
Abstract
The rolling circle (RC) mechanism of DNA replication generating single-stranded DNA (ssDNA) intermediates is common in various high-copy circular plasmids in Streptomyces, and the ssDNA released after leading strand synthesis is converted to its double-stranded form (dsDNA) by the host proteins. The in vivo and in vitro lagging strand syntheses from ssDNA replicative intermediates of RC plasmid pSN22 in Streptomyces lividans was characterized. The presence or absence of the single-strand origin (sso), the replication initiation site of lagging strand synthesis, did not significantly affect the copy numbers of pSN22 derivatives. In vivo lagging strand synthesis was not affected by the rifampicin inhibition of S. lividans RNA polymerase. Likewise, in vitro lagging strand synthesis using cell-free extracts revealedsso-independent, rifampicin-resistant lagging strand synthesis in S. lividans. Although all four dNTPs are usually required for the initiation of such synthesis, the presence of only one NTP was sufficient to carry outlagging strand synthesis in vitro. Interestingly, the cell-free extract of exponential-phase cells required less ATP than that of stationary-phase cells. These results reveal a predominant RNA polymerase-independent priming system in S. lividans that may be a result of the stabilization of RC plasmids lacking sso in S. lividans.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
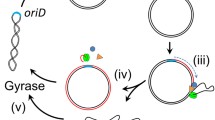
Rolling circle (RC) replication is the replication mechanism of a large number of small circular plasmids isolated from gram-positive bacteria (Novick 1989; Gruss and Ehrlich 1989; Jannière et al. 1993; Khan 1997; Leenhouts et al. 1991; del Solar et al. 1993). RC replication yields strand-specific single-stranded circular plasmid DNA (ssDNA) replication intermediates (te Riele et al. 1986a,b), whose conversion from ssDNA to a double-stranded (ds) plasmid molecule through lagging strand synthesis is initiated at the single strand origin (sso), also called the minus origin (M-O). The replication mechanisms of small circular plasmids in the genus Streptomyces have been investigated in order to develop vectors useful in the genetic manipulation of this industrially important microorganism. Studies showed that most small circular plasmids replicate via the RC mechanism (Deng et al. 1988; Hagège et al. 1993; Kataoka et al. 1994b; Muth et al. 1995; Pigac et al. 1988; Servín-González et al. 1995; Yokoyama et al. 1996), with Streptomyces lividans being the major host strain used.
sso-initiated lagging strand replication is a problem in the initiation of RC replication. As for other RC plasmids from gram-positive bacteria, the lack of sso decreases the efficiency of initiating lagging strand replication in S. lividans, resulting in the accumulation of a large number of ssDNAs in host cells (Deng et al. 1988; Pigac et al. 1988). S. lividans plasmid pIJ101 (Kieser et al. 1982) lacking sso leads to a decrease in plasmid copy number without causing instability by plasmid segregation (Deng et al. 1988). However, the copy number of its derivative, pIJ702 (Katz et al. 1983), is more than 100 molecules per chromosomal DNA (Zaman et al. 1993) and the plasmids are stable in S. lividans cells. In contrast, the decrease in RC plasmid copy number to less than ten molecules per cell in Staphylococcus aureus (Gruss et al. 1987) due to the lack of the sso sequence, which causes plasmid segregation, indicates the suppression of the initiation of complementary strand synthesis at random nucleotide sequences of the ssDNA replication intermediate in S. aureus (Boe et al. 1989). However, the lack of the sso sequence does not affect the copy number of RC plasmids in Bacillus subtilis although there is an accumulation of ssDNA replication intermediates in B. subtilis cells (Gruss et al. 1987). S. lividans and B. subtilis thus appear to be efficient in terms of initiation mechanisms at random DNA sequences on ssDNA replication intermediates even without sso sequences.
In this report, the in vivo and in vitro sso-independent lagging strand replications of a pSN22-derived RC plasmid in S. lividans were studied. The 11-kbp pSN22 is a high-copy-number, broad-host-range, and self-transmissible plasmid isolated from Streptomyces nigrifaciens SN22 (Kataoka et al. 1991). pSN22 is stable in S. lividans and replicates by the RC mechanism (Kataoka et al. 1994b). The effect of sso sequence on the copy number of pSN22 derivatives was examined. In vitro conversion of ssDNA to dsDNA using cell-free extract of S. lividans TK21, a plasmid-free strain, revealed that S. lividans has a rifampicin-resistant sso-independent lagging strand synthesis activity. The S. lividans extract was characterized in order to determine the optimal conditions for random sso-independent lagging strand synthesis.
Materials and methods
Bacterial strains, plasmids and media
Escherichia coli strain JM109 was used as host for pUC19 (Yanisch-Perron et al. 1985) and pBluescript II SK + (Stratagene), E. coli JM105 as a host for M13mp18 and M13mp19 (Yanisch-Perron et al. 1985), and E. coli BL21 (Studier and Moffatt 1986) for cell-free extract (fraction II) preparation. S. lividans TK21 (Kieser et al. 1982) was the host strain for Streptomyces plasmids and was used for cell-free extract preparation. E. coli cultures were grown in 2xYT medium (Sambrook et al. 1989). For the in vivo experiments of plasmid replication, S. lividans cultures were grown in liquid YEME medium I (Hopwood et al. 1985). When required, thiostrepton was added at 5 μg/ml. To determine the role of RNA polymerase in the conversion of ssDNA to dsDNA, protein synthesis was blocked by adding 100 μg erythromycin/ml to the medium and RNA synthesis was blocked by adding 100 μg rifampicin/ml. For preparation of the cell-free extract, S. lividans was cultured in liquid YEME medium II (Thompson et al. 1984) containing 0.5% (w/v) polyethyleneglycol (PEG) 6000 instead of sucrose. The plasmids used in this study are listed in Fig. 1.

Plasmids used in this study. sso activity indicates the in vivo activity of pRTS020N and pRTS020O (Suzuki et al. 1997a). pRTS020N and pRTS020O harbor the PstI–BglII fragment of pSN22-containing sso1 inserted in the normal (5′ to 3′) and opposite (3′ to 5′) orientations, respectively, in accordance with the double-strand origin (dso)-rep of pSN22 (Kataoka et al. 1994a,b; Suzuki et al. 1997b). Thiostrepton-resistance gene (tsr) from pIJ6 (Thompson et al. 1982) served as a selection marker. pSS020 and pSS021 harbor the PstI–BglII fragment with or without sso1, respectively, on a single-stranded phagemid
DNA manipulations
Plasmid and bacteriophage DNA isolation, in vitro DNA manipulations, and E. coli transformations were done as described by Sambrook et al. (1989). S. lividans TK21 protoplasts were prepared and transformed as described by Hopwood et al. (1985). The enzymes used for DNA manipulations were purchased from TAKARA (Kyoto, Japan).
Detection of ssDNA accumulation
Total DNA was isolated from S. lividans transformants as described by Hopwood et al. (1985). To detect both ds and ss plasmid molecules, 1.6 μg total DNA was digested with EcoRI, applied to a 1% agarose gel, and fractionated by electrophoresis. The denatured DNA was transferred onto nylon membranes (Hybond-N, Amersham) and the plasmid molecules were detected by Southern hybridization (Southern 1975) using a DIG luminescent detection kit (Boehringer Mannheim) with DIG-labeled pBluescript II SK + as probe. To detect ssDNA molecules only, 1.6 μg total DNA was electrophoresed and then transferred onto the nylon membrane without prior denaturation (te Riele et al. 1986a).
Preparation of cell-free extract
Cell-free extracts of S. lividans were prepared from the plasmid-free strain S. lividans TK21. Cells were grown at 30°C in 6 l of YEME medium II using a 10-l jar fermentor stirred at 200 rpm and 1 vvm. Mycelia from the mid-exponential phase (12 h) or stationary phase (36 h) were harvested by filtration (Thompson and Cundliffe 1981) at room temperature and then stored at −80°C. The cells were resuspended in S30 buffer (50 mM HEPES–KOH pH 7.5, 60 mM NH4OAc, 10 mM Mg(OAc)2, 5 mM 2-mercaptoethanol, and 10% (v/v) glycerol) based on 2.5 ml buffer per gram wet weight. The cell suspension was subjected to a French press at 70–80 MPa, and the lysate obtained was centrifuged for 30 min at 39,000×g at 4°C. Endogenous DNA in the lysate was removed by streptomycin sulfate precipitation as described by Conrad and Campbell (1979). To 1.0 ml of the supernatant, 0.1 ml of a 33% (w/v) streptomycin sulfate solution was added, and the mixture was stirred for 30 min at 0°C. The collected supernatant after 30-min centrifugation at 39,000×g at 4°C was subjected to ammonium sulfate precipitation at 70% saturation and stirred for 30 min at 0°C. The precipitate obtained was suspended in S30 buffer and the suspension was dialyzed against the same buffer for 3 h at 4°C, after which the volume of the dialyzed material was adjusted to the initial lysate volume. The protein concentration of the prepared cell-free extract, determined using Protein Assay Kit I (Bio-Rad) with IgG as standard, was 6.4 mg/ml. The cell-free extract was subdivided into small aliquots and stored at −80°C.
ssDNA preparation
M13mp18 ssDNA was isolated as described by Sambrook et al. (1989). Recombinant pBluescript II SK + ssDNA was prepared according to the protocol provided by Stratagene using the helper phage VCSM13. The ssDNAs were further purified by alkaline agarose gel electrophoresis through electroelution. The presence of residual small oligonucleotide fragments in the purified ssDNA solution was tested by measuring the incorporation of [5-3H]dCTP using the Klenow fragment. The incorporation reaction mixture, carried out at 37°C for 30 min in a total volume of 12.5 μl, contained 20 mM Tris–HCl (pH 8.0), 10 mM MgCl2, 50 μM of each dATP, dGTP, and dTTP, 1 μl (4 μM, 25 Ci/mmol)[5-3H]dCTP, 0.5 μg ssDNA and 0.16 U Klenow fragment/ml . Termination of the reaction and measurement of [5-3H]dCTP incorporation were carried out as described below. After alkaline agarose gel electrophoresis, the incorporation of [5-3H]dCTP was reduced 55-fold to 0.06 pmol per 0.5 μg M13mp18 ssDNA.
RNase treatment of cell-free extract and template ssDNA
To eliminate possible contaminating endogenous RNA that might prime lagging strand synthesis, the cell-free extracts and template ssDNAs were treated with RNase A and RNase T1 (90 u/ml each) for 1 h at 30°C and then passed through a NAP-10 column (Pharmacia) to remove the digested ribonucleotides.
Assay for in vitro ssDNA replication
The standard reaction mixtures (12.5 μl) contained 50 mM HEPES–KOH pH 7.5, 60 mM NH4OAc, 7.5 mM Mg(OAc)2, 5 mM 2-mercaptoethanol, 10% (v/v) glycerol, 5 μg rifampicin/ml, 2 mM ATP, 50 μM each of dATP, dGTP, and dTTP, 1.0 μl (4 μM, 25 Ci/mmol)[5-3H]dCTP or 0.5 μl (0.132 μM, 3,000 Ci/mmol) of [α-32P]dCTP, 0.5 μg ssDNA, and 2.0 μl cell-free extract. When using 0.5 μg poly(dT) (Pharmacia) as a template, 0.5 μl (0.132 μM, 3, 000 Ci/mmol) [α-32P]dATP was added and the three dNTPs were omitted. Incubation was carried out at 30°C. Reactions using [5-3H]dCTP were terminated by adding 500 μl of cold 10% (w/v) trichloroacetic acid–0.1 M sodium diphosphate (TCA–NaPPi) solution. The precipitates were collected on glass-fiber filters as described by Inuzuka and Helinski (1978), and radioactivity was determined using a liquid scintillation counter. Reactions involving [α-32P]dATP and poly(dT) were terminated and analyzed as described above. Reactions using [α-32P]dCTP were terminated by adding 1.25 μl 500 mM EDTA. The samples were fractionated by 0.7% agarose gel electrophoresis. The reaction products were identified by autoradiography of dried gels.
Preparation of E. coli cell-free extracts
E. coli cell-free extracts (fraction II) were prepared from the plasmid-free strain BL21 as described by Fuller et al. (1981). The in vitro lagging strand synthesis reaction mixtures (12.5 μl) contained 50 mM HEPES–KOH (pH 7.5), 60 mM NH4OAc, 7.5 mM Mg(OAc)2, 5 mM 2-mercaptoethanol, 10% (v/v) glycerol, 50 μM each of dATP, dGTP, and dTTP, 1.0 μl[5-3H]dCTP, 0.5 μg M13mp18 ssDNA, and 12.8 μg cell-free extract proteins. The mixture were incubated for 30 min at 37°C.
Results
Lack of sso has no effect on copy number of pSN22 derivatives in S. lividans
The lack of the sso sequence does not cause significant segregational instability of the pSN22 derivatives, but leads to the accumulation of ssDNA molecules in S. lividans cells (Kataoka et al. 1994a). Hence, either sso deletion does not influencethe copy number of the plasmid or pSN22 has a specific segregation mechanism. To assess the former possibility, the copy numbers of the two pSN22 derivatives (Suzuki et al. 1997a), pRTS020N (carrying sso1 in active orientation) and pRTS020O (carrying sso1 in inactive orientation), with and without the sso sequence, respectively, were determined. As shown in Fig. 2, the intensity ratios of both plasmids compared to the chromosome showed no significant differences, indicating that the lack of sso1 does not affect the plasmid copy number of the derivatives in S. lividans. Alternatively, the sso-independent lagging strand synthesis of pSN22 derivatives is effective in preserving the homeostasis of plasmid copy number.

Copy number of pSN22 derivatives in Streptomyces lividans. EcoRI-digested total DNA (1.6 μg) was fractionated by 1% agarose gel electrophoresis and visualized by ethidium bromide staining. Lanes 1,, 2 pRTS020N (carrying sso1 in the active orientation); lanes 3’, 4 pRTS020O (carrying sso1 in the inactive orientation)
Effect of rifampicin on lagging strand synthesis of pSN22 derivatives in S. lividans
It has been postulated that lagging strand synthesis is initiated by a primer RNA synthesized from sso by RNA polymerase (RNAP) (Birch and Khan 1992; Dempsey et al. 1995; Kramer et al. 1997,1998). To determine the involvement of RNAP in the lagging strand synthesis of pSN22 derivatives in S. lividans, the effect of the RNAP inhibitor rifampicin on ssDNA conversion was examined. S. lividans TK21 did not grow on the agar plate containing 50 μg rifampicin/ml (data not shown). Two strains harboring pRTS020N or pRTS020O were examined. Cultures were grown to the mid-exponential phase, and 100 μg rifampicin/ml (final) was added to half of the cultures. Simultaneously, 100 μg erythromycin/ml (final)was added to all of the cultures to inhibit the initiation of plasmid replication. The cells were incubated at 30°C for 30–120 min after which DNA was extracted, and plasmid DNA molecules were determined by Southern hybridization using DIG-labeled pBluescript II SK + as a probe. Interestingly, in contrast with the lagging strand synthesis of RC plasmids in S. aureus cells, in which the amount of ssDNA molecules increases (Boe et al. 1989), rifampicin had no effect on ssDNA and dsDNA with either of the plasmids (Fig. 3), indicating that replication from a ss plasmid in S. lividans is independent of RNAP. However, both first and lagging strand syntheses might have been inhibited simultaneously by rifampicin.

Effect of rifampicin on lagging strand synthesis of pSN22 derivatives in S. lividans. After adding 100 μg rifampicin/ml, total DNA was isolated from S. lividans transformants harboring pRTS020N (active sso1) or pRTS020O (inactive sso1) at the indicated times. Erythromycin (100 μg/ml) was added simultaneously to all the samples to further inhibit protein synthesis and thus another cycle of replication. The electrophoresed total DNA (1.6 μg) was detected by Southern hybridization as described in “Materials and methods”. Arrows indicate the positions of EcoRI-linearized ds plasmid and ss plasmid (circular or linear) molecules as marked. Rif rifampicin. a Southern blotting of denatured DNA. b Southern blotting without prior denaturation. The ss plasmid molecules are observed as two signals, probably indicating circular and linear forms, since the circular ss plasmid could be linearized during the DNA extraction process
sso-independent lagging strand synthesis using cell-free extracts of S. lividans
As shown above, sso1 exerts no effect on the copy number of pSN22 derivative in S. lividans, implying that lagging strand synthesis in S. lividans can be initiated without the sso sequence. The in vitro lagging strand synthesis using the cell-free extract of the plasmid-free strain S. lividans TK21 confirmed likewise that the rates and total amounts of incorporated [5-3H]dCTP on the ssDNA template were similar in the presence or absence of sso1 (Fig. 4). In addition, a similar [5-3H]dCTP incorporation rate was obtained for M13 mp18 ssDNA, which harbors no DNA fragment from pSN22 (data not shown). These results show that lagging strand synthesis is initiated on a random nucleotide sequence, but they do not explain the in vivo result that plasmid withoutsso1 accumulates in its ss form. The factor necessary for efficient lagging strand synthesis of the sso1 sequence is still unknown.

Time course of lagging strand synthesis using cell-free extract from exponential-phase cells of S. lividansTK21. The reaction mixture (300 μl) described in “Materials and methods“ was incubated under standard assay conditions with [5-3H]dCTP, and 12.5-μl aliquots were sampled, acid-precipitated, and assayed at different time intervals. Twelve μg ssDNA, pSSO021 (active sso1, open circles), or pSSO020 (inactive sso1, closed circles) were used as templates. Open triangles No ssDNA template was added to the reaction mixture
sso-independent lagging strand synthesis analyzed from cell-free extracts prepared from exponential-phase S. lividans cells showed relatively sharp bands consistent with DNA synthesis on ssDNA templates (Fig. 5a). To exclude any influence of sso, M13mp18 ssDNA was used as the template. Similar to in vivo experiments, the addition of 5 μg rifampicin/ml had no effect on the amount of ssDNA (Fig. 5a, lanes 2, 4), suggesting that initiation of lagging strand synthesis of the RC plasmid is independent of RNAP in S. lividans. Furthermore, the presence of all four NTPs is not a prerequisite for sso-independent lagging strand synthesis, as the process proceeded well even with ATP alone (Fig. 5a, lanes 3, 4). Moreover, it should be noted that the incorporation of labeled dCTP was observed in reactions without NTPs but with Mg2+ (Fig. 5a, lanes 5, 6, and Table 1). No incorporation of [α-32P]ATP was observed even with addition of all four NTPs when the cell-free extract from exponential-phase cells was used (data not shown). Likewise, rifampicin-resistant lagging strand synthesis was observed in the extract from stationary-phase cells (Fig. 5b, lane 1). However, in contrast to the extract prepapred from exponential-phase cells, the absence of ATP impeded lagging strand synthesis even when Mg2+ was added to the reaction mixture (Fig. 5b, lane 3, and Table 1). GTP could substitute for ATP (Fig. 5b, lane 2) or any other NTP (Table 1). These results indicate that lagging strand synthesis in cell-free extracts prepared from stationary-phase mycelium is dependent on at least one NTP. Lagging strand synthesis on a random nucleotide sequence was also confirmed using poly (dT) as a template, and Mg2+-dependent incorporation of [α-32P] dATP was observed (Table 2).

Effects of rNTPs, rifampicin, and Mg2+ on sso-independent lagging strand synthesis using cell-free extracts obtained from different growth phases of S. lividans. Cell-free extract was prepared from a exponential-phase or b stationary-phase mycelium. Products obtained from standard reaction mixtures (12.5 μl), containing alkaline-gel-purified M13mp18 ssDNA as template and [α-32P]dCTP, and incubated at 30°C for 30 min, were separated by agarose gel electrophoresis. [α-32P]dCTP incorporation was detected by autoradiography. The presence or absence of NTPs, rifampicin, and Mg2+ in the reactions is indicated. The components of the reaction mixture are as follows: NTPs 2 mM ATP and 500 μM each of CTP, GTP, and UTP; ATP 2 mM ATP only; GTP 2 mM GTP only; Rif 5 μg rifampicin/ml ; Mg 2+ 7.5 mM Mg(OAc)2. Arrows indicate the positions of circular ds and ss plasmid molecules as marked
The incorporation of dNTPs in exponential-phase cells was absolutely dependent on the presence of Mg2+ (Fig. 5, lane 6, and Table 1) at an optimal concentration of 7.5 mM (Fig. 6a). The incorporation of [5-3H]dCTP was optimal at pH 7.5 (Fig. 6b) and the maximum incorporation was at 0.5 μg template ssDNA (Fig. 6c) and 12.8 μg cell-free-extract proteins (2 μl; Fig. 6d).

Effects of a Mg2+, b pH, c amount of ssDNA template, and d protein concentration on in vitro lagging strand synthesis using S. lividans extracts. Standard mixtures (12.5 μl) contain Mg(OAc)2 at various concentrations, or HEPES–KOH buffer at various pH, or M13mp18 ssDNA or various amounts of cell-free extract prepared from exponential-phase cells. The mixtures were incubated at 30°C for 30 min
Comparison of in vitro lagging strand synthesis between S. lividans and E. coli extracts
Results of the in vitro study suggested the similarity of sso-independent lagging strand synthesis in S. lividans cell-free extract to the “general priming” reaction of E. coli. The general priming reaction is well characterized in E. coli ssDNA phages such as ϕX174 (Kornberg and Baker 1992), which are rifampicin-resistant, sso-independent, and require ATP to assemble the protein complex for primer synthesis (“primosome” )(Arai and Kornberg 1981b; Arai et al. 1981). In the absence of three NTPs but in the presence of ATP and four dNTPs, the primosome synthesizes deoxyribonucleotide primers under the condition of the general priming in vitro (Arai et al. 1981). The sso-independent lagging strand synthesis in S. lividans was compared with that in E. coli cell-free extract using alkaline-gel-purified M13mp18 ssDNA as a template. The sso of M13mp18 (minus-strand origin) is recognized by host-encoded RNAP in E. coli (Kaguni and Kornberg 1982). Table 3 shows that rifampicin decreased the incorporation of [5-3H]dCTP in the E. coli extract to 37%, which further decreased to 21% upon the elimination of GTP, UTP, and CTP. Conversely, the withdrawal of three NTPs did not change the level of [5-3H]dCTP incorporation by S. lividans extract from exponential-phase mycelium. The residual rifampicin-resistant and NTP-independent (ATP-dependent) activity of the E. coli extract, which was relatively lower than that of the S. lividans extract, corresponds to general priming. These results suggest that sso-independent lagging strand synthesis in S. lividans differs from general priming in terms of the NTP requirement for primer synthesis.
Discussion
Copy number control of pSN22
The maintained plasmid copy number could have induced segregational stability of pSN22 derivatives in S. lividans even without the sso, although there was an accumulation of ssDNA. Lagging strand synthesis of the RC plasmid is initiated by the host-encoding factor (Gruss and Ehrlich 1989), and it was shown that sso-independent primer synthesis in S. lividans is sufficiently efficient to maintain plasmid copy number. In RC replication, the ssDNA intermediate is produced from a ds plasmid molecule by Rep. In sso-dependent lagging strand synthesis, ssDNA is immediately converted to dsDNA; thus, there is no accumulation of ssDNA. If the plasmid lacks sso, ssDNAs accumulate since sso-independent primer synthesis is less efficient than sso-dependent primer synthesis. In contrast, the difference in the efficiency of primer synthesis in the presence or absence of sso did not affect the copy number of pSN22 derivatives, suggesting that the amount of ds plasmid molecules is limited whether the plasmid carries sso or not. Different from what was observed in pSN22, the deletion of sso was previously shown to decrease the plasmid copy number of pIJ101 derivatives to 50–100 copies from the expected 40–300 copies in S. lividans (Deng et al. 1988). It should be noted that the copy number of pSN22 is approximately 60 (unpublished data), which is lower than that of the pIJ101 derivatives without sso. These observations suggest that sso-independent primer synthesis in S. lividans is able to convert 50–100 copies of ssDNA to dsDNA, which is sufficient for maintaining the copy number of pSN22 derivatives, but not that of pIJ101 derivatives.
Rifampicin effect
Different from lagging strand synthesis in Staphylococcus aureus (Birch and Khan 1992; Boe et al. 1989; Dempsey et al. 1995) rifampicin failed to inhibit both sso-dependent and sso-independent lagging strand replications in S. lividans, though the strain was found to be rifampicin-sensitive and most probably have a rifampicin-sensitive RNAP. The only known rifampicin-resistant lagging strand replication in gram-positive bacteria occurs in pWVO1 in Lactococcus lactis, in which the activation of the sso of the plasmid has been suggested to involve two different routes; RNAP-dependent and RNAP-independent (Leenhouts et al. 1991; Seegers et al. 1995) routes. If RNAP is required for the preferential lagging strand replication at sso, rifampicin should have inhibited the lagging strand replication of the pSN22 derivative containing sso leading to ssDNA accumulation. However, our results suggest otherwise, as RNAP is not required for both in vivo sso-dependent and sso-independent lagging strand syntheses, suggesting the plausible existence of a dominant RNAP independent priming system in S. lividans.
Lagging strand replication in vitro
While sso hampered the accumulation of ss plasmid molecules in the cells during in vivo lagging strand synthesis (Fig. 3), there was no difference in complementary strand synthesis in the presence or absence of sso during in viitro synthesis (Fig. 4). These results indicate that the cell-free extract might have been deficient in factors that drive in vivo sso-dependent lagging strand synthesis. In E. coli, a single-stranded DNA binding protein (SSB) is involved in recognition of the signal sequence for primer synthesis of the RC-replicating bacteriophage (Arai and Kornberg 1979). E. coli SSB (Stratagene) was added to the reaction mixture of in vitro lagging strand synthesis using S. lividans extract, but there was no significant effect on sso recognition (data not shown). Further studies are required to characterize the factors initiating lagging strand synthesis at the sso of RC plasmids in S. lividans.
Figure 5 shows the in vitro replication product of lagging strand synthesis in the exponential and stationary phases of S. lividans. Although M13mp18 RF has two ClaI sites, digestion by the restriction enzyme failed to produce the expected fragments (data not shown), indicating that the products were neither initiated at a specific site nor completely replicated. It should be noted that M13mp18 cloned into pSN22 derivatives in different orientations all showed ssDNA accumulation, and consequently, no sso activity in S. lividans in vivo (unpublished data). Lagging strand synthesis on a random nucleotide sequence was also confirmed using poly(dT) as template (Table 2). In addition, eliminating other dNTPs in the reaction induced no dCTP incorporation (Table 1), implying that dCTP incorporation is independent of nonspecific dNTP incorporation into ssDNA such as mediated by terminal transferase activity. Thus, the molecular structure of the products is partly assumed to be that of dsDNA consisting of short DNA fragments (primers) on template ssDNA. Further studies are required to determine the precise molecular structure of the in vitro replication products.
As priming activity in the extract was resistant to rifampicin and all four dNTPs, and only one NTP was sufficient to carry out lagging strand synthesis in vitro, RNAP is not involved in lagging strand initiation of RC plasmids in S. lividans. It is therefore possible that in vitro priming synthesis in S. lividans is similar to that in E. coli primase. E. coli primase and DnaB require ATP and all four dNTPs for the general priming system in vitro (Arai and Kornberg 1981b), and ATP is required for the assembly of the primase complex onto template ssDNA (Arai and Kornberg 1981a). Similarly, ATP or any one of the NTPs, but not rNTPs, was required for efficient, Mg2+-dependent lagging strand synthesis in cell-free extract from stationary-phase cells. However, lagging strand synthesis differs from the “general priming” synthesis in cell-free extract from exponential-phase cells, where ATP was not an absolute requirement. If the primase complex present in E. coli is involved in lagging strand synthesis in S. lividans, the complex could have been activated during exponential growth. In addition, a similar RNAP-independent lagging strand replication was reported in B. subtilis, where synthesis was attributed to the presence of primosome assembly involving DnaE (Bruand et al. 1995). It appears then that the sso-independent, rifampicin-resistant lagging strand replication mechanism in S. lividans is not unique but is found in a wide variety of gram-positive bacteria.
The specific requirement of ATP or any one of the NTPs for efficient lagging strand synthesis in S. lividans, particularly in cell-free extracts prepared from stationary-phase mycelium, suggests a role for ATP as an activator during such synthesis. The difference in NTP requirement between exponential-phase and stationary-phase extracts was not due to contamination by endogenous NTPs because lagging strand synthesis was also observed without NTPs in further purified fractions of the exponential-phase extract using three steps of liquid chromatography, i.e. gel filtration, Q-Sepharose, and heparin (data not shown). The difference in the efficacy of ATP stimulation between exponential-phase and stationary-phase mycelia is indicative of the difference in the components of the replicating pool at each phase, that is, ATP stimulation is dependent on the growth phase of S. lividans. Signal transduction, including the protein phosphorylation system, regulating aerial mycelial development and secondary metabolism in Streptomyces (Beppu 1995) may contribute to DNA replication in Streptomyces.
References
Arai K, Kornberg A (1979) A general priming system employing only dnaB protein and primase for DNA replication. Proc Natl Acad Sci USA 76:4308–4312
Arai K, Kornberg A (1981a) Mechanism of dnaB protein action. III. Allosteric role of ATP in the alteration of DNA structure by dnaB protein in priming replication. J Biol Chem 256:5260–5266
Arai K, Kornberg A (1981b) Mechanism of dnaB protein action. IV. General priming of DNA replication by dnaB protein and primase compared with RNA polymerase. J Biol Chem 256:5267–5272
Arai K, Low RL, Kornberg A (1981) Movement and site selection for priming by the primosome in phage ϕX174 DNA replication. Proc Natl Acad Sci USA 78:707–711
Beppu T (1995) Signal transduction and secondary metabolism: prospects for controlling productivity. Trends Biotechnol 13:264–269
Birch P, Khan SA (1992) Replication of single-stranded plasmid pT181 DNA in vitro. Proc Natl Acad Sci USA 89:290–294
Boe L, Gros MF, te Riele H, Ehrlich SD, Gruss A (1989) Replication origins of single-stranded-DNA plasmid pUB110. J Bacteriol 171:3366–3372
Bruand C, Ehrlich SD, Jannière L (1995) Primosome assembly site in Bacillus subtilis. EMBO J 11:2642–2650
Conrad SE, Campbell JL (1979) Characterization of an improved in vitro DNA replication system for Escherichia coli plasmids. Nucl Acids Res 6:3289–3303
Dempsey LA, Zhao AC, Khan SA (1995) Localization of the start sites of lagging-strand replication of rolling-circle plasmids from gram-positive bacteria. Mol Microbiol 15:679–687
Deng ZT, Kieser T, Hopwood DA (1988) “Strong incompatibility” between derivatives of the Streptomyces multi-copy plasmid pIJ101. Mol Gen Genet 214:286–294
Fuller RS, Kaguni JM, Kornberg A (1981) Enzymatic replication of the origin of the Escherichia coli chromosome. Proc Natl Acad Sci USA 78:7370–7374
Gruss A, Ehrlich SD (1989) The family of highly interrelated single-stranded deoxyribonucleic acid plasmids. Microbiol Rev 53:231–241
Gruss A, Ross HF, Novick RP (1987) Functional analysis of a palindromic sequence required for normal replication of several staphylococcal plasmids. Proc Natl Acad Sci USA 84:2165–2169
Hagège J, Pernodet JL, Friedmann A, Guérineau M (1993) Mode and origin of replication of pSAM2, a conjugative integrating element of Streptomyces ambofaciens. Mol Microbiol 10:799–812
Hopwood DA, Bibb MJ, Chater KF, Kieser T, Bruton CJ, Kieser HM, Lydiate DJ, Smith CP, Ward JM, Schrempf H (1985) Genetic manipulation of Streptomyces: a laboratory manual. The John Innes Foundation, Norwich, UK
Inuzuka M, Helinski DR (1978) Replication of antibiotic resistance plasmid R6K DNA in vitro. Biochemistry 17:2567–2573
Jannière L, Gruss A, Ehrlich SD (1993) Plasmids. In: Sonenshein AL, Losick R, Hoch JA (eds) Bacillus subtilis and other gram positive bacteria: biochemistry, physiology, and molecular genetics. American Society for Microbiology, Washington DC, pp 625–644
Kaguni JM, Kornberg A (1982) The ρ subunit of RNA polymerase holoenzyme confers specificity in priming M13 viral DNA replication. J Biol Chem 257: 5437–5443
Kataoka M, Seki T, Yoshida, T (1991) Five genes involved in self-transmission of pSN22, a Streptomyces plasmid. J Bacteriol 173:4220–4228
Kataoka M, Kiyose YM, Michisuji Y, Horiguchi T, Seki T, Yoshida T (1994a) Complete nucleotide sequence of the Streptomyces plasmid, pSN22; genetic organization and correlation with genetic properties. Plasmid 32:55–69
Kataoka M, Kuno N, Horiguchi T, Seki T, Yoshida T (1994b) Replication of the Streptomyces plasmid pSN22 through single-stranded intermediates. Mol Gen Genet 242:130–136
Katz E, Thompson CJ, Hopwood, DA (1983) Cloning and expression of the tyrosinase gene from Streptomyces antibioticus in Streptomyces lividans. J Gen Microbiol 129:2703–2714
Khan SA (1997) Rolling-circle replication of bacterial plasmids. Microbiol Mol Biol Rev 61:442–455
Kieser T, Hopwood DA, Wright HM, Thompson CJ (1982) pIJ101, a multi-copy broad host-range Streptomyces plasmid: functional analysis and development of DNA cloning vectors. Mol Gen Genet 185:223–238
Kornberg A, Baker TA (1992) DNA replication, 2nd edn. WH Freeman & Co, New York
Kramer MG, Khan SA, Espinosa M (1997) Plasmid rolling circle replication: identification of the RNA polymerase-directed primer RNA and requirement for DNA polymerase I for lagging strand synthesis. EMBO J. 16:5784–5795
Kramer MG, Espinosa M, Misra TK, Khan SA (1998) Lagging strand replication of rolling-circle plasmids: specific recognition of the ssoA-type origins in different gram-positive bacteria. Proc Natl Acad Sci USA 95:10505–10510
Leenhouts KJ, Tolner B, Bron S, Kok J, Venema G, Seegers JFML (1991) Nucleotide sequence and characterization of the broad-host-range lactococcal plasmid pWVO1. Plasmid 26:55–66
Muth G, Farr M, Hartmann V, Wohlleben W (1995) Streptomyces ghanaensis plasmid pSG5: nucleotide sequence analysis of the self-transmissible minimal replicon and characterization of the replication mode. Plasmid 33:113–126
Novick RP (1989) Staphylococcal plasmids and their replication. Annu Rev Microbiol 43:537–565
Pigac J, Vujaklija D, Toman Z, Gamulin V, Schrempf H (1988) Structural instability of a bifunctional plasmid pZG1 and single-stranded DNA formation in Streptomyces. Plasmid 19:222–230
te Riele H, Michel B, Ehrlich SD (1986a) Single-stranded plasmid DNA in Bacillus subtilis and StaphylococcuStaphylococcus aureus. Proc Natl Acad Sci USA 83:2541–2545
te Riele, H, Michel B, Ehrlich SD (1986b) Are single-stranded circular intermediates involved in plasmid DNA replication? EMBO J 5:631–637
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory, Cold Spring Harbor, New York
Seegers JFML, Zhao AC, Meijer WJJ, Khan SA, Venema G, Bron S (1995) Structural and functional analysis of the single-strand origin of replication from the lactococcal plasmid pWVO1. Mol Gen Genet 249:43–50
Servín-González L, Sampieri AIII, Cabello J, Galván L, Juárez V, Castro C (1995) Sequence and functional analysis of the Streptomyces phaeochromogenes plasmid pJV1 reveals a modular organization of Streptomyces plasmid that replicate by rolling circle. Microbiol 141:2499–2510
del Solar GH, Moscoso M, Espinosa M. (1993) Rolling-circle replicating plasmids from gram-positive and gram-negative bacteria: a wall falls. Mol Microbiol 8:789–796
Southern EM (1975) Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol 98:503–517
Studier FW, Moffatt BA (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol 189:113–130
Suzuki I, Kataoka M, Seki T, Yoshida T (1997a) Three single strand origins located on both strands of the Streptomyces rolling circle plasmid pSN22. Plasmid 37:51–64
Suzuki I, Seki T, Yoshida T (1997b) Nucleotide sequence of a nicking site of the Streptomyces plasmid pSN22 replicating by the rolling circle mechanism. FEMS Microbiol Lett 150:283–288
Thompson J, Cundliffe E (1981) Purification and properties of an RNA methylase produced by Streptomyces azureus and involved in resistance to thiostrepton. J Gen Microbiol 124:291–297
Thompson CJ, Ward JM, Hopwood DA (1982) Cloning of antibiotic resistance and nutritional genes in Streptomycetes. J Bacteriol 151:668–677
Thompson J, Rae S, Cundliffe E (1984) Coupled transcription-translation in extracts of Streptomyces lividans. Mol Gen Genet 195:39–43
Yanisch-Perron C, Vieira J, Messing J (1985) Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119
Yokoyama E, Doi K, Kimura M, Ogata S (1996) Detection of the single-stranded DNA of Streptomyces plasmid pSA1.1 and a binding histone-like protein. FEMS Microbiol Lett 138:197–200
Zaman S, Radnedge L, Richards H, Ward, JM (1993) Analysis of the site for second-strand initiation during replication of the Streptomyces plasmid pIJ101. J Gen Microbiol 139:669–676
Acknowledgments
We greatly thank to Drs. T. Kieser, H. Araki and T. Ito for valuable discussions; Dr E. Ko-Mitamura for editorial suggestions and manuscript corrections; and Dr H. Masai for valuable comments on the general priming of E. coli.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Suzuki, I., Kataoka, M., Yoshida, T. et al. Lagging strand replication of rolling-circle plasmids in Streptomyces lividans: an RNA polymerase-independent primer synthesis. Arch Microbiol 181, 305–313 (2004). https://doi.org/10.1007/s00203-004-0656-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-004-0656-6