detection of TOD genes declined during the process, and eventually they were non-detectable by the final treatment, suggesting that methanotrophs displaced the TOD gene containing heterotrophs. Active transcription of sMMO and TOD was evidenced by hybridization to mRNA. These analyses combined with results showing the concomitant decline in TCE concentrations, increases in chloride concentration and increases in methanotroph viable counts, provide multiple lines of evidence that TCE remediation was caused specifically by methanotrophs. Our results suggest that sMMO genes are responsible for most, if not all, of the observed biodegradation of TCE. This study demonstrates that the use of nucleic acid analytical methods provided a gene specific assessment of the effects of in situ treatment technologies.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Trichloroethylene (TCE) and tetrachloroethylene (PCE) are primarily volatile organic anthropogenic compounds present commonly in soil and groundwater. TCE is introduced in the environment, as a result of its manufacture and use as an ingredient in adhesives, paint removers, typewriter correction fluid. Exposure to TCE and PCE over extended periods has been known to cause impaired heart function, nausea, dizziness, unconsciousness and nerve, liver and kidney damage (http://www.dhs.ca.gov/ohb/HESIS/tce.htm). Recent studies performed with mice and rats have further suggested that high levels of TCE may lead to cancer of the liver, kidney, or lung. Due to these reasons, the US Environmental Protection Agency has regulated a maximum contaminant level of 5 ppb of both TCE and PCE in drinking water in the United States (http://www.atsdr.cdc.gov/tfacts19.html).
Microbial bioremediation is an effective tool in removing several volatile organic compounds including TCE and PCE from contaminated soil and groundwater (Shannon et al. 1995). It has been previously suggested that the aerobic bioremediation of TCE is efficiently mediated by the ubiquitous methane-oxidizing methanotrophs (Bowman et al. 1993). Testing the feasibility of this process, Semprini et al. (1992) reported enhanced in situ biotransformation of TCE by stimulating the indigenous methanotrophs via injection of methane in water into an aquifer at the Moffett Naval Air Station, California. The U.S. Department of Energy (U.S. DOE) utilized direct methane/air injection in a similar strategy in a large-scale demonstration at an aquifer extensively contaminated with TCE and PCE at the Savannah River Site (SRS) near Aiken, South Carolina (Hazen 1999). Methanotrophs like Methylosinus trichosporium that have been known to be capable of TCE degradation (Burrow et al. 1984; Oldenhuis et al. 1989) oxidize methane catalyzed by the enzyme methane monooxygenase (MMO), two forms of which have been known to exist, the soluble methane monooxygenase (sMMO) and the particulate methane monooxygenase (pMMO). Further, TCE degradation can also be mediated by the enzyme toluene dioxygenase (TOD) present in some heterotrophic bacteria, for example, the Pseudomonas species (Van Hamme et al. 2003). These two enzymes, MMO and TOD, are well known for their broad substrate specificity as well as high rates of TCE degradation (Burrow et al. 1984; Zylstra et al. 1989; Oldenhuis and Janssen 1993). To monitor the biodegradation of TCE in our field studies, we used DNA gene probes of sMMO and TOD genes as well as of methanol dehydrogenase (MDH). MDH is believed to be highly conserved in methanotrophs and serves as a positive control for this group of organisms (McDonald and Murrell 1997; McDonald et al. 1997). In addition, mRNA was isolated from a subpopulation of soil samples and probed for the presence of sMMO and TOD transcripts as an indicator of the degradative activity of those genes (Sayler et al. 2001). PCE, while not degraded by methanotrophs, is easily degraded to TCE by anaerobic reductive dechlorination. This was demonstrated to occur in the large number of anaerobic microniches created in the subsurface by the biostimulation of methanotrophs (Phelps et al. 1990; Enzien et al. 1994; Pfiffner et al. 1997; Hazen 1999) in bulk aerobic environments. Thus, stimulation of methanotrophs will result in significant biodegradation of PCE. An added advantage of co-metabolic aerobic biodegradation of chlorinated solvents over strictly anaerobic reductive dechlorination is that it does not require depletion of competing terminal electron acceptors (e.g. O2, NO3, and SO4), it does not produce toxic daughter products (e.g., vinyl chloride), and it does not alter redox conditions so that toxic metals become more soluble.
In this article, we present the results of using DNA probes to monitor the frequency, distribution, and shifts of co-metabolic genes relevant to TCE degradation at the TCE contaminated SRS test site. The specific objective of this study was to explore the use of nucleic acid analytical methods to provide a gene specific assessment of the effects of the in situ treatment technology.
For our study at the SRS test site, two horizontal wells were installed (AMH-1 and AMH-2) as outlined in Fig. 1. One of them (AMH-1) was used for the injection of air, methane, and nutrients, while the second well (AMH-2) was used for vacuum extraction. The nutrients triethyl phosphate (TEP, 0.007%), and nitrous oxide (N2O, 0.07%) were injected together in a gaseous form when necessary (referred as N + P in the rest of the article), since bioremediation rates can usually be significantly enhanced by increasing the supply of nutrients for the indigenous contaminant-degrading microorganisms. Sediment core samples were taken prior to and after each treatment from the several monitoring boreholes drilled for evaluating the efficacy of the techniques being tested. The detailed process and the success of the previous stage of the project- the air-stripping phase has been detailed by Hazen (1999).

Location of pre-test and treatment borings at the IDS (integrated demonstration site). The horizontal wells include AMH-1 (injection well) and AMH-2 (extraction well)
Results and discussion
Site characteristics
Table 1 summarizes the different treatments performed at the monitoring wells of the site. Figure 1 shows the location of the various monitoring boreholes drilled, and the horizontal injection/extraction wells installed during the bioremediation process. As shown in the figure, core samples were collected from the wells designated for the different treatments. Four lithostratigraphic zones were identified for the core samples, namely, the Upland Unit (surface to 12 m deep), Tobacco Road Sands (12–20 m), Dry Branch formation (20–33 m), and Santee formation (33–46 m). The total organic content (TOC) was highest in shallow sediments (2 m depth) and decreased with depth. Comparison of pre-test samples to post-air samples indicated a decrease in the median TOC levels that could be attributed to the microbial utilization of available carbon (Eddy et al. 1993). The nitrite and nitrate were found to be below detection limits (<1 mg/kg). Total nitrogen as measured by the Kjeldahl method was also very low (<20 mg/kg). Orthophosphate was readily detectable in sediment samples; however, the form which it was present in the sediment was largely insoluble apatite and therefore most likely biologically unavailable.
Sediment microbial population
Bacterial populations in the sediment samples ranged mostly between 103 and 106 CFU/g (Fig. 2). The low population densities observed could be attributed to the low organic and nutrient levels in the sediments (Eddy et al. 1993; Pfiffner et al. 1997). The TVC values corresponded well with biomass values as calculated from phospholipid fatty acid (PLFA) abundance analyses (data not shown). Regression analyses of TVC indicated that a bimodal pattern occurred across the depth profile. The bacterial populations were higher in the Upland Unit (formation below soil) and in the Santee formation (which includes the capillary fringe and upper aquifer) as compared to samples from the Tobacco Road Sands and the Dry Branch formation during the various treatments. This bimodality was not believed to be related to clay content and the pattern became somewhat less pronounced following the methane:air treatments. Overall, TVC values increased from pre-test samples peaking after the 1% methane:air treatment (Fig. 2). Contingency analyses showed that methylotrophic and methanotrophic MPN populations in individual samples were strongly co-associated with each other and thus seem to occupy small niches throughout the various sediment layers. Methanotroph MPN values were found to be highest after the 4% methane:air treatment (Fig. 2). Subsequent to the 4% methane:air:N + P treatment, a second- to third-order of magnitude increase in MPNs corresponding to methylotroph and methanotroph was observed in samples primarily near the capillary fringe (45–47 m depth). Similar large increases in bacterial populations were also reported as measured by acridine orange direct counting after the 1% methane:air campaign at the capillary fringe. In other areas of the vadose zone, no such stimulation was apparent.

The effect of treatment regimes on total viable heterotrophic counts (TVC) and methylotroph and methanotroph most probable numbers (MPN) categorized by lithostratigraphic zones. The methylotroph and methanotroph MPN were non-detectable for the pre- and post-air campaigns
DNA probe hybridization
Because of the great variability of the hybridization data, it was judged expedient to express the DNA gene probing data qualitatively as either present or absent. For each well site, the data are presented as the percent of the total number of samples from that site that yielded a detectable hybridization signal. On the basis of the standard curve included on each slot blot, the minimum detection limit was determined to be approximately 10 pg of target sequence, which is equivalent to 1 × 107 copies of a 1-kb target sequence. With one-fifth of the DNA from a 50 g sample loaded per slot, the detection level for a single copy gene would therefore be approximately 1 × 106 copies/g sediment. Strong DNA hybridization signals were observed for some samples with the sMMO probe located specially at the capillary fringe coinciding with the pattern seen for the TVC and MPN results (Fig. 3). Since probing of sediments from the 4% methane, nutrient regime yielded a high proportion of sMMO positively probing DNA samples and the highest proportion of MDH probing samples, suggesting an increase in methanotrophs (Fig. 3), mRNA was extracted from these sediments and probed for sMMO transcripts. These samples were also probed for TOD mRNA. Interestingly, the sediment samples that yielded detectable TOD and sMMO mRNA yielded lower corresponding DNA probe results. While TOD and sMMO mRNA was found at the 12.2–33.5 m lithostratigraphic zone (Table 2), sMMO DNA occurrence was highest in the 33–46 m zone, and TOD DNA was virtually undetectable with the 4% methane, nutrient treatment (Fig. 3). One possible explanation for this anomaly is that the specific activity sMMO RNA probes may have been higher than the corresponding DNA probe, or that the RNA extraction method was more efficient compared to the DNA extraction method. Concerning the RNA values, another source of error may have been introduced by normalizing the mRNA to the rRNA content. It is now generally accepted that rRNA content is not a constant value but varies as a function of metabolic activity. The detection of sMMO mRNA and DNA at levels corresponding to much higher cell densities than the methanotrophic MPN results reported here and elsewhere (Brockman et al. 1995) suggests that culture methods for methanotrophs can greatly underestimate actual population densities. This may be in part due to inefficiency in disrupting sediment-associated methanotroph microcolonies into component cells prior to setting up MPN dilutions. Interestingly, sMMO was detected in several samples that did not produce a detectable methanotroph MPN result. This is perhaps due to the existence of biases in culturing these microbes. The data support the premise that gene probing, while a less sensitive assay compared to MPNs provides critical complementary information on microbial populations in environmental samples. To provide for unbiased conclusions from gene probing results, especially in heterogeneous places like the test site, analyses need to be pursued on a broad scale to obtain statistically significant trends. Factors affecting such analyses would partially be dictated by the physical dimensions of the contaminant plume and the various geological and hydrological unconformities that may be present at the site.

The effect of treatment regimes on DNA gene hybridization incidence across the IDS. Numbers on bars are percent values indicating the proportion of positive DNA gene probe results for samples taken from a particular core sample. Values in parentheses indicate only samples from the Upland Unit and/or Tobacco Road sands were examined for the particular core
Incidence of target genes in the test site
Of 367 DNA samples taken during various treatments 107 (30%), 98 (28%), and 85 (24%) showed presence of sMMO, MDH, and TOD genes, respectively. DNA extraction efficiency has been reported to be lower in high clay sediments (Frostegard et al. 1999). However, in this study, the incidence of positive hybridization with each probe was not significantly affected by clay content category (0–20%, 20–40%, and so on; data not shown), or by concentrations of total organic carbon (TOC), TCE, and PCE in the samples. Further, the pH or water content (data not shown) also did not seem to affect positive hybridization results. Given these observations, the percentage of samples positively hybridizing to each of the gene probes in relation to the location of wells and lithostratigraphic zone as a function of experimental protocol was calculated by dividing the number of positive samples for a given target gene over the total number of appropriate samples. The location of the wells did not appear to be an important factor for determining the distribution of positive hybridizations (Fig. 3). Lithostratigraphic zone appeared to be a major determining factor for hybridization incidence (Fig. 4). In support of this conclusion, non-parametric Runs test showed that hybridization data taken for the post-air, 4% methane:air and (v/v) methane:air:N + P campaign samples were significantly non-random (P < 0.01).

The effect of treatment regimes on DNA gene probe hybridization incidence across the depth profile, categorized by lithostratigraphic zones. ND non-detectable
Shifts of target genes induced by treatments
The number of samples showing positive DNA sMMO hybridization signals was significantly increased by the initial air treatment campaign. The total average incidence increased from 16 ± 5% in pre-air samples to 40 ± 15% in post-air samples. Only one monitoring well, MHT-11C, located near the extraction well had a sMMO gene incidence less than pre-test samples. All well samples showed a low sMMO incidence following the 4% methane:air pulse, averaging 6 ± 6%. This result appears to be in contrast to the MPN values. However, it is possible for culturability to increase while gene densities decrease due to competitive interactions between microbial types and dynamic changes in community structure and cell physiological state (Pfiffner et al. 1997). Injection of nutrients appeared to stimulate the population of the methanotroph community based upon an increase in the sMMO gene incidence to 28 ± 12%. Since the MDH DNA probe was used as an indicator of the total methylotrophic community (both methane- and methanol-assimilators), MDH genes were expected to be either at an equal or greater incidence than the sMMO genes. This was generally true, with much higher incidences of MDH genes compared to sMMO genes in the last two campaigns. However, the incidence of sMMO genes was generally much higher than MDH genes during the 1% methane:air campaign (Fig. 3). A possible explanation is that major shifts in methylotrophic subpopulations occurred during the demonstration, and the moxF gene used as the MDH probe may not hybridize to all potential MDH targets due to genetic diversity (Bowman et al. 1993). However, the MDH probe used in this study was very poor; under conditions of a 35% mismatch only 4 of 25 type II methanotrophs probed positive with the moxF gene probe (Bowman et al. 1993). Recent work has shown that the methanol dehydrogenase gene is very diverse. A study that used a PCR primer set designed and validated using fourteen mxaF (methanol dehydrogenase) containing methylotrophs has detected greater diversity over earlier primer sets derived from only several methylotrophs (Neufeld et al. 2007). Considering the fact that the moxF probe was designed from a single sequence (Methylobacterium oganophilium) and that more recent environmental studies have demonstrated great diversity in the methanol dehydrogenase gene the anomaly between MDH and sMMO is explained.
In comparison, the sMMO probes used in this study may be retrospectively judged very good. While both of the sMMO probes, an mmoYBZC end labeled fragment or a PCR amplified mmoB fragment, were derived solely from M. trichosporium, subsequent studies demonstrated that the mmoB gene is highly conserved. Sequencing of M. capsulatus Bath and M. tichosporium (McDonald 1995), Methylocystis sp. Strain W114 (Grosse et al. 1999), Methylocystis sp. Strain M (McDonald et al. 2008) and Methylomonas species (Shigematsu et al. 1999) led to the development of a number of different primer sets for the amplification of the mmoX gene from methanotrophs. All the environmental studies using these primer sets suggest that the sMMO genes have limited diversity (McDonald et al. 2008). Therefore, it is reasonable to assume that the sMMO probing data presented here are representative of the actual sMMO activity at the site.
The present study did not target the particulate methane monooxygenase (pMMO) which is found in all methanotrophs with the exception of the genus Methylocella (Theisen 2005) but has a limited substrate range (Hanson and Hanson 1992). In comparison, sMMO is found only in several methanotrophs and has a much broader substrate range. The fact that sMMO expressing cells can degrade chlorinated ethenes at a much higher rate compared to pMMO expressing cells suggests that sMMO expressing cells always outperform pMMO expressing cells. For example, considering the degradation of TCE by M. trichosporium OB3b, the V max for sMMO was greater than 100× that for pMMO (Lee et al. 2006). The innate superiority of sMMO activity is disputed in a recent paper which suggests that at high concentrations of solvents, pMMO expressing cells may be preferred over sMMO expressing cells (Lee et al. 2006). In this study, at concentrations of 100 μM of VC, DCE and TCE pMMO expressing cells degraded the solvent mixtures in 50% less time compared with sMMO expressing cells. However, even at the 100 μM level, sMMO expressing cells degraded the same percentage of TCE compared with pMMO expressing cells (Lee et al. 2006). Considering the impact of the relative rates of the two enzyme systems on the interpretation of the data in the present study, since the concentration of TCE or PCE did not exceed 25 μM, the rate advantage of pMMO cells over sMMO cells at high solvent concentrations need not be considered here. Additionally, considering the higher degradative rate of sMMO cells compared to pMMO cells at the site solvent concentrations in this study, to have an realistic effect on the interpretation of the data presented here, the population of pMMO expressing cells would have to be 100× greater compared to sMMO expressing cells.
TOD DNA gene frequency showed no apparent stimulation following the initial air injection of the in situ bioremediation. Following methane injection, TOD hybridization incidence showed a general decline throughout the site in all well locations and lithostratigraphic zones and finally it was non-detectable. Sample by sample contingency analysis showed the sMMO and TOD genes at the site to be significantly associated (contingency coefficient = 0.9) before methane injection, suggesting organisms associated with the two genes tend to occupy the same niches at the site. Chi-squared analysis also agreed with the above conclusion (P < 0.05). The rapid drop in TOD gene frequency after the methane pulse invalidated further contingency testing. PLFA analysis of total microbial communities in the post-air sediment samples showed presence of relatively high trans/cis and cyclopropyl/monoenoic ratios. This suggests that the microbial community was undergoing stress (Hazen 1999), which could be attributed to lack of nutrients (namely nitrogen and phosphorus). To corroborate this finding, addition of gaseous nitrogen and phosphorus resulted in a stimulation of the methylotrophic populations of the microbial community as observed by increases in sMMO and MDH DNA hybridization incidence (Figs. 3, 4). Similar recovery was not observed when the TOD gene probe was used. This could be due to the fact that TOD genotypes were unable to effectively compete for or assimilate the nutrients, or the fact that their recovery could be significantly slower than the methylotrophic community.
TCE degradation
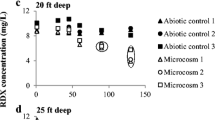
TCE degradation was observed in situ in almost all the depths throughout the test site (Fig. 5a, b). Overall, all reductions were highly significant in both the sediment and the groundwater, although contaminated groundwater outside the zone of influence continued to infiltrate the treatment area. In addition, the modeling of all variables in comparison to air stripping alone showed that the dominant process was biodegradation of TCE by methanotrophs (Brockman et al. 1995; Pfiffner et al. 1997; Travis and Rosenberg 1997; Hazen 1999). The levels of TCE decreased rapidly following the air injection treatment, and after the 1% methane:air treatment, it was as low as almost 10 ppb. The in situ degradation of TCE was mediated by the resident microbial flora. In support of this, the TVC peaked following the 1% methane:air treatment (Fig. 2) when the levels of TCE were at the lowest. In addition, the detection of sMMO genes (indicator of methanotrophs) also increased at that time period (Fig. 3).

The distribution of the levels of TCE in the test plot. As indicated, the color intensity corresponds to the concentration of TCE and the axes to the actual latitude (SRS units), longitude (SRS units), and depth in feet above mean sea level
Conclusions
TCE was degraded at the Savannah River test site to very low levels. The degradation of TCE seems to have been mediated mainly by the resident methanotrophic microbial population as evidenced by the DNA hybridization results. The DNA extraction and slot blot hybridization methodology used in this study was sufficiently sensitive to demonstrate gene specific assessment of the effects of addition of air, methane and gaseous nutrients on the degradation of TCE and PCE (70–90%) (Hazen 1999). The incidence of detectable hybridization for all the three kinds of gene-probes tested was related to the treatment procedure and to the lithostratigraphy of the test zone. It was observed that hybridization to the probes was not influenced by location of well, clay content, sediment organic carbon content and the concentration of the organic contaminant. The Santee formation (33–46 m) had the highest incidence of hybridization to the probes especially at the capillary fringe and at the upper part of the aquifer.
The incidence of sMMO genes increased with addition of air—only and with 1% methane:air, while the incidence of MDH genes increased with the 4% methane:air:N + P treatment. In contrast, the TOD genes decreased upon the addition of methane to the site and were not detected after the 4% methane:air:N + P treatment. However, the detection of sMMO genes in several samples did not correspond to the analogous methanotroph MPN data. Further, sMMO mRNA and DNA was detected at levels corresponding to much higher cell densities than the MPN results depicted, suggesting that culture-based methods for enumeration of methanotrophs often underestimate actual population densities. The data support the hypothesis that gene probing, although a less sensitive assay compared to the MPN method, provides critical complementary information on microbial populations in environmental samples. This technique aides to develop a greater understanding of the factors influencing the abundance, distribution, and dynamics of organisms and genes that may be amendable to enhanced in situ bioremediation. Our results also suggest that bacteria capable of degrading organic contaminant like TCE can be stimulated in situ by the addition of relatively simple nutrients like methane, air, phosphorous and nitrogen as gasses, a much more efficient injection strategy for biostimulation. The trend observed in gene distribution induced by various treatment protocols employed in this study demonstrate that in situ bioremediation treatment needs to be carefully drafted in accordance with a site’s geochemical profile. Further, the nutritional requirements and subsequent amendments for the targeted indigenous microbial community warrant careful considerations for successful biostimulation and bioremediation of contaminants.
Experimental procedures
Core sediment samples
Samples were collected at various depths ranging from 0 to 43 m as described earlier (Hazen 1991, 1999). Samples were collected before, during and after each treatment, i.e. air injection, after 1% methane in air continuous injection, after 4% methane in air continuous injection and after 4% methane in air pulsed with N + P. The treatments were performed in sequence as above and lasted 14 months. Immediately after collection, samples were stored at 4°C for culture-based measurements and at −80°C for nucleic acid probe analyses.
Microbial enumeration
Total viable counts were estimated by diluting 1 g of sediment samples in 9 ml of PBS buffer (8.3 mM Na2HPO4, 16 mM NaH2PO4 and 0.15 M NaCl at pH 7.6) and then plating 0.1 ml of a serial dilution of the sample onto yeast extract-glucose (YEPG) agar plates. YEPG medium consisted of 0.2 g yeast extract, 2 g peptone, 1 g d-glucose, 0.2 g NH4NO3, and 15 gl−1 agar in distilled water. Plates were incubated for 7 days at 25°C. Population densities were calculated as colony forming units (CFU). The most probable number (MPN) procedure (Brockman et al. 1995) was used to enumerate methylotrophs and methanotrophs in the site samples. For MPN enumeration, modified Pfennig SL-9 mineral salts medium (10 ml) was used. This medium consisted of 0.9 g NaCl, 0.1 g MgSO4·7H2O, 0.5 g NaNO3, 0.044 g NaH2PO4, 0.26 g K2HPO4, 10 ml mineral solution A, and 1 ml vitamin solution in 1 l distilled water. Mineral solution A consisted of 1.5 g nitriloacetic acid, 0.2 g FeCl2·4H2O, 0.1 g MgCl2·6H2O, 0.02 g Na2HPO4·2H2O, 0.1 g MnCl2·4H2O, 0.1 g CaCl2·2H2O, 0.05 g ZnCl2, 0.002 mg CuCl2, 0.002 mg H2BO3, 0.01 g Na2MoO4·2H2O, 0.017 g Na2SeO3, and 0.024 g NiCl2·6H2O in 1 l of distilled water. The vitamin solution consisted of 0.02 g biotin, 0.02 g folic acid, 0.1 g pyridoxine, 0.05 g thiamine-HCl, 0.05 g riboflavin, 0.05 g niacin, 0.05 g nicotinic acid, 0.05 g pantothenic acid, 0.001 mg cyanocobalamin, 0.05 g p-aminobenzoic acid, 0.05 g lipoic acid in 1 l distilled water. To prepare samples from MPN analyses, 1 g of homogenized sediment was added to each of two sets of triplicate tubes. These initial tubes were vortexed well forming a slurry. Each set of the sediment slurries were then serially diluted and sealed with butyl rubber stoppers. For enumeration of methylotrophs, methanol (0.2% v/v final concentration) was added as the carbon source and for methantrophs, enough methane was added to create a 1:9 methane: air atmosphere. Tubes were incubated for 4–6 weeks at room temperature. Further, transfers were performed to fresh media tubes to confirm growth.
DNA-targeted probes
For samples from cores MHT-1C, MHT-6C, and MHT-9B, DNA probes were mmoYBZC (2.2 kb EcoRI fragment previously cloned into pBlueScript II SK) (Cardy et al. 1991; Tsien and Hanson 1992) encoding sMMO, moxF (2.5 kb SmaI fragment from pUS325) (Bowman et al. 1993; Ogram et al. 1995) encoding MDH, and todC1C2BA (2.5 kb SmaI-NotI fragment from pDTG601) (Shannon et al. 1995) encoding TOD. Plasmid DNA for the target sequences was isolated and used as standards on each slot blot. Each membrane therefore included a standard curve that allowed the detection limit of each probe to be estimated. Probes were cut from plasmids using the appropriate restriction enzyme and purified by agarose gel electrophoresis. The fragments were labeled with 32P-dCTP (ICN Biochemicals, Costa Mesa, CA, USA) using a nick translation kit (Gibco BRL, Gaithersburg, MD, USA). Unincorporated nucleotides were removed using a size exclusion column (Stratagene, La Jolla, CA, USA). The incorporated efficiency was estimated using a scintillation counter (Beckman, Hayward, CA, USA). Samples from MHB-1 V, MHB-5T, MHB-2T, MHB-3T, MHB-7T, MHT-9B, MHT-10B, and MHT-11C were probed using a 0.4 kb fragment of mmoB (Cardy et al. 1991), a 2.5 kb SmaI fragment containing moxF, and a 2.5 kb PstI fragment containing todC2BAD. The mmoB fragment was generated by PCR in a reaction with 100 ng M. trichosporium OB3b (ATCC 35070) DNA, 1.25 × PCR buffer, 2.5 mM MgCl2, 1.25 mM nucleotides, 2.5 units of Taq polymerase and 50 pmol forward primer 5′-ATGTCCAGCGCTCATAAC-3′ and reverse primer 5′-TCAGATGTCGGTCAGGGC-3′. Cycling conditions were as follows: denaturation at 94°C for 2 min followed by 38 cycles with denaturation at 94°C for 0.5 min, annealing at 50°C for 1 min, and elongation at 72°C for 1 min. A final elongation at 72°C for 5 min was performed after temperature cycling. A labeled mmoB probe was generated by asymmetric PCR using 32P-dCTP. Probes moxF and todC2BAD were cut from plasmids pUS325 and pDTG601, respectively, using the appropriate restriction enzyme, purified using agarose gel electrophoresis, and labeled with 32P-dCTP using a random priming kit (Stratagene, La Jolla, CA, USA). Unincorporated nucleotides were removed and incorporation efficiency determined as described above. All remaining samples were analyzed using PCR-generated probes: the mmoB probe as described above, a 1.0 kb moxF probe internal to the SmaI pUS325 fragment, and a 0.9 kb todC1 fragment. Forward and reverse primers for the 1.0 kb moxF probe were 5′-TGGAACGAGACCATGCGT and 3′ and 5′-CTTGGAGGCCGACTTCCA 3′. The forward and reverse primers for the 0.9 kb todC1 probe were 5′-ATGAATCAGACCGACACATC 3′- and 5′-AGACGGTCATGTGCTCGACCACTAGTTTCG-3′. PCR conditions utilized were the same as described earlier.
RNA-targeted probes
An antisense mmoB RNA probe (specific activity of 109 cpm/ml) was synthesized from the T7 promoter in pJF15, which contained a cloned 0.4 kb mmoB gene fragment, using an in vitro transcription kit (Stratagene, La Jolla, CA, USA) and 32P-dUTP. To determine the mass of mmoB mRNA extracted from sediment samples, mmoB mRNA (sense strand) was synthesized from the pJF15 SP6 promoter using the same kit. The mass of synthesized mmoB mRNA was calculated as described in the ribonuclease protection assay (RPA) II manual (Ambion, Austin, TX, USA) and a standard curve was constructed. To provide for normalization of signal obtained from hybridizations to extracted mRNA, a 16S RNA-targeted DNA probe (0.5 kb) was synthesized by PCR using universal primers for the 16S rRNA gene. The PCR reaction included 10–100 ng E. coli HB101 DNA, 1.25 × PCR Buffer II, 2.5 mM MgCl2, 10 mM dNTPs, 0.22 mM {32P}-dCTP, forward primer (5′-AGAGTTTGATCMTGGCTCAC-3′) 50 pmol, reverse primer (5′-GWATTACCGCGGCKGCTG-3′) 50 pmol and 2.5 units Taq polymerase. Cycling conditions were as described for the DNA-targeted probes.
DNA extraction and hybridization
Whole community DNA from 367 sediment samples was obtained using a modification of the direct extraction procedure described by Ogram et al. (1995). A single DNA extraction was performed on each sediment sample. Briefly, 50 g sediment was incubated in 0.12 M Na2HPO4 (pH 8.0) and 5% (w/v) sodium dodecyl sulfate at 70°C for 1 h. Cells were lysed by bead beating for 5 min (BioSpec Prod, Bartlesville, OK, USA) after the addition of an equal amount of glass beads (0.1 mm in diameter). Samples were then washed three times by centrifugation at 16,000×g with 0.12 M Na2HPO4 with the supernatants subsequently pooled and precipitated overnight using 0.5 vol polyethylene glycol. The precipitate was purified by two phenol extractions and one chloroform/isoamyl alcohol (24:1, v/v) extraction. DNA was precipitated overnight with 0.1 vol of 2 M sodium acetate and 2 vol absolute ethanol at −20°C. The precipitate was dried under vacuum and resuspended in TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). DNA samples (300 μl) and six different concentrations of DNA homologous to the probe were denatured in 0.5 ml of 0.4 N NaOH, boiled for 10 min, and applied via vacuum to a slot blot apparatus (Bio Rad, Richmond, CA, USA) using Biotrans nylon membranes (ICN Biochemicals, Costa Mesa, CA). After drying, nylon membranes were washed with 0.5 M NaOH and baked at 80°C for 1 h. The membranes were finally rinsed with 2× SSC (1× SSC, 0.015 trisodium citrate, 0.15 M NaCl, pH 7.0) and stored at room temperature. Membranes were prehybridized in 7% SDS, 1 mM EDTA, and 0.5 M Na2HPO4 for 10 min. Probe was added and hybridized overnight, followed by 3 × 30 min washes in a high stringency buffer composed of 10 mM NaCl, 20 mM Tris, 1 mM EDTA, 17 mM SDS (pH 8). All operations were carried out at 65°C unless stated otherwise. Membranes were exposed to autoradiography film (Kodak) for up to 14 days to detect low signal intensities. The amount of target gene DNA included on the slot blots to generate a standard curve was determined by multiplying the total plasmid DNA concentration by the ratio of the insert to total plasmid size. The detection limit of each gene was estimated based a comparison between the sediment sample slot intensity and the standard curve slot intensities. The gene probing results are expressed as the proportion of positive gene probes for samples taken from a particular core sample.
RNA extraction, hybridization, and quantification
SRS samples showing positive signals for mmoB were chosen for total RNA extraction. Frozen samples were thawed briefly at room temperature. Samples (4 g) were extracted in 50 ml disposable centrifuge tubes with 15 ml preheated (60°C) solution consisting of 0.2 M NaHPO4, 0.1 M EDTA (pH 8.0) and 10 ml chloroform, shaken briefly, and incubated for 10 min at 60°C. The tubes were agitated vigorously for 5 min using a wrist-action shaker and centrifuged at 1 K rpm for 10 min in a Beckman TJ-6 centrifuge. The upper aqueous phase was transferred to fresh tubes and the nucleic acid material was then precipitated overnight at −20°C using 0.1 volume 3 M sodium acetate/0.8 volume isopropanol. Tubes were centrifuged at 10 K RPM [8.2K × g (ave)] for 25 min at room temperature using a Beckman JA-21 centrifuge. The resulting pellet was resuspended in 0.1–0.5 ml DEPC (diethylpyrocarbonate)-treated water and then treated with DNase. The DNase treatment consisted of adding 0.1 volume 10× DNase I buffer (Ambion, TX, USA) and 1 μl DNase I (Ambion, TX, USA) to the resuspended pellet, incubating at 37°C for 30 min. The aqueous phase was then extracted with phenol (500 μl)/chloroform(500 μl) and precipitated overnight at −20°C using 0.1 volume 3 M sodium acetate/0.8 volume isopropanol. While the soil RNA extraction efficiency was not determined in these experiments, a later report using similar soils reported an RNA final % recovery for this extraction method as 6% (Ogram et al. 1995). Duplicate samples were treated with NaOH to hydrolyze RNA as negative control to insure that positive probing slot blots were truly indicative of RNA and not due to DNA hybridization or non-specific hybridization. The integrity of the RNA samples as inferred by the relative concentrations of ribosomal subunits was not determined by gels prior to slot blotting because of the very low amounts of RNA obtained. However, a subsequent comparison of soil RNA extraction procedures demonstrated that this extraction method yielded intact RNA (Ogram et al. 1995). Aliquots of each sediment RNA were denatured in a solution of 50% (v/v) formamide and 7% (v/v) formaldehyde in 1× SSC for 15 min at 68°C. Samples were then cooled on ice and 2 vol of 20× SSC was added. The denatured RNA samples were applied to 0.2 μm nylon membranes using a BioRad slot blot apparatus. The blot was air dried, fixed by UV light from a trans-illuminator and baked at 80°C for 30 min. Prehybridization was carried out for 2 h in 30 ml 5× Denhardt’s solution (Shingleton et al. 1998). The labeled mmoB, todC1 and the 16S rRNA gene probes were added to the prehybridization mixture and hybridized for 18 h at 55, 42, and 42°C, respectively. The blots were then washed with 2× SSC containing 0.1% (w/v) sodium dodecyl sulfate (SDS) vigorously at room temperature for 2 × 15 min, followed by washing with 0.1× SSC containing 0.1% (w/v) SDS for 15 min twice. The final wash for the mmoB probe blots was performed at 65°C while the final wash for the other blots was at 55°C. The membranes were then placed on X-ray film (Kodak X-Omat RP) with intensifying screens at −80°C. After development, the autoradiogram was scanned with a Visage 110 computer assisted imager (Millipore, Bedford, MA, USA) and the integrated optical density (IOD) of each slot band was determined.
Statistical analysis
The relationship of TVC and gene probe results to measurable factors including sample depth, treatment campaign, and sediment composition were determined using regression analysis. Randomness or lack of randomness of gene distribution was tested by the non-parametric Runs tests, while the spatial association of genes in the samples was tested by determining the contingency coefficient using chi-squared analysis. Analyses were performed using the computer software: SSPS for Windows v.6 (Marija J. Norusis/SSPS Inc., Chicago, IL, USA) and Microsoft-Excel (Microsoft Corp., Redmond, WA, USA). Three-dimensional representations of TCE concentrations in the sediment were done using earthVision software (Dynamic Graphics, Alameda, CA, USA).
References
Bowman JP, Jiménez L, Rosario I, Hazen TC, Sayler GS (1993) Characterization of the methanotrophic bacterial community present in a trichloroethylene-contaminated subsurface groundwater site. Appl Environ Microbiol 59:2380–2387
Brockman FJ, Payne W, Workman DJ, Soong A, Manley S, Hazen TC (1995) Effect of gaseous nitrogen and phosphorus injection on in situ bioremediation of a trichloroethylene-contaminated site. J Hazard Mater 41:287–298
Burrow KJ, Cornish A, Scott D, Higgins IJ (1984) Substrate specificities of the soluble and particulate methane monooxygenase of Methylosinus trichosporium OB3b. Mol Microbiol 5:3327–3333
Cardy DLN, Laidler V, Salmond GPC, Murrell JC (1991) The methane monooxygenase gene cluster of Methylosinus trichosporium: cloning and sequencing of the mmoC gene. Arch Microbiol 156:477–483
Eddy CA, Looney BB, Hazen TC, Nichols RL, Fliermans CB, Parker WH et al. (1993) Post-test evaluation of the geology, geochemistry, microbiology, and hydrology of the in situ air stripping demonstration site at the Savannah River Site. WSRC-TR-93-369. Westinghouse Savannah River Company, Aiken, SC
Enzien MV, Picardal F, Hazen TC, Arnold RG, Fliermans CB (1994) Reductive dechlorination of trichloroethylene and tetrachloroethylene under aerobic conditions in a sediment column. Appl Environ Microbiol 60:2200–2204
Frostegard A, Courtois S, Ramisse V, Clerc S, Bernillon D, Le Gall F et al (1999) Quantification of bias related to the extraction of DNA directly from soils. Appl Environ Microbiol 65:1248–5409
Grosse S, Laramee L, Wendlandt KD, McDonald IR, Miguez CB, Kleber HP (1999) Purification and characterization of the soluble methane monooxygenase of the type II methanotrophic bacterium Methylocystis sp strain WI 14. Appl Environ Microbiol 65:3929–3935
Hazen TC (1991) Test plan for in situ bioremediation demonstration of the Savannah River Integrated Demonstration Project DOE/OTD TTP No.: SR 0566–01
Hazen TC (1999) Case study: full-scale in situ bioremediation demonstration (methane biostimulation) of the Savannah River Site Integrated Demonstration Project. In: Adriano DC, Bollag JM (eds) Bioremediation of contaminated soils
Lee SW, Keeney DR, Lim DH, Dispirito M, Semrau JD (2006) Mixed pollutant degradation by Methylosinus trichosporium OB3b expressing either soluble or particulate methane monooxygenase: can the tortoise beat the hare? Appl Environ Microbiol 72:7503–7509
McDonald IR, Murrell JC (1997) The methanol dehydrogenase structural gene mxaF and its use as a functional gene probe for methanotrophs and methylotrophs. Appl Environ Microbiol 63:3218–3224
McDonald IR, Kenna EM, Murrell JC (1995) Detection of methanotrophic bacteria in environmental samples with the PCR. Appl Environ Microbiol 61:116–121
McDonald IR, Uchiyama H, Kambe S, Yagi O, Murrell JC (1997) The soluble methane monooxygenase gene cluster of the trichloroethylene-degrading methanotroph Methylocystis sp. strain M. Appl Environ Microbiol 63:1898–1904
McDonald IR, Bodrossy L, Chen Y, Murrell JC (2008) Molecular ecology techniques for the study of aerobic methanotrophs. Appl Environ Microbiol 74:1305–1315
Neufeld JD, Schafer H, Cox MJ, Boden R, McDonald IR, Murrell JC (2007) Stable-isotope probing implicates Methylophaga spp and novel Gammaproteobacteria in marine methanol and methylamine metabolism. ISME J 1:480–491
Ogram A, Sun WH, Brockman FJ, Fredrickson JK (1995) Isolation and characterization of RNA from low-biomass deep-subsurface sediments. Appl Environ Microbiol 61:763–768
Oldenhuis R, Janssen DB (1993) Degradation of trichloroethylene by methanotrophic bacteria. In: Murrell JC, Kelly DP (eds) Microbial growth on C1 compounds. Intercept Ltd, Andover, pp 121–133
Oldenhuis R, Vink LJM, Janssen DB, Witholt B (1989) Degradation of chlorinated hydrocarbons by Methylosinus trichosporium OB3b expressing soluble monooxygenase. Appl Environ Microbiol 55:2819–2826
Pfiffner SM, Palumbo AV, Phelps TJ, Hazen TC (1997) Effects of nutrient dosing of subsurface methanotrophic populations and trichloroethylene degradation. J Ind Microbiol Biotechnol 18:204–212
Phelps TJ, Niedzielski JJ, Schram RM, Herbes SE, White DC (1990) Biodegradation of trichloroethylene in continuous-recycle expanded-bed bioreactors. Appl Environ Microbiol 56:1702–1709
Sayler GS, Fleming JT, Nivens DE (2001) Gene expression monitoring in soils by mRNA analysis and gene lux fusions. Curr Opin Biotechnol 12:455–460
Semprini L, Hopkins GD, Roberts PV, McCarty PL (1992) Pilot scale field studies of in situ bioremediation of chlorinated solvents. J Hazard Mater 32:145–162
Shannon D, Lawson S, Macy JM (1995) Bioremediation consortium to tackle chlorinated solvents Bioremediation of selenite in oil refinery waste-water. Environ Sci Technol 29:762–765
Shigematsu T, Hanada S, Eguchi M, Kamagata Y, Kanagawa T, Kurane R (1999) Soluble methane monooxygenase gene clusters from trichloroethylene-degrading Methylomonas sp strains and detection of methanotrophs during in situ bioremediation. Appl Environ Microbiol 65:5198–5206
Shingleton JT, Applegate BM, Nagel AC, Bienkowski PR, Sayler GS (1998) Induction of the tod operon by trichloroethylene in Pseudomonas putida TVA8. Appl Environ Microbiol 64:3445–5049
Theisen AR et al (2005) Regulation of methane oxidation in the facultative methanotroph Methylocella silvestris BL2. Mol Microbiol 58:682–692
Travis BJ, Rosenberg ND (1997) Modeling in situ bioremediation of TCE at Savannah River: effects of product toxicity and microbial interactions on TCE degradation. Environ Sci Technol 31:3093–3102
Tsien HC, Hanson RS (1992) Soluble methane monooxygenase component B gene probe for identification of methanotrophs that rapidly degrade trichloroethylene. Appl Environ Microbiol 58:953–960
Van Hamme JD, Singh A, Ward OP (2003) Recent advances in petroleum microbiology. Microbiol Mol Biol Rev 67:503–549
Zylstra GJ, Wackett LP, Gibson DT (1989) Trichloroethylene degradation by Escherichia coli containing the cloned Pseudomonas putida f1 toluene dioxygenase genes. Appl Environ Microbiol 55:3162–3166
Acknowledgments
This work was supported by the U.S. Department of Energy under Contract Nos. DE-AC02-05CH11231 and DE-AC06-76RLO-1830, and contracts from Westinghouse Savannah River Co. to the University of Tennessee. John P. Bowman was supported by the Environics Directorate, Armstrong Laboratories, Tyndall AFB, Panama City, FL, U.S. Air Force Office of Scientific Grants. We would like to thank Bruce Applegate for valuable technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Erko Stackebrandt.
Rights and permissions
About this article
Cite this article
Hazen, T.C., Chakraborty, R., Fleming, J.M. et al. Use of gene probes to assess the impact and effectiveness of aerobic in situ bioremediation of TCE. Arch Microbiol 191, 221–232 (2009). https://doi.org/10.1007/s00203-008-0445-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-008-0445-8