
Abstract
Brevibacillus laterosporus G4, which was isolated from soil sample, kills free-living nematodes (Panagrellus redivius) and plant-parasite nematodes (Bursaphelenchus xylophilus) and degrades their cuticle in previous bioassay. Our works for B. laterosporus G4 had demonstrated that an extracellular alkaline protease BLG4 played a key role as a pathogenic factor in infection against nematode. In this study, the nematicidal activity of BLG4 was further verified by an in vitro assay with purified recombinant BLG4. The encoding gene of BLG4 was cloned and showed high degree of homology with the subtilisin subclass of serine protease gene and another reported cuticle-degrading protease gene from nematophagous bacterium Bacillus sp. B16. Deletion of BLG4 by homologous recombinant had a significant effect on the pathogenicity of B. laterosporus. In infection assays the BLG4-deficient strain (BLG4-6) lost about 50% of its nematocidal activity and in toxicity tests the mortality rate of nematodes decreased with ∼56% in comparison to wild-type strain. This is the first report analyzing the function of a subtilisin enzyme involved in bacterium against nematode at the molecular level, and it is possible to use B. laterosporus as a model to study host-parasite interaction and to gain detailed knowledge of the infection process.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The cuticle of nematode almost completely surrounds the animal and is a very rigid but flexible multilayered extracellular exoskeleton that prevents the organism from being environmentally damaged (Cox et al. 1981; Bird and Bird 1991; Maizels et al. 1993). Additionally, the cuticle surface may be covered with a proteinaceous membrane, collagen is the main structural component of the cuticle and some other proteins are essential for the integrity of the cuticle (Bird and Bird 1991; Åhman et al. 1996). If these structural components are affected, the cuticle is easily destroyed. It has been assumed on basis of studies with entomopathogen fungi that penetration of the cuticle by fungi is due to a combination of mechanical force and hydrolytic enzymes (St Leger et al. 1992; St Leger 1995; Zhao et al. 2004). Extracellular enzymes, especially proteases, were revealed to contribute to the process of infection by solubilizing the cuticle, digesting the host tissue and releasing nutrients for pathogen growth (Tunlid and Jansson 1991; St Leger 1995; Zhao et al. 2004).
In recent years, growing number of novel pathogenic proteases were identified and cloned from nematophagous fungi but information about the molecular background of nematode infection by fungi remained to be elucidated (Åhman et al. 2002; Yang et al. 2005). Majority of these works were focused on the purification, characterization and gene cloning of proteases. Among them, PII from Arthrobotrys oligospora is the only pathogenic protease whose role in the infection against nematodes was well studied at molecular level. Heterologous expression experiments revealed that PII served as an important pathogenic factor in fungal penetration of cuticle, but assay of PII-deficient strain in A. oligospora showed limited effect on nematicidal activity (Åhman et al. 1996, 2002).
It has been reported that bacterial protease and collagenase could degrade and destroy the inner cuticle layer and lining of the pharynx, suggesting involvement of bacterial protease in penetration of nematode cuticle in natural environment (Decraemer et al. 2003; Huang et al. 2004, 2005a; Niu et al. 2006). An alkaline protease BLG4 with strong nematocidal activity was purified from Brevibacillus laterosporus strain G4 to further elucidate its role in the infection process against nematode. Histological observation demonstrated the destruction of nematode cuticle and eventually followed by digestion of the host, suggesting the involvement of hydrolytic proteases as potential nematotoxic components in the bacterial infection of nematodes (Huang et al. 2005b).
To get more detailed molecular information about the bacterial infection against nematode, the encoding gene of BLG4 was cloned and deleted. The BLG4-deficient mutant showed a sharp drop in nematocidal and cuticle-degrading activities. Furthermore, the encoding gene of BLG4 was successfully expressed in Bacillus subtilis and recombinant BLG4 has nematotoxic activity in vitro. To our knowledge, this is the first report analyzing the function of a subtilisin enzyme involved in bacterium against nematode at the molecular level
Materials and methods
Bacterial strains, plasmids, and cultivation conditions
Brevibacillus laterosporus strain G4 (CCTCC, M203045) was maintained on YPD agar for bioassays and on LB agar for genetic manipulation. B. subtilis strain WB600, which is a six-extracellular-protease-deficient strain (Westers et al. 2004), was used for heterologous expression of B. laterosporus BLG4. The nematode Panagrellus redivius was grown axenically for 4–7 days at 28°C in oat medium (oat 20 g; water 10 ml). The plasmids used in this study were listed in Table 1.
Cloning of the BLG4 gene
Genomic DNA of B. laterosporus strain G4 was isolated using the Wizard genomic DNA purification kit for Gram-positive bacteria (Promega, Madison, WI, USA). Primers to get a 1,146 bp fragment including sequences encoding the signal peptide, propeptide and mature enzyme of BLG4 were designed on basis of the gene sequence of Bacillus amyloliquefaciens BPN′ precursor (Subtilisin Novo, La Jolla, CA, USA) (ID: P00782): I-1, 5′-ggagaggataaagagtgagaggcaaaa; I-2, 5′-ctgagctgccgcctgtacgt (Wells et al. 1983).
The BLG4 gene covering the entire coding gene and its promoter and terminator was obtained from a B. laterosporus genomic library, which was constructed by ligating 1–3 kb Sau3AI-digested fragments of genomic DNA into BamHI-digested pUC19. Screening was performed using PCR with primers I-1 and I-2 that were designed for cloning the BLG4 gene. Transformants in different panes were pooled and used as PCR templates. A second round of PCR was done for the positive panes to screen for single colonies. The plasmids were extracted and confirmed by enzyme-cut and sequence analysis, until a desired plasmid containing 1,600–1,700 bp was selected, designed pAP2000. The nucleotide sequence was analyzed using DNAman software package and the promoter was predicted using the BDGP Neural network promoter prediction interface (http://www.fruitfly.org/seq_tools/promoter.html) (Reese 2001).
Heterologous expression of BLG4
To validate the role of BLG4 involved in infection against nematodes, the BLG4 gene was expressed in B. subtilis WB600. pHY300PLK, a shuttle vector for B. subtilis and Escherichia coli with ampicillin and tetracycline resistance, was digested by BamHI/HindIII and ligated with a 1.7 kb fragment from pAP2000 resulting in pHYAP2000. This vector (pHYAP2000) was transformed into B. subtilis WB600 according to the method described by Spizizen (1958). Transformants with tetracycline resistance were selected and protease activity were determined in superrich medium (2% Yeast extract, 2.5% Tryptone, 0.3% K2HPO4, and 3% Glucose) supplemented with 100 μg/ml tetracycline resistance and used for purifying recombinant BLG4 according to previous described procedure (Huang et al. 2005b).
Construction of a BLG4-deficient mutant
To disrupt the BLG4 gene, a 156-bp gene fragment, which is identical with the middle part (indicates as orf2) of the encoding gene of mature protease BLG4, was amplified from G4 chromosomal DNA using two primers containing PstI sites (underlined): 164-1 (5′-aactgcagcccaacgcatctctttac) and 164-2 (5′-acgctgcagaaccagaaggtccgc). The PCR-product was recovered from the agarose gel after electrophoresis and subsequently digested with PstI in parallel with vector pBGSC (Bacillus Genetic Stock Center Columbus OH, BGSC). After precipitation, the fragment and plasmid were mixed and ligated (molar ratio: 3–7:1). The ligation mixture was used to transform competent cells of E. coli. Recombinant plasmids designed as pAP164 were determined by restriction analysis and sequenced before transforming into B. laterosporus strain G4 according to the modified methods described by Chang and Cohen (1979). Transformants were selected on LB agar containing 3 μg/ml chloramphenicol and were determined by western blot to ensure the disruption of protease BLG4 in B. laterosporus strain G4.
Peptide sequencing and western blot
The N-terminal amino acid sequence of the purified protease BLG4 was determined on an ABI precise 491-protein sequencer. The N-terminal sequence of the first ten amino acids was used as a query for blast in GenBank.
Antisera were generated in mice against recombinant BLG4, which had purified to homogeneity. The antisera containing polyclonal antibodies against BLG4 were directly used for western blot analysis to detect the protease BLG4 in wild-type B. latersporus G4 and mutant strain. After SDS-PAGE and electrotransfer of the protein to a PVDF-membrane, the membranes were blocked by incubation with TBS containing 5% non-fat dry milk for 30 min at room temperature, and subsequently incubated with the antisera for 1 h at shaker. After three cycles of washing with TBS solution, the membrane was incubated with goat anti-mouse IgG-AP. Immunostaining was performed using the NBT/BCIP kit (Sino-American Biotechnology Co., Beijing, China) according to manufacturer’s instructions.
Bioassays
Free-living nematode (P. redivius) was selected for the bioassay. Infection assay using a cellophane paper technique was employed (Huang et al. 2005b). After 2 days of bacterial growth, the infection experiments were started by adding 200–300 nematodes per plate. After another 5 days, the nematodes were washed and dead nematodes were counted under a light microscope. The toxicity effects of crude extracts from wild-type B. latersporus G4 and mutant strain on P. redivivus were also tested according to the method described by Huang et al. (2005b). The numbers of dead and cuticle-degraded nematodes were determined under a light microscopy. The effects of the recombinant protease BLG4 against the cuticle of nematodes were examined by SEM 48 h after treatment. The bioassay experiments were performed in triple parallels and repeated at least five times.
Results
Cloning and analysis of BLG4 gene
The N-terminal amino acid sequence of the purified protease BLG4 was AQSVPYGVSQ, which has 100% homology to Subtilisin BPN′ precursor (Subtilisin Novo) of B. amyloliquefaciens. The full-length gene was cloned by PCR with primers based on the published sequences of Subtilisin BPN′ (ID: P00782) (Wells et al. 1983). The nucleotide sequence of an amplified 1,146-bp fragment was determined and placed in the public domain (ID: AY720895). The deduced protein consisted of a signal peptide of 30 amino acids, a propeptide of 77 amino acids and mature protease of 275 residues. The previously sequenced N-terminal amino acids were found to be located at amino acids 108–118 of Subtilisin BPN′, which was consistent with the beginning of the encoded protein BLG4. Biochemical experiments have previously indicated that BLG4 is a serine protease (Huang et al. 2005a, b). This was confirmed by inspection of the deduced amino acid sequence of BLG4, which has a catalytic triad center containing Asp32, His64, and Ser221. GenBank database search showed that the nucleotide and amino acid sequence of BLG4 shares extensive similarities with members of the subtilisin family of serine proteases. BLG4 exhibited 99.7% homology with another cuticle-degrading protease from Bacillus sp. B16, and showed 69–97% sequence identity with other subtilisins produced by bacteria. However, BLG4 shared low homology (less than 35%) with protease K and other cuticle-degrading proteases from nematode-trapping fungi. Moreover, a high degree of homology between the sequences was found in regions containing the active site residues Asp-His-Ser and the two blocks of side-chains residues Ser-Leu-Gly-Gly and Ala-Ala-Gly that form the sides of the substrate-binding pockets in subtilisin (Siezen and Leunissen 1997; Niu et al. 2006), which is consistent with their similar biochemical traits and mutual action against nematodes.
To amplify entire BLG4 gene including 5′ and 3′ flanking regions, a B. laterosporus genomic library was constructed. The library was differentially screened using PCR. A total of about 7,000 clones were determined and seven positive clones were identified, and a genomic clone (pAP2000) contained a complete ORF, promoter and terminator (ID: AY720895) was selected. A putative ribosomal binding site with the sequence GGAGAG can be identified 8 bp upstream of GTG. At −95 bp (the first G in GTG being +1), the sequence GGTCTA indicates a potential −35 region and a putative −10 region can be identified at −73 bp. The stem-loop structure and poly T region of the potential transcription terminator can be identified 4 bp downstream from the termination codon (TAA).
Heterologous expression of BLG4
The B. subtilis expression vector pHYAP2000 was constructed by inserting the entire BLG4 gene with its promoter and terminator sequences in the pHY300PLK vector. After transforming into B. subtilis, all the tetracycline resistant colonies showed significant protease activity compared to control strain containing pHY300PLK. SDS-PAGE analysis revealed that pHYAP2000 transformant 1–3 (lane 4 in Fig. 1a) had an additional protein band of approximately 28.7 kDa, which was not present in the control (Fig. 1a). The recombinant BLG4 was purified according to previously described method (Huang et al. 2005b). There were no significant differences between the recombinant and native BLG4 with regard to biochemical properties. Both of them were inhibited by PMSF and degraded a broad range of substrates (Fig. 2a). Western-blot analysis showed one band hybridizing to supernatant from B. laterosporus strain G4 and to pHYA2000 transformants 1–3 (Fig. 1b). No band was present after immunostaining with negative controls.

SDS-PAGE and western blotting analysis of heterologous expression of BLG4. a SDS-PAGE analysis. Line 1: marker; lines 2–4: supernatant for 24 h from Bacillus subtilis, pHY300PLK transformant and pHYAP2000 transformant 1–3, respectively. Lines 5–7: supernatant for 48 h from Bacillus subtilis, pHY300PLK transformant and pHYAP2000 transformant 1–3, respectively. b Western blotting analysis. Line 1: supernatant from Brevibacillus laterosporus G4; line 2: marker; lines 3–6: supernatant from pHY300PLK transformant (control) and pHYAP2000 transformant 1–3 for 24 and 48 h, respectively

Assays of protease activity of recombinant BLG4 and mutant BLG4-6. a Assays of protease activity of purified recombinant BLG4 at pH 7.0, 37°C. Protease activity of purified recombinant BLG4 against casein was 100%. b Assays of protease activity of mutant BLG4-6 at pH 7.0, 37°C. Protease activity of supernatant from Brevibacillus laterosporus G4 against casein was 100% at pH 7.0, 37°C. c Assays of protease activity of mutant BLG4-6 at pH 10.0, 50°C. Protease activity of supernatant from Brevibacillus laterosporus G4 against casein was 100% at pH 10.0, 50°C
Construction of BLG4-deficient mutants
The gene disruption vector pAP164 was transformed into wild-type B. laterosporus and 16 different clones were examined for proteolytic activity when grown on LB agar supplemented with 0.5% casein. Six clones had significantly reduced proteolytic activity, and one of them showed elevated levels of protease activity. Western blot analysis was performed to verify whether the 30 kDa BLG4 protein was present or not. The wild-type strain contained one hybridizing band of 30 kDa (Fig. 3b, lane 3), which was not present in mutant BLG4-6. This pattern of hybridization is expected and indicates that the BLG4 gene was properly mutated by insertion of vector pAP164 (Fig. 3a) (Vosman et al. 1986; Vgner et al. 1998). There was no difference in the phenotype when the mutant and the wild type grew in LB and YPD medium. The protease activity of mutant BLG4-6 was further examined and compared to the wild-type strain. Under the condition of pH 7.0, 37°C, the activity of mutant BLG4-6 was decreased by 77% (the wild-type protease activity was 100%). The serine protease inhibitor PMSF inhibited about 73% of the protease activity in wild-type strain, but did not have a significant effect on the protease activity of the mutant. However, EDTA inhibited almost all of the residual activity of mutant BLG4-6 and reduced the activity of wild-type strain by 21% (Fig. 2b). Under the condition of pH10, 50°C, mutant BLG4-6 almost lost all protease activity and PMSF also completely inhibited the activity of wild-type strain (Fig. 2c).

Integration of the disruption vector pAP164 into the target gene and western blot analysis of mutant BLG4-6. a orf1, orf3 segments of BLG4 gene are indicated as white boxes. Hatched box corresponds to the internal 156-bp segment or orf2 of the target gene. The vector pAP164 is integrated in orf2 by a single cross-over recombination event. Arrows indicate direction of transcription. Chl R indicates the chloramphenicol resistance gene. Amp R indicates the ampicillin resistance gene. b Western blot analysis of mutant BLG4-6. Line 1: marker; line 2: mutant BLG4-6; line 3: Brevibacillus laterosporus G4. Brevibacillus laterosporus G4 contained one hybridizing band of 30 kDa. The mutant BLG4-6 produced one band, which translates from orf1 to orf2 gene fragment (about 18 kDa), indicating that the BLG4 disruption vector pAP164 was successfully inserted in the BLG4 gene of Brevibacillus laterosporus as shown in (a)
Bioassay
The pathogenicity of mutant BLG4-6, pHYAP2000 transformant 1–3 (heterologous expression), B. laterosporus strain G4, B. sutilis strain WB600 and E. coli DH5α were examined against the nematode P. redivius. After the nematodes were rinsed with distilled water, the nematodes were examined under a light microscope to count the mortality rate and the percentage of nematodes of which the cuticle was degraded. In two of the control experiments with the non-pathogenic bacteria strains WB600 and E. coli DH5α, the numbers of nematodes increased 5–20-folds. Majority of the tested nematodes grew well and the percentage of dead nematodes remained below 10% within 5 days, no degraded nematode was found. In contrast, the B. laterosporus strain G4 killed about 93% of the nematodes, and majority of dead nematodes (91%) were degraded and digested. Mutant BLG4-6 had a significant reduced activity against nematodes; only 43% of the nematodes were killed and in 22% of the cuticles of dead nematodes were degraded (but not completely digested). These results indicate that BLG4 has a significant effect on nematocidal activities of B. laterosporus and degradation of nematode cuticles.
Transformant 1–3 (the strain heterologous expressing BLG4) also showed significant nematocidal activity in infection experiments. About 68% of the nematodes were killed and 51% of the dead nematodes were degraded, confirming the nematocidal activity and cuticle-degrading function of BLG4 in vitro. However, the effect of pHYAP2000 transformant 1–3 against nematodes was lower than wild-type strain. There are several possibilities to explain this: (1) lower protease efficient in the supernatant of pHYAP2000 transformant 1–3. By assaying the protease activity of the purified recombinant BLG4 showed lower activity against different substrates, including extracted nematode cuticle (recombinant BLG4 showed 7% activity whereas native BLG4 showed 14.6% activity. The activity against casein was 100%) (Fig. 2a). (2) Alteration in the modification pattern of the protein affects the nematocidal activity of BLG4 against nematodes. (3) The lack of other pathogenic factors in pHYAP2000 transformant 1–3 could also decrease the nematocidal activity.
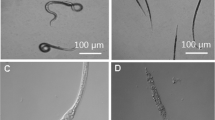
The toxicity effects of mutant BLG4-6 and transformant 1–3 against nematodes were also determined and compared to B. laterosporus strain G4 and negative controls (Table 2). As shown in Table 2, crude extracellular protein extract from supernatant of B. laterosporus strain G4 showed high toxicity activity toward the free-living nematodes P. redivius. The transformant 1–3 had significant nematotoxic activity, about 77% of nematodes were killed and 70% of the dead nematodes were degraded within 48 h. Histopathological changes of the cuticle of nematodes after being treated with crude extracellular protein extract from pHYAP2000 transformants 1–3 were examined with scanning electron microscopy (SEM) (Fig. 4). After being treated with crude extracellular protein extract for 48 h, the cuticle of the nematodes was degraded and exfoliated. Significant flaws can be found in some nematode bodies, while nematodes treated with control extract had a smooth surface with distinct striaes and lateral lines. After disruption of BLG4 in B. laterosporus, the mutant BLG4-6 had a limited toxicity effect on the nematodes. Only 44% of nematodes were killed within 48 h and majority of dead nematodes (82%) was not degraded, indicating an important role for BLG4 in killing and digesting nematodes.

The action of recombinant protease BLG4 against the nematode Panagrellus redivius observed with SEM. a, b Nematodes were treated by boiled purified recombinant protease for 48 h, majority of nematodes (more than 80%) were viable and their cuticles were intact. c, d Nematodes were treated with the purified recombinant protease BLG4 for 48 h, majority of nematodes (68%) were dead and their cuticles were degraded and destroyed, and the outer layer of the nematode cuticle was exfoliated
Discussion
As a pathogen, B. laterosporus has a broad spectrum of activities against invertebrates and its toxicity effects against nematodes have been described in detail (Singer 1996, 1973; Singer et al. 1997). In these papers, the observed toxicity was reported against the nematode phytoparasite Heterodera glycines and zooparasite Trichostrongylus colubriformi (De Oliveira et al. 2004). However, there is little information about the pathogenic mechanism of B. laterosporus on invertebrates. In our recent reports, the function of the extracellular protease BLG4 from B. laterosporus strain G4 was demonstrated to serve as a pathogenic factor in the infection against nematode without forming parasporal crystals (Huang et al. 2005b). Genetic diversity among the various B. laterosporus strains can explain the different virulence factors present among these strains (Zahner et al. 1999; De Oliveira et al. 2004).
The roles of cuticle-degrading proteases in the infection against nematodes have been studied extensively in nematophagous fungi (Segers et al. 1994; Bonants et al. 1995; Åhman et al. 2002). The previous biochemical analysis of BLG4 isolated from B. laterosporus strain G4 indicated that it is similar to cuticle-degrading proteases from fungi, including maximum activity in an alkaline situation and a high temperature, inhibition by PMSF, broad range of substrates and similar actions against the nematode cuticle. The reasons that the infection against invertebrates by fungi is more effective than by bacteria may be related to the involvement of mechanic forces and other enzymes such as chitinase during the infection process (Tikhonov et al. 2002).
The BLG4 gene encodes a mature peptide containing 275 amino acids with a calculated molecular mass of 28.7 kDa. The electrophoretic mobility of the recombinant protein on the denaturing gel is in accordance with this calculated mass although it migrates slightly faster than native BLG4 (30 kDa) from B. laterosporus strain G4. These differences are probably related to protein modification. In protease activity assays, native BLG4 from B. laterosporus strain G4 showed 14.6% activity against cuticle of nematodes and only 7% activity was measured for recombinant BLG4 expressed in B. subtilis. Similar observations were found when studying PII from nematophagous fungi A. oligospora. The electrophoretic mobility of the recombinant PII was slightly different from the native enzyme and the specific activity of the recombinant enzyme was less efficient than the native enzyme (0.8 vs. 1.9 U/mg, respectively). These observations suggest the pathogenicity of these enzymes may be improved by altering the modification patterns after translation (Åhman et al. 2002).
In recent years, some nematophagous bacteria were reported (Huang et al. 2005b; Niu et al. 2006). Involvement of extracellular protease in the infection against nematode was confirmed by degrading of nematodes cuticle. The further works to analyze the mode of the infection and the function of subtilisin enzyme from bacterium involved in infection against nematode at the molecular level were done with protease BLG4 from B. laterosporus strain G4. Bioassays indicated that recombinant BLG4 has nematotoxic activity in vitro. Moreover, mutant BLG4-6 showed a sharp drop in nematocidal activity and cuticle-degrading activity. These results demonstrate the important role of BLG4 in destroying the nematode and the key role it plays in penetrating the nematode cuticle by bacteria. In addition, Mutant BLG4-6 remained 43% mortality of the nematodes and 22% degradation activity against nematode cuticle (not completely digested), suggesting extracellular protease BLG4 play more important role in degradation of nematode cuticle than mortality of nematodes and other pathogenic factors such as other proteases or toxin involved in infection against nematodes in B. laterosporus strain G4.
Compared to nematophagous fungi, there are several advantages for bacteria to be developed as a model to study the function of proteases in infection against nematode. First, bacteria do not form trapping device and rely on the involvement of mechanic force to attack nematodes. Only enzymes are involved in the penetration of nematode cuticle during bacterial infection. Therefore, deletion of pathogenic subtilisin in B. laterosporus strain G4 had a significant effect on nematicidal activity. Second, there is a substantial array of molecular biological methods available for bacteria and abundant knowledge about bacterial subtilisin is available. It is therefore possible to investigate the functional protein domains responsible for the cuticle degrading-activity and/or killing of nematodes by genetic engineering of the subtilisin enzyme. Being amenable to genetic manipulation, higher nematocidal activity and a soil pathogen, B. laterosporus strain G4 is an ideal host to introduce different pathogenic genes to construct a strain with even more toxicity to improve the biocontrol of nematodes.
References
Åhman J, Ek B, Rask L, Tunlid A (1996) Sequence analysis and regulation of a gne encoding a cuticle-degrading serine protease from the nematophagous fungus Arthrobotrys oligospora. Microbiology 142:1605–1616
Åhman J, Johansson T, Olsson M, Punt PJ, van den Hondel CA, Tunlid A (2002) Improving the pathogenicity of a nematode-trapping fungus by genetic engineering of a subtilisin with nematotoxic activity. Appl Environ Microbiol 68:3408–3415
Bird AF, Bird J (1991) The structure of nematode. 2nd edn. Academic Press, San Diego, CA
Bonants PJ, Fitters PF, Thijs H, den Belder E, Waalwijk C, Henfling JW (1995) A basic serine protease from Paecilomyces lilacinus with biological activity against Meloidogyne hapla eggs. Microbiology 141:775–784
Chang S, Cohen SH (1979) High frequency transformation of Bacillus subtulis protoplasts by plasmid DNA. Mol Gen Genet 168:111–115
Cox GN, Kusch M, Edgar RS (1981) Cuticle of Caenorhabditis elegans: its isolation and partial characterization. J Cell Biol 90:7–17
Decraemer W, Karanastasi E, Brown D, Backeljau T (2003) Review of the ultrastructure of the nematode body cuticle and its phylogentic interpretation. Biol Rev 78:465–510
De Oliveira EJ, Rabinovitch L, Monnerat RG, Passos LK, Zahner V (2004) Molecular characterization of Brevibacillus laterosporus and its potential use in biological control. Appl Environ Microbiol 70:6657–6664
Huang XW, Zhao NH, Zhang KQ (2004) Extracellular enzymes serving as virulence factors in nematophagous fungi involved in infection of the host. Res Microbiol 155:811–816
Huang XW, Niu QH, Zhou W, Zhang KQ (2005a) Bacillus nematocida sp. Nov., a novel bacterial strain with nematotoxic activity isolated from soil in Yunnan, China. Syst Appl Microbiol 28:323–327
Huang XW, Tian BY, Niu QH, Yang JK, Zhang LM, Zhang KQ (2005b) An extracellular protease from Brevibacillus laterosporus G4 without parasporal crystal can serve as a pathogenic factor in infection of nematodes. Res Microbiol 156:719–727
Maizels RM, Blaxter ML, Selkirk ME (1993) Forms and functions of nematode surfaces. Exp Parasitol 77:380–384
Niu QH, Huang XW, Tian BY, Yang JK, Liu J, Zhang L, Zhang KQ (2006) Bacillus sp. B16 kills nematodes with a serine protease identified as a pathogenic factor. Appl Microbiol Biotechnol 69:722–730
Reese MG (2001) Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput Chem 26:51–56
Segers R, Butt TM, Kerry BR, Peberdy JF (1994) The nematophagous fungus Verticillium chlamydosporium Goddard produces a chymoelastase-like protease which hydrolyses host nematode proteins in situ. Microbiology 140:2715–2723
Siezen RJ, Leunissen JAM (1997) Subtilase: the superfamily of subtilisin-like serine proteases. Protein Sci 6:501–523
Singer S (1973) Insecticidal activity of recent bacterial isolates and their toxins against mosquito larvae. Nature 244:110–111
Singer S (1996) The utility of strains of morphological group II Bacillus. Adv Appl Microbiol 42:219–261
Singer S, van Fleet AL, Viet JJ, Genevese EE (1997) Biological control of the zebra mussel Dreissena polymorpha and the snail Biomphalaria glabrata, using Gramicidin S and D and molluscicidal strains of Bacillus. J Ind Microbiol Biotechnol 18:226–231
Spizizen J (1958) Transformation of biochemically deficient strains of Bacillus subtulis by deoxyribonucleate. Proc Natl Acad Sci USA 44:1072–1075
St Leger RJ (1995) The role of cuticle-degrading proteases in fungal pathogenesis of insects. Can J Bot 73:1119–1125
St Leger RJ, Frank DC, Roberts DW, Staples RC (1992) Molecular cloning and regulatory analysis of the cuticle-degrading-protease structure gene from the entomopathogenic fungus Metarhizium anisopliae. Eur J Biochem 204:991–1001
Tikhonov VE, Lopez-Llorca LV, Salinas J, Jansson HB (2002) Purification and characterization of chitinases from the nematophagous fungi Verticillium chlamydosporium and Verticillium suchlasporium. Fungal Genet Biol 35:67–78
Tunlid A, Jansson S (1991) Proteases and their involvement in the infection and immobilization of nematodes by the nematophagous fungus Arthrobotrys oligospora. Appl Environ Microbiol 57:2868–2872
Vgner V, Dervyn E, Ehrlich SD (1998) A vector for systematic gene inactivation in Bacillus subtilis. Microbiology 144:3097–3104
Vosman B, Kooistra J, Olijve J, Venema G (1986) Integration of vector-containing Bacillus subtilis chromosomal DNA by a campbel-like mechanism. Mol Gen Genet 204:524–531
Wells JA, Ferrari E, Henner DJ, Estell DA, Chen EY (1983) Cloning, sequencing, and secretion of Bacillus amyloliquefaciens subtilisin in Bacillus subtilis. Nucleic Acids Res 11:7911–7925
Westers L, Westers H, Quax WJ (2004) Bacillus subtilis as cell factory for pharmaceutial proteins: a biotechnological approach to optimize the host organism. Biochim Biophys Acta 1694:299–310
Yang JK, Huang XW, Tian BY, Wang M, Niu QH, Zhang KQ (2005). Isolation and characterization of a serine protease from the nematophagous fungus Lecanicillium psalliotae, displaying nematicidal activity. Biotechnol Lett 27:1123–1128
Zahner V, Rabinovitch L, Suffys P, Momen H (1999) Genotypic diversity among Bacillus laterosporus strains. Appl Environ Microbiol 65:5182–5185
Zhao ML, Mo MH, Zhang KQ (2004) Characterization of a neutral serine protease and its full-lenth cDNA from the nematode-trapping fungus Arthrobotrys oligospora. Mycologia 96:16–22
Acknowledgments
We thank Dr. Xiaowei Huang, Dr. Minghe Mo, Dr. Hong Luo, Dr. Hui Sun, Dr Linqian Dong, and Ms Qiuhong Niu, Kaihui Liu, Yanfang Liu for their help and advice in our studies. We also thank Yongjun Liu of the Yunnan University for his help in taking SEM-pictures and Professor Lan Ma of the Yunnan University for preparation of antisera. We are also indebted to Wei Zhou for her help during all the work and W. G. Dilantha Fernando for preparing this manuscript. The work was funded by projects from National Natural Science Foundation (approved No. 30500338) and Department of Science and Technology of Yunnan Province (approved Nos. 2005NG05 and 2005NG03).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tian, B., Li, N., Lian, L. et al. Cloning, expression and deletion of the cuticle-degrading protease BLG4 from nematophagous bacterium Brevibacillus laterosporus G4. Arch Microbiol 186, 297–305 (2006). https://doi.org/10.1007/s00203-006-0145-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-006-0145-1