
Abstract
Cadmium is a widespread pollutant that has been associated with oxidative stress, but the mechanism behind this effect in prokaryotes is still unclear. In this work, we exposed two glutathione deficient mutants (ΔgshA and ΔgshB) and one respiration deficient mutant (ΔubiE) to a sublethal concentration of cadmium. The glutathione mutants show a similar increase in reactive oxygen species as the wild type. Experiments performed using the ΔubiE strain showed that this mutant is more resistant to cadmium ions and that Cd-induced reactive oxygen species levels were not altered. In the light of these facts, we conclude that the interference of cadmium with the respiratory chain is the cause of the oxidative stress induced by this metal and that, contrary to previously proposed models, the reactive oxygen species increase is not due to glutathione depletion, although this peptide is crucial for cadmium detoxification.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cadmium is a relatively abundant heavy metal that in the last decades has been utilized in large scale and has been listed as a priority pollutant by the US Environmental Protection Agency (Waisberg et al. 2003). Among its biological effects are inhibition of DNA repair, interference with the cellular antioxidant system, inhibition of DNA methylation, disruption of cell adhesion and induction of apoptosis (Stohs and Bagchi 1995; Waisberg et al. 2003).
Cadmium induces specific alterations in mitochondria, which are the main source of reactive oxygen species (ROS) in eukaryotic cells. In guinea pig liver cells, it interferes with the electron transport chain leading to the accumulation of semiubiquinones. These molecules are unstable and prone to donating electrons directly to molecular oxygen, generating superoxide radicals (Wang et al. 2004).
Cadmium-induced ROS lead to the oxidation of lipids, which results in the permeabilization of the plasma membrane (Gadd 1993; Howlett and Avery 1997). Increased lipid peroxidation in the presence of Cd2+ was indeed detected in Saccharomyces cerevisiae (Vido et al. 2001) and in several rat tissues (Manca et al. 1991; Stohs and Bagchi 1995).
It is known that Cd2+ interacts with thiol groups of proteins resulting in structural modification and/or in their inactivation (Chrestensen et al. 2000; Thévenod and Friedmann 1999).
Complex formation between Cd2+ and glutathione or phytochelatins and sequestration by metallothioneins are the general mechanisms by which eukaryotes detoxify this metal (Adamis et al. 2004; Hatcher et al. 1995; Li et al. 1997; Perego and Howell 1997). The formation of the complex with GSH leads to a depletion of the cytoplasmatic concentration of this molecule. In S. cerevisiae, the cadmium–glutathione complex is transported to the vacuole and thus removed from the cytoplasm (Li et al. 1996, 1997).
Since cadmium is not a redox-active metal, it was hypothesized that it could induce oxidative stress indirectly by displacement of redox-active metals, by the depletion of endogenous radical scavengers (e.g., GSH) or by affecting the activity of antioxidant enzymes. Glutathione depletion is pointed out to be the cause of generation of ROS and oxidative stress (Almazan et al. 2000; Avery 2001; Ercal et al. 2001; Liu et al. 2005; Rikans and Yamano 2000; Stohs et al. 2001; Wolf and Baynes 2007).
Most of the works on the effects of heavy metals in prokaryotes published to date have focused on long-term resistance mechanisms and still relatively few on stress response (Ackerley et al. 2006; Banjerdkij et al. 2003, 2005; Ferianc et al. 1998; Hu et al. 2005; Kershaw et al. 2005; Lee et al. 2005; Puškárová et al. 2002; Wang and Crowley 2005).
Previous works, on E. coli, reported that exposure to low Cd2+ concentrations (3 μM) induced a temporary growth stasis. During this phase, Cd-induced damage is repaired and cell physiology is adjusted to limit the distribution of the ion within the cell (Mitra et al. 1975; Mitra and Bernstein 1977). It was also observed that, during stasis, there was loss of cell viability, although synthesis of some specific proteins was increased (Ferianc et al. 1998; Khazaeli and Mitra 1981; Shapiro and Keasling 1996) and the repair of DNA damage was concomitant with recovery of viability (Mitra 1984; Mitra and Bernstein 1978).
Proteomic analysis in E. coli (Ferianc et al. 1998; VanBogelen et al. 1987) confirmed the activation of the SOS, heat shock and oxidative stress regulons, although they were only a minor part of the response to cadmium.
Copper is a metal with important biological functions, but, being a redox-active metal, when present in excess it can react with H2O2 generating hydroxyl radicals and cellular damage (Rensing and Grass 2003). Less is known on the mechanisms of toxicity of Ag+, Hg2+ and Zn2+, but data point to their interaction with the respiratory chain (Bragg and Hou 1968; Bragg and Rainnie 1974; Kasahara and Anraku 1972; Kim and Bragg 1971; Poole et al. 1989).
This study was designed to understand the mechanisms involved in cadmium toxicity in E. coli and assess the roles of respiration, GSH and oxidative stress in the process.
Materials and methods
Strains and culture conditions
Escherichia coli K-12 BW25113, the deletion mutants derived from this strain ΔgshA, ΔgshB, ΔubiE (obtained from the Keio Collection, Keio University, National BioResource Project, NIG, Japan; Baba et al. 2006) and Escherichia coli K-12 MG1655 (obtained from the E. coli Genetic Stock Center, University of Yale, USA) were used throughout this study.
The minimal medium was constituted of: NaCl 0.5 g l−1, NH4Cl 1.0 g l−1, MgSO4·7H2O 49.2 mg l−1, K2SO4 48 mg l−1, K2HPO4·3·H2O 46 mg l−1 (to give a final concentration of 200 μM of phosphate), micronutrient solution (Tuovinen and Kelly 1973) 2 ml l−1, 3-(N-Morpholino)propanesulfonic acid (MOPS) 40 mM pH 7.4 and glucose 20 mM. The concentration of phosphate in the medium was minimized to avoid cadmium precipitation as Cd phosphate.
Cultures in aerobic conditions were inoculated from overnight pre-cultures and grown in an orbital shaker (130 rpm) at 37°C to OD420 = 0.5 (UVmini-1240, Shimadzu). For cultures grown in fermentative conditions, Erlenmeyer flasks with a magnetic bar were filled almost to the top with sterile medium that was stripped of oxygen by bubbling with a nitrogen stream. To avoid contact with air, sterile SubaSeal® rubber stoppers were used.
Kanamycin (25 mg l−1) was added to the mutants pre-cultures.
Effect of Cd on cell growth and culturability
To assess the effect of Cd on cell growth, OD420 was monitored in control cells (untreated) and cultures treated with 30 μg ml−1 Cd2+ (273 μM CdCl2), a concentration used in previous works on cadmium stress in E. coli K-12 (Ferianc et al. 1998). To test the effect of glutathione on Cd-induced growth arrest, exogenous GSH and GSSG were added to a final concentration of 4 mM, 5 min prior to Cd exposure.
Culturability assays were performed by drawing aliquots at different time points (0, 30 and 60 min after cadmium was added), serial diluting sterile saline solution and plating onto LB agar. Culturability was expressed as the percentage of colony-forming units (CFU).
Assessment of oxidative damage
Protein oxidation was assessed by immunodetection of protein carbonyls. Control and Cd-treated cells were collected by centrifugation for 10 min at 20,000×g at 4°C, then washed twice in cold potassium phosphate buffer 50 mM pH 7.0 and used immediately or stored at −80°C for later usage.
The pellets were resuspended in potassium phosphate buffer (at) 50 mM, pH 7.0, containing protease inhibitors (Complete™ Mini EDTA-free Protease cocktail inhibitor, Roche). The cells were then disrupted by sonication on ice with a Branson Sonifier 250 using two cycles of 15 s (50% duty cycle, output 3) intercalated with one cycle of 1 min off duty and centrifuged for 10 min at 16,000×g at 4°C, keeping the supernatants. Protein concentration was measured by the BCA Protein Assay Kit (Pierce) according to the manufacturer’s instructions. Proteins were derivatized with 2,4-dinitrophenylhydrazine (DNPH) as described by Levine et al. (1990), separated by SDS-PAGE and stained with Coomassie blue or blotted onto a Hybond-ECL membrane (GE Healthcare, UK). Immunodetection was performed using a rabbit anti-dinitrophenyl (DNP) IgG (Dako Cytomation, Glostrup, Denmark) at 1:5,000 dilution as primary antibody and goat anti-rabbit IgG peroxidase conjugate (Sigma, St Louis, USA), at 1:5,000 dilution as secondary antibody. Detection was performed by chemiluminescence using the kit ECL Western blotting (GE Healthcare, UK). Analysis of SDS-PAGE gels and carbonyl immunodetection films was performed using the Quantity One® programme version 4.5 (Bio-Rad).
The determination of lipid peroxidation was performed according to Steels et al. (1994) with adaptations: E. coli cell pellets obtained from 50 ml cultures were resuspended in 250 μl potassium phosphate buffer 50 mM pH 7.0 and sonicated as described above. To each sample, 28 μl of trichloroacetic acid (TCA) was added and the mixture was vortexed at a maximum speed for 2 min. Extracts were then centrifuged for 15 min at 2,000×g at 4°C and to 100 μl supernatant, 100 μl EDTA 0.1 M plus 600 μl of a solution of 2-thiobarbituric acid (TBA) 1% (w/v), NaOH 50 mM and 2,6-di-tert-Butyl-p-cresol (BHT) 0.025% (w/v) were added. Samples were kept in boiling water for 15 min and, after cooling, the A 532 was measured.
Catalase activity and glutathione measurement
For the determination of catalase activity, E. coli extracts were prepared as described in “Assessment of oxidative damage”. Samples were dialyzed overnight against cold potassium phosphate buffer at 50 mM, pH 7.8, EDTA 0.1 mM. Protein concentration was determined as described in “Assessment of oxidative damage”. Catalase activity was measured as described in Beers and Sizer (1952) following the decrease of A 240 due to H2O2 disappearance. One unit of catalase is defined as the quantity of enzyme needed to degrade 1 μmol of H2O2 per minute at 25°C.
For glutathione determination, cells were collected by centrifugation and washed as described in “Assessment of oxidative damage”. The cell pellets were resuspended in 200 μl phosphate buffer at 100 mM, EDTA at 2 mM, pH 7.4, and 200 μl HClO4 at 2 M was added. Then they were sonicated as described in “Assessment of oxidative damage” and centrifuged for 10 min at 16,000×g at 4°C. The extracts were neutralized to pH 6–7 using 200 μl KOH at 2 M and MOPS at 0.3 M and spun at 16,000×g at 4°C for 2 min. Total intracellular glutathione was determined by the DTNB-GSSG reductase recycling method as described by Akerboom and Sies (1981). The levels of extracellular glutathione were determined in culture supernatants. For this purpose, 20 ml aliquots of the supernatants were collected, frozen to −80°C and lyophilized to dryness (Owens and Hartman 1986). The lyophilized supernatants were resuspended in phosphate buffer at 100 mM, EDTA at 2 mM, pH 7.4, and HClO4 at 2 M (in equal volumes) and neutralized to pH 6–7 as described above. Total glutathione was determined using the same method as described for the cell pellets.
Respiration measurement
Cells were grown to an OD420 = 0.5–0.7 and harvested at 4,500×g at 4°C, washed twice in 5 ml MOPS buffer at 40 mM, pH 7.4, and resuspended in the same buffer. Respiration rates were determined polarographically for suspensions with final OD420 = 2 in 1.5 ml working volume in a Clark-type oxygen electrode (Oxygraph, Hansatech Instruments, UK) after the addition of either glucose, glycerol, succinate, lactate, fumarate or pyruvate at 2 mM final concentration. In inhibition tests, Cd2+ 30 μg ml−1 was added in the absence or presence of GSH at 2 mM or GSSG at 2 mM. The results were normalized with respect to dry weight.
Quantification of intracellular ROS
E. coli suspensions were incubated with dihydrorhodamine-123 (DHR), 0.025 μg μl−1 (Molecular Probes, Eugene, Oregon, USA) at 37°C shaking for 2 h in the dark using 500 μl of culture in 2 ml microtubes. The treated samples received Cd2+ 30 μg ml−1, 1 h after the addition of DHR, so that they were exposed to Cd2+ for 1 h. Control samples received an equivalent volume of sterile distilled water. The samples were then diluted 1:20 in PBS (NaCl 8% (w/v); KCl 0.02% (w/v); Na2HPO4 0.18% (w/v) pH 7.4). Cell-associated fluorescence was measured by flow-cytometry with a Becton Dickinson FACSCalibur (San Jose, California, USA). For each sample, 10,000 cells were acquired and results were analyzed using the CellQuest programme version 3.3.
Statistical data treatment
All results in this study are expressed as means of at least three independent replicates with the associated standard deviation. Differences between treatments were considered statistically significant when Student’s t-test was <0.05.
Results
Aiming to assess the effect of cadmium on E. coli cells, cultures were grown to early logarithmic phase and exposed to 30 μg ml−1 of cadmium (273 μM of CdCl2), a sublethal concentration able to induce growth arrest The results show that cadmium treatment induced a stasis in the growth of aerobic cultures (Fig. 1a, b) accompanied by a decrease in cell viability (CFUs; Fig. 2), as expected. On the other hand, the effect of Cd2+ on growth and culturability was much less pronounced in cultures grown by fermentation. These results are consistent with published data suggesting that ROS associated with aerobic metabolism contribute to Cd-induced cell death in S. cerevisiae (Brennan and Schiestl 1996).

Effect of 30 μg ml−1 Cd2+ on cell growth. a Untreated culture (open square), wild-type + Cd (filled square), ΔgshA + Cd (filled triangle), ΔgshB + Cd (filled circle). The untreated culture growth curve shown is representative of the curves obtained for all the untreated cultures of the strains used in the experiments: wild-type BW25113, ΔgshA and ΔgshB. b ΔubiE untreated (open square), ΔubiE + Cd (filled square)

Effect of 30 μg ml−1 Cd2+ on culturability: wild type (filled bar), ΔubiE (diagonal stripe bar), ΔgshA (open bar), ΔgshB (gray bar). The absolute values (CFU ml−1) corresponding to 100% (T 0) are: wild type = 1.41E + 08 ± 1.90E + 07, ΔubiE = 1.74E + 08 ± 3.03E + 07, ΔgshA = 1.53E + 08 ± 3.67E + 07 and ΔgshB = 1.86E + 08 ± 2.01E + 07. Statistically significant differences between Cd-treated and untreated cultures are identified: * (P < 0.05) and ** (P < 0.01)
Since glutathione depletion has been pointed out to be the cause of oxidative stress in Cd-exposed cells, we analyzed the effect of its absence on toxicity by using glutathione mutants. GshA catalyzes the first step in glutathione biosynthesis, whereas GshB converts gamma-glutamylcysteine into glutathione. The growth stasis of the mutants ΔgshA and ΔgshB was similar to that seen in the wild-type strain (Fig. 1a). However, GshA deficiency significantly decreased the culturability of cadmium-treated cells: the ΔgshA and ΔgshB strains showed 33 and 54% culturability, respectively, compared to 63% in the wild-type strain (Fig. 2). These results are in agreement with a very important role of glutathione in cadmium detoxification.
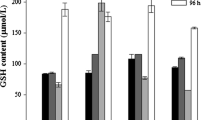
In previous works, the depletion of glutathione has been pointed out as the direct cause for the rise of ROS levels in cadmium-exposed cells. In E. coli MG1655 cultures treated with cadmium, we observed a 72% depletion of intracellular glutathione, which was accompanied by a rise in the extracellular concentration of this molecule (3.4 times; Fig. 3). Extracellular glutathione has a protective role as the Cd-dependent growth-arrest effect was not observed when exogenous GSH (but not GSSG) was added to the growth medium (data not shown). To investigate if glutathione deficiency increased Cd-induced production of ROS, we used DHR 123 to assess the amount of peroxides formed in the different strains. As shown in Fig. 4, exposure of the wild-type cells to cadmium increased by twofold the levels of ROS. Accordingly, we detected a 100% increase in catalase activity in Cd-treated cultures (data not shown). When ΔgshA and ΔgshB mutant cells were exposed to the heavy metal, the levels of ROS increased by about 40%. This result clearly indicates that intracellular glutathione depletion cannot be the cause of the Cd-induced production of ROS.

Effect of 30 μg ml−1 Cd2+ on total glutathione: intracellular concentration (nmol (GSH + 2GSSG) mg−1 protein; open bar), extracellular concentration (μM (GSH + 2GSSG); filled bar). * (P < 0.05) and ** (P < 0.01) express significant differences between Cd-treated (30 and 60 min) and untreated (0 min) samples

ROS detection by flow-cytometry: wild type (filled bar), ΔubiE (diagonal stripe bar), ΔgshA (open bar), ΔgshB (gray bar). The absolute values (arbitrary fluorescence units) corresponding to 100% (T 0) are: wild type = 14.99 ± 6.85, ΔubiE = 27.09 ± 2.92, ΔgshA = 4.82 ± 1, ΔgshB = 8.08 ± 2.01. * (P < 0.05) and ** (P < 0.01) mean that there is a significant difference between the control and treated samples
It has been suggested that the toxic effect of Cd and other metals such as zinc, silver and mercury involves interference with the respiratory chain. To test this hypothesis, we analyzed the effect of cadmium on oxygen consumption and Cd toxicity in ΔubiE mutant cells that have a deficient respiratory chain due to lack of ubiquinone and menaquinone (Gennis and Valley 1987). In wild-type cells of MG1655 strain, cadmium inhibited oxygen consumption and this effect was reverted by the exogenous addition of GSH, but not GSSG (Fig. 5). In ΔubiE mutant cells, oxygen consumption was 38% of that observed in its isogenic wild-type strain (data not shown). Notably, this reduction in aerobic metabolism significantly decreased the growth stasis induced by cadmium. Indeed, cell growth was resumed after just 2 days of Cd exposure (vs. 5 days in the wild type; Fig. 1b). In addition, the culturability of Cd-treated ΔubiE cells was higher compared to the wild type (Fig. 2). Oxidative stress markers were analyzed in the ΔubiE mutant cells to test the link between the toxicity of cadmium and the respiratory chain. The constitutive levels of ROS were higher in this strain than in the wild-type: this fact can be justified by the deficient flow of electrons in the respiratory chain, which probably leads to the accumulation of unstable reduced intermediates of the chain. However, Cd-induced ROS production was suppressed in this mutant (Fig. 4), whereas the direct exposure to 10 mM H2O2 still increased intracellular oxidation (data not shown).

Effect of 30 μg ml−1 Cd2+ on respiration rate. The absolute value (nmol O2 min−1) corresponding to 100% is 98 ± 7.9. Significant differences between the control and the addition of Cd or GSH + Cd or GSSG + Cd are represented: * (P < 0.05) and ** (P < 0.01)
ROS production is usually associated with oxidative damage in proteins and lipids. For this reason, we analyzed the levels of protein carbonylation and lipid peroxidation in the wild-type and in the ΔubiE mutant. In wild-type cells, the exposure to cadmium increased protein oxidation (Fig. 6a) and lipid peroxidation (Fig. 6b). In the ΔubiE strain, the constitutive levels of protein and lipid oxidation were higher, compared to those observed in the untreated wild-type cells, which is in accordance with the higher constitutive ROS levels detected in this mutant. Notably, Cd-induced protein carbonylation was significantly reduced: the increase in protein carbonyl content was 278% in the wild-type cells and just 70% in the ΔubiE mutant cells (Fig. 6a). In addition, no increase in lipid peroxidation was detected in ΔubiE cells treated with cadmium (Fig. 6b).

Effect of 30 μg ml−1 Cd2+: a on protein carbonyl levels. SDS-PAGE is shown as loading control. b) on lipid peroxidation levels: wild type (solid bar), ΔubiE (diagonal stripe bar). The absolute values (μmol MDA mg−1 prot) corresponding to 100% are: wild type = 6.60 ± 0.29, ΔubiE = 10.68 ± 0.99. In the wild type there is significant difference between the control and treated samples * P < 0.05)
All these results support the hypothesis that Cd-induced ROS production is associated with the activity of the respiratory chain.
Discussion
Cadmium is a relatively abundant heavy metal and its toxic effects have been associated with the induction of ROS production and mutagenesis (Clark and Kunkel 2004; Pathak and Khandelwal 2006). Although the effects of cadmium have been widely described, the molecular mechanisms underlying its toxicity are still unclear. In order to investigate these mechanisms and their relationship with oxidative stress in E. coli, K-12 strains were submitted to a sublethal concentration of the metal that is able to induce growth arrest. Similar to observations from previous reports, growth inhibition was accompanied by a decrease in culturability and by an increase in oxidative stress markers. Indeed, cadmium increased ROS production and induced catalase activity. Concomitantly, there was a Cd-dependent depletion of intracellular glutathione and a rise in its external concentration. These observations are in accordance with the general mechanism by which cadmium is detoxified in E. coli, whereby the bis(glutathionato)cadmium complex is formed and presented to the ZntA pump that excretes the metal from the cell (Blencowe et al. 1997; Li et al. 1997; Rensing et al. 1997; Sharma et al. 2000). The rise in the extracellular concentration of glutathione can constitute a further defence mechanism of the cell to prevent the entrance of the metal. Owens and Hartman (1986) reported GSH export in E. coli and some years later a bacterial transporter of reduced glutathione was identified (Pittman et al. 2005).
The depletion of glutathione by cadmium has been pointed out as the cause of the oxidative stress status in the presence of this metal. To clarify the contribution of glutathione depletion in cadmium toxicity in E. coli, we used ΔgshA and ΔgshB mutants that lack glutathione. The results obtained reveal that the effects of cadmium exposure in these mutants were similar to those observed in the wild type because a rise of ROS is detected whether GSH is present in the cells or not. Our data clearly show that the generation of ROS in cadmium-exposed cells is completely independent of the presence of glutathione. Despite this fact, glutathione mutants seem to recover more slowly from cadmium stress, which confirms the importance of GSH in cadmium detoxification. The ΔgshB strain was probably less affected by Cd because the first step in GSH biosynthesis still occurs in this mutant and the dipeptide gamma-glutamylcysteine may result in partial protection against cadmium (Cruz-Vásquez et al. 2002).
We also observed that cadmium inhibits oxygen consumption and that fermenting cultures are less sensitive to this metal. A reduction in cadmium toxicity has also been observed in fermenting S. cerevisiae (Brennan and Schiestl 1996; Vido et al. 2001). Our results indicate that cadmium toxicity is correlated with ROS production by the respiratory chain. In agreement with this hypothesis, the detrimental effects of cadmium on growth and culturability were significantly reduced in ΔubiE cells that show diminished respiration rates. Furthermore, although ROS levels were constitutively higher in ΔubiE cells, no increase was detected after exposure to the heavy metal.
Other studies have shown an interaction of cadmium with mitochondrial respiration (Wang et al. 2004) and of other heavy metal ions (Ag+, Hg2+ and Zn2+) with the bacterial respiratory chain (Bragg and Rainnie 1974; Kasahara and Anraku 1972; Kim and Bragg 1971).
Wang and Crowley (2005) reported in their transcriptome study on E. coli K-12 that cadmium affects the expression of genes associated with protein synthesis, energy metabolism and cell rescue. The up-regulation of genes associated with anaerobic metabolism and the shutdown of all high-energy consumption processes such as the biosynthesis of amino acids suggests that, when exposed to cadmium, cells switch to an energy conservation mode. Our hypothesis whereby cadmium poisons the respiratory chain is in accordance with these observations. These authors conclude that ROS are not a direct cause of Cd2+ toxicity, because the base excision DNA repair system and OxyR were not induced and because the genes affected by cadmium are quite a different subset from those affected by superoxide. However, it should be noted that the concentration of cadmium used in their study was very low, 1 μg ml−1, a level at which we were not able to detect any effect on growth kinetics.
We propose the following model for Cd toxicity in respiring E. coli cells: cadmium enters the cell through one of the essential metal transporters, like ZupT, a zinc transporter (Grass et al. 2002). Here it poisons the respiratory chain leading to the accumulation of unstable reduced intermediates (Messner and Imlay 1999) that reduce molecular oxygen to ROS. Although the cell defenses are activated to eliminate the ROS, they are not enough to completely prevent oxidative damage. The poisoning of the respiratory chain leads to a depletion of the ATP stock and to growth arrest. Cadmium is detoxified through the excretion from the cell by the ZntA pump (Blencowe et al. 1997; Rensing et al. 1997; Sharma et al. 2000) and through the formation of a complex with the GSH excreted. Although glutathione plays a crucial role in detoxifying the heavy metal ion, clearly its depletion is not the cause of the ROS burst resulting from exposure to cadmium.
Abbreviations
- BHT:
-
2,6-Di-tert-Butyl-p-cresol
- DHR:
-
Dihydrorhodamine 123
- DNPH:
-
2,4-Dinitrophenylhydrazine
- DTNB:
-
5,5′-Dithiobis (2-nitrobenzoic acid)
- GSH:
-
Reduced glutathione
- GSSG:
-
Oxidized glutathione
- MDA:
-
Malondialdehyde
- MOPS:
-
3-(N-morpholino)propanesulfonic acid
- ROS:
-
Reactive oxygen species
- TBA:
-
2-Thiobarbituric acid
- TCA:
-
Trichloroacetic acid
References
Ackerley DF, Barak Y, Lynch SV, Curtin J, Matin A (2006) Effect of chromate stress on Escherichia coli K-12. J Bacteriol 188:3371–3381
Adamis PD, Gomes DS, Pinto ML, Panek AD, Eleutherio EC (2004) The role of glutathione transferases in cadmium stress. Toxicol Lett 154:81–88
Akerboom TP, Sies H (1981) Assay of glutathione, glutathione disulfide, and glutathione mixed disulfides in biological samples. Methods Enzymol 77:373–382
Almazan G, Liu HN, Khorchid A, Sundararajan S, Martinez-Bermudez AK, Chemtob S (2000) Exposure of developing oligodendrocytes to cadmium causes HSP72 induction, free radical generation, reduction in glutathione levels, and cell death. Free Radic Biol Med 29:858–869
Avery SV (2001) Metal toxicity in yeasts and the role of oxidative stress. Adv Appl Microbiol 49:111–142
Baba T, Ara T, Hasegawa M et al (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006, 0008
Banjerdkij P, Vattanaviboon P, Mongkolsuk S (2003) Cadmium-induced adaptive resistance and cross-resistance to zinc in Xanthomonas campestris. Curr Microbiol 47:260–262
Banjerdkij P, Vattanaviboon P, Mongkolsuk S (2005) Exposure to cadmium elevates expression of genes in the OxyR and OhrR regulons and induces cross-resistance to peroxide killing treatment in Xanthomonas campestris. Appl Environ Microbiol 71:1843–1849
Beers RF Jr, Sizer IW (1952) A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J Biol Chem 195:133–140
Blencowe DK, Marshall SJ, Morby AP (1997) Preliminary characterization of zntA gene, which encodes a Zn(II)/Cd(II)-export protein in Escherichia coli. Biotechnol et alia 1–6
Bragg PD, Hou C (1968) Oxidative phosphorylation in Escherichia coli. Can J Biochem 46:631–641
Bragg PD, Rainnie DJ (1974) The effect of silver ions on the respiratory chain of Escherichia coli. Can J Microbiol 20:883–889
Brennan RJ, Schiestl RH (1996) Cadmium is an inducer of oxidative stress in yeast. Mutat Res 356:171–178
Chrestensen CA, Starke DW, Mieyal JJ (2000) Acute cadmium exposure inactivates thioltransferase (Glutaredoxin), inhibits intracellular reduction of protein-glutathionyl-mixed disulfides and initiates apoptosis. J Biol Chem 275:26556–26565
Clark AB, Kunkel TA (2004) Cadmium inhibits the functions of eukaryotic MutS complexes. J Biol Chem 279:53903–53906
Cruz-Vásquez BH, Díaz-Cruz JM, Ariño C, Esteban M, Tauler R (2002) Study of Cd2+ complexation by the glutathione fragments Cys-Gly (CG) and γ-Glu-Cys (γ-EC) by differential pulse polarography. Analyst 127:401–406
Ercal N, Gurer-Orhan H, Aykin-Burns N (2001) Toxic metals and oxidative stress part I: mechanisms involved in metal-induced oxidative damage. Curr Top Med Chem 1:529–539
Ferianc P, Farewell A, Nyström T (1998) The cadmium-stress stimulon of Escherichia coli K-12. Microbiol 144:1045–1050
Gadd GM (1993) Interactions of fungi with toxic metals. New Phytol 124:25–60
Gennis RB, Valley S (1987) Respiration. In: Ingraham JL, Neidhardt FC (eds) Escherichia coli and Salmonella typhimirium: celular and molecular biology, vol. 1. ASM, Washington DC, pp 217–261
Grass G, Wong MD, Rosen BP, Smith RL, Rensing C (2002) ZupT is a Zn(II) uptake system in Escherichia coli. J Bacteriol 184:864–866
Hatcher EL, Chen Y, Kang YJ (1995) Cadmium resistance in A549 cells correlates with elevated glutathione content but not antioxidant enzymatic activities. Free Radic Biol Med 19:805–812
Howlett NG, Avery SV (1997) Induction of lipid peroxidation during heavy metal stress in Saccharomyces cerevisiae and influence of plasma membrane fatty acid unsaturation. Appl Environ Microbiol 63:2971–2976
Hu P, Brodie EL, Suzuki Y, McAdams HH, Andersen GL (2005) Whole-genome transcriptional analysis of heavy metal stresses in Caulobacter crescentus. J Bacteriol 187:8437–8449
Kasahara M, Anraku Y (1972) Inhibition of the respiratory chain of Escherichia coli by zinc ions. J Biochem (Tokyo) 72:777–781
Kershaw CJ, Brown NL, Constantinidou C, Patel MD, Hobman JL (2005) The expression profile of Escherichia coli K-12 in response to minimal, optimal and excess copper concentrations. Microbiol 151:1187–1198
Khazaeli MB, Mitra RS (1981) Cadmium-binding component in Escherichia coli during accommodation to low levels of this ion. Appl Environ Microbiol 41:46–50
Kim IC, Bragg PD (1971) Some properties of the succinate dehydrogenase of Escherichia coli. Can J Biochem 49:1098–1104
Lee LJ, Barrett JA, Poole RK (2005) Genome-wide transcriptional response of chemostat-cultured Escherichia coli to zinc. J Bacteriol 187:1124–1134
Levine RL, Garland D, Oliver CN et al (1990) Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol 186:464–478
Li ZS, Lu YP, Zhen RG, Szczypka M, Thiele DJ, Rea PA (1997) A new pathway for vacuolar cadmium sequestration in Saccharomyces cerevisiae: YCF1-catalyzed transport of bis(glutathionato)cadmium. Proc Natl Acad Sci USA 94:42–47
Li ZS, Szczypka M, Lu YP, Thiele DJ, Rea PA (1996) The yeast cadmium factor protein (YCF1) is a vacuolar glutathione S-conjugate pump. J Biol Chem 271:6509–6517
Liu J, Zhang Y, Huang D, Song G (2005) Cadmium induced MTs synthesis via oxidative stress in yeast Saccharomyces cerevisiae. Mol Cell Biochem 280:139–145
Manca D, Ricard AC, Trottier B, Chevalier G (1991) Studies on lipid peroxidation in rat tissues following administration of low and moderate doses of cadmium chloride. Toxicology 67:303–323
Messner KR, Imlay JA (1999) The identification of primary sites of superoxide and hydrogen peroxide formation in the aerobic respiratory chain and sulfite reductase complex of Escherichia coli. J Biol Chem 274:10119–10128
Mitra RS (1984) Protein synthesis in Escherichia coli during recovery from exposure to low levels of Cd2+. Appl Environ Microbiol 47:1012–1016
Mitra RS, Bernstein IA (1977) Nature of the repair process associated with the recovery of Escherichia coli after exposure to Cd2+. Biochem Biophys Res Commun 74:1450–1455
Mitra RS, Bernstein IA (1978) Single-strand breakage in DNA of Escherichia coli exposed to Cd2+. J Bacteriol 133:75–80
Mitra RS, Gray RH, Chin B, Bernstein IA (1975) Molecular mechanisms of accommodation in Escherichia coli to toxic levels of Cd2+. J Bacteriol 121:1180–1188
Owens RA, Hartman PE (1986) Export of glutathione by some widely used Salmonella typhimurium and Escherichia coli strains. J Bacteriol 168:109–114
Pathak N, Khandelwal S (2006) Influence of cadmium on murine thymocytes: Potentiation of apoptosis and oxidative stress. Toxicol Lett 165:121–132
Perego P, Howell SB (1997) Molecular mechanisms controlling sensitivity to toxic metal ions in yeast. Toxicol Appl Pharmacol 147:312–318
Pittman MS, Robinson HC, Poole RK (2005) A bacterial glutathione transporter (Escherichia coli CydDC) exports reductant to the periplasm. J Biol Chem 280:32254–32261
Poole RK, Williams HD, Downie JA, Gibson F (1989) Mutations affecting the cytochrome d-containing oxidase complex of Escherichia coli K-12: identification and mapping of a fourth locus, cydD. J Gen Microbiol 135:1865–1874
Puškárová A, Ferianc P, Kormanec J, Homerová D, Farewell A, Nyström T (2002) Regulation of yodA encoding a novel cadmium-induced protein in Escherichia coli. Microbiology 148:3801–3811
Rensing C, Grass G (2003) Escherichia coli mechanisms of copper homeostasis in a changing environment. FEMS Microbiol Rev 27:197–213
Rensing C, Mitra B, Rosen BP (1997) The zntA gene of Escherichia coli encodes a Zn(II)-translocating P-type ATPase. Proc Natl Acad Sci USA 94:14326–14331
Rikans LE, Yamano T (2000) Mechanisms of cadmium-mediated acute hepatotoxicity. J Biochem Mol Toxicol 14:110–117
Shapiro N, Keasling JD (1996) The recA gene and cadmium toxicity in Escherichia coli K-12. Microbios 86:23–26
Sharma R, Rensing C, Rosen BP, Mitra B (2000) The ATP hydrolytic activity of purified ZntA, a Pb(II)/Cd(II)/Zn(II)-translocating ATPase from Escherichia coli. J Biol Chem 275:3873–3878
Steels EL, Learmonth RP, Watson K (1994) Stress tolerance and membrane lipid unsaturation in Saccharomyces cerevisiae grown aerobically or anaerobically. Microbiology 140:569–576
Stohs SJ, Bagchi D (1995) Oxidative mechanisms in the toxicity of metal ions. Free Radic Biol Med 18:321–336
Stohs SJ, Bagchi D, Hassoun E, Bagchi M (2001) Oxidative mechanisms in the toxicity of chromium and cadmium ions. J Environ Pathol Toxicol Oncol 20:77–88
Thévenod F, Friedmann JM (1999) Cadmium-mediated oxidative stress in kidney proximal tubule cells induces degradation of Na+/K+-ATPase through proteasomal and endo-/lysosomal proteolytic pathways. Faseb J 13:1751–1761
Tuovinen OH, Kelly DP (1973) Studies on the growth of Thiobacillus ferrooxidans. I. Use of membrane filters and ferrous iron agar to determine viable numbers, and comparison with 14CO2 -fixation and iron oxidation as measures of growth. Arch Mikrobiol 88:285–298
VanBogelen RA, Kelley PM, Neidhardt FC (1987) Differential induction of heat shock, SOS, and oxidation stress regulons and accumulation of nucleotides in Escherichia coli. J Bacteriol 169:26–32
Vido K, Spector D, Lagniel G, Lopez S, Toledano MB, Labarre J (2001) A proteome analysis of the cadmium response in Saccharomyces cerevisiae. J Biol Chem 276:8469–8474
Waisberg M, Joseph P, Hale B, Beyersmann D (2003) Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology 192:95–117
Wang A, Crowley DE (2005) Global gene expression responses to cadmium toxicity in Escherichia coli. J Bacteriol 187:3259–3266
Wang Y, Fang J, Leonard SS, Rao KM (2004) Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radic Biol Med 36:1434–1443
Wolf MB, Baynes JW (2007) Cadmium and mercury cause an oxidative stress-induced endothelial dysfunction. Biometals 20:73–81
Acknowledgments
The authors wish to thank Vítor Costa, M. Amélia Amorim, João Vieira, Fernando Tavares, Perpétua do Ó, Margarida Duarte, Salomé Gomes, Simon Monard and Susana Lousada (IBMC, Porto), Nuno Mateus (FCUP, Porto), Félix Carvalho (FFUP, Porto) and Etelvina Figueira (Aveiro, Portugal) for their technical support and/or useful suggestions and criticism. C.C.P. was supported by a Ph.D. grant from FCT, Portugal (SRFH/BD/12771/2003). P.D.M. was the beneficiary of a post-doc grant from FCT, Portugal (SRFH/BPD/20577/2004).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by John Helmann.
Rights and permissions
About this article
Cite this article
Pacheco, C.C., Passos, J.F., Castro, A.R. et al. Role of respiration and glutathione in cadmium-induced oxidative stress in Escherichia coli K-12. Arch Microbiol 189, 271–278 (2008). https://doi.org/10.1007/s00203-007-0316-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-007-0316-8