
Abstract
One of the most serious healthcare problems in the world is bone loss and fractures due to a lack of physical activity in elderly people as well as in bedridden patients or otherwise inactive youth. Crucial here are the osteocytes. Buried within our bones, these cells are believed to be the mechanosensors that stimulate bone formation in the presence of mechanical stimuli and bone resorption in the absence of such stimuli. Intercellular signaling is an important physiological phenomenon involved in maintaining homeostasis in all tissues. In bone, intercellular communication via chemical signals like NO plays a critical role in the dynamic process of bone remodeling. If bones are mechanically loaded, fluid flows through minute channels in the bone matrix, resulting in shear stress on the cell membrane that activates the osteocyte. Activated osteocytes produce signaling molecules like NO, which modulate the activity of the bone-forming osteoblasts and the bone-resorbing osteoclasts, thereby orchestrating bone adaptation to mechanical loading. In this review, we highlight current insights in the role of NO in the mechanical adaptation of bone mass and structure, with emphasis on its role in local bone gain and loss as well as in remodeling supervised by osteocytes. Since mechanical stimuli and NO production enhance bone strength and fracture resistance, these new insights may facilitate the development of novel osteoporosis treatments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
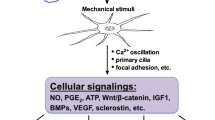
Bone is a dynamic tissue that constantly renews and adapts to its local loading environment. Mechanical loading results in changes in bone size and structure that strengthen the bone. The mechanical adaptation of bone starts with mechanotransduction, the process wherein resident bone cells perceive a mechanical stimulus and translate it into a biochemical response. This involves pathways within the cell membrane and cytoskeleton of the osteocytes, the professional mechanosensor cells of bone. During the last decade, we have come to understand the crucial role of the lacuno-canalicular porosity as the structure that mediates osteocyte mechanosensing. Strain-derived fluid flow of interstitial fluid through the lacuno-canalicular porosity seems to mechanically activate the osteocytes as well as ensures transport of signaling molecules, nutrients, and waste products. Mechanotransduction is followed by the signaling from the sensor cells (osteocytes) to the effector cells (osteoblasts or osteoclasts), resulting in bone formation or resorption. One factor that may be crucial in this signaling is nitric oxide (NO).
NO plays a critical role in the neuronal system, during inflammation, in blood pressure control, and bone mass regulation [1–5]. The importance of NO in skeletal tissue has been known for more than a decade [3, 6–10]. NO is a water-soluble, gaseous, short-lived radical molecule, that by its nature is both able to easily diffuse and highly reactive. It moves freely across cell membranes. Its broad ranging actions are determined largely by the site, the rate, and the quantity of NO generated, and by the nature of the environment into which it is released. NO is an important regulator of intracellular and intercellular homeostatic processes. NO is synthesized by NO synthase (NOS). It is produced by the oxidation and cleavage of one of the terminal nitrogen atoms of the amino acid l-arginine. The reaction is dependent on electrons donated by the cofactor NADPH and requires oxygen, yielding l-citrulline as a coproduct. NO can be generated independently from NOS as well. NO can be produced by reduction of nitrite, which can occur spontaneously under hypoxic and/or acidic conditions, such as may pertain in the osteocyte lacuna. Enzymes such as xanthine oxidase and cytochrome oxidase-c can also mediate reduction of nitrite [11, 12]. NO acts frequently by S-nitrosylation of specific proteins, a reaction that has been shown to be reversible through the action of denitrosylases [13], thereby releasing NO.
Three different NOS isoforms known are encoded by three distinct genes. Neuronal NOS (nNOS) and endothelial NOS (eNOS) are calcium-dependent constitutively expressed enzymes, involved in physiological processes as regulation of blood pressure. They are characterized by highly regulated, rapid, but low-output NO production that has a tonic physiological function [14]. Inducible NOS (iNOS) is a calcium-independent inducible enzyme that is upregulated and activated during inflammatory processes and is responsible for the production of very high amounts of NO. In contrast to rapid activation and deactivation of eNOS and nNOS, iNOS expression is induced over several hours, requiring transcription of mRNA and de novo protein synthesis. Once activated, iNOS is capable of generating sustained high levels of NO locally over many hours [14]. In these circumstances, NO contributes to localized cell and tissue damage [14]. The tissue specificity of the different NOS isotypes originally suggested by the NOS nomenclature is not absolute, and the constitutive NOS enzymes have since been found to have a much wider tissue distribution [3]. All three NOS isoforms, iNOS, eNOS, and nNOS, are expressed in bone tissue [3, 6–9]. A role for NO in bone metabolism has been confirmed in intact animals with single gene deletions of NOS isoforms [15–18]. Targeted deletion of the eNOS isoform in mice leads to an osteoblast-driven mild osteoporotic bone phenotype with female mice showing a blunted response to estrogen, suggesting that the anabolic effect of estrogen is mediated, in part, by eNOS [15–18]. Studies in iNOS knockout mice have illustrated that iNOS-derived NO activates osteoclasts in inflammatory bone disease [15] and plays a role in the catabolic response of bone to lowered estrogen levels [19]. iNOS-derived NO also stimulates fracture healing as well as recovery of bone mass after unloading-induced bone loss in the mouse tail suspension model [20]. Interestingly, iNOS null mice do not show a bone phenotype unless challenged by mechanical or inflammatory factors [15]. Nowadays, it has become clear that nNOS null mice do not have a low bone mass but rather a high bone mass phenotype [18]. The triple eNOS, iNOS, and nNOS null mouse shows a phenotype similar to that of the single nNOS knockout with a sex-dependent, low turnover, high bone mass phenotype [21]. The effect of nNOS in vivo may largely be indirect, i.e., it is mediated via the central nervous system.
The importance of NO for the skeleton is demonstrated by the use of NO donors such as nitroglycerine and nitrates, which have been used as therapeutic agents for the past century. When the body cannot generate adequate amounts of NO for its biological homeostasis such as in osteoporosis patients, administration of exogenous NO is a practical method of supplementation. NO has an estrogen-like effect and can in part replace the beneficial effect of estrogen in bone [16, 17]. Also, postmenopausal NO deficiency is rectified by hormone replacement therapy, enhancing local NO production [22]. This has important therapeutic implications, i.e., NO donor therapy is a most attractive novel therapy for the prevention and treatment of osteoporosis in men and women.
Osteocyte mechanosensation
Increased mechanical loading results in bone mass gain and increased mineral density in vivo, while unloading decreases bone formation, reduces bone mineral content, and reduces bone matrix protein production [23]. Since bone is so stiff, the direct strains that osteocytes receive from bone loading are very small. This has emphasized the role of osteocytes as the “professional” mechanosensor cells of bone and the lacuno-canalicular porosity as the structure that mediates mechanosensing [24]. The osteocytes likely sense the strain-derived flow of interstitial fluid through this porosity (Fig. 1) [25, 26]. Increased loading as well as disuse and overuse produce abnormal canalicular flow, which can be translated into different production of signaling molecules such as NO (Fig. 2). Osteocytes might also sense hydrostatic pressure. Recently, a computational study has predicted that the hydrostatic pressure in osteocyte lacunae would be sufficient to activate osteocytes in vivo when compared with hydrostatic pressures needed in vitro [27].

Mechanical loading of bone results in deformation of load-bearing bone matrix and interstitial fluid flow around osteocytes

Osteocytes in situ respond to mechanical stimulation by upregulation of NO production. Fluorescence of the chromophore DAR-4M AM [45] shows the NO response of osteocytes in a mechanically loaded mouse fibula. a Control fibula and b mechanically loaded fibula
The rate of the applied loading stimulus correlates to bone formation rather than the magnitude of strain. Low magnitude (<10 με), high frequency (10–100 Hz) loading can stimulate bone growth and inhibit disuse osteoporosis [28]. Notably high-magnitude, low-frequency stimuli are quite rare in the activities of daily life, whereas high-frequency, low-magnitude stimuli are quite common [28–30]. High rates of loading also increase bone mass and strength after jumping exercises in middle-aged osteopenic ovariectomized rats [31]. The rate of loading seems to be a decisive factor in bone formation and maintenance. Bacabac et al. [32] found a linear correlation between increased amounts of NO released by bone cells and the rate of fluid shear stress. Since NO mediates bone formation in vivo [33], this supports the concept that bone formation in vivo is stimulated by dynamic rather than static loads [34] and that low-magnitude, high-frequency mechanical stimuli may be as stimulatory as high-amplitude, low-frequency stimuli. This rate-dependent response was found to occur, provided that the cells are “kicked” in a preconditioned state [35]. The finding that the bone cell’s response to fluid shear stress is rate dependent provides an explanation why adaptive bone formation can occur despite the sporadic occurrence of high-amplitude strains in daily life [29].
The excitation mechanism of osteocytes might be due to a unique strain amplification upon fluid flow that results from the interaction of the pericellular matrix and the cell process cytoskeleton [36, 37]. This also provides an explanation for sustained bone formation despite the sporadic occurrence of high-amplitude strains in normal physiological loading conditions. Studies on the osteogenic activity of bone cells investigated the effects of stress using varying techniques, such as fluid flow, substrate strain, hydrostatic pressure, and vibration stress [38]. The magnitude and rate of stress were shown to correlate with the cellular response, which was likely cell deformation-dependent. Relating the effects of fluid flow and substrate straining have shown that the former induces higher release of signaling molecules [39]. McGarry et al. [39] reported large differences in NO release by primary human bone cells (obtained as outgrowing cells from small bone fragments) stimulated with 0.6 Pa fluid shear stress and 1,000 με substrate strain due to the differences in cellular deformation that both stimuli cause. The higher NO response due to fluid shear stress could be due to higher membrane stress because it has been suggested that mechanically induced formation of NO results from activation of eNOS in bone cells [6, 8, 40]. eNOS is an enzyme bound to the plasma membrane that may be rendered susceptible to activation by increased stressing of the cell membrane. Osteocytes respond to an increase in fluid flow rate in vitro with a linear increase in cell strain as well as intracellular NO and calcium concentrations [41]. eNOS as well as nNOS are dependent on calcium for their activity. It has been shown that mechanical loading of osteocytes in vitro leads to NO production in a calcium-dependent manner [42]. A possible mechanism for NO release after mechanical loading is that membrane stress opens stretch-activated ion channels, resulting in an increase in intercellular calcium permitting NO release. The cytoskeleton is crucial to the working of stretch-activated calcium channels. The cytoskeleton, which can be considered a composite gel-like material, is the scaffold determining cellular shape and stiffness [43].
The cytoskeleton plays a crucial role in a multitude of cellular processes, i.e., migration, differentiation, mechanosensing, and even cell death, and largely determines the stiffness of the cell. McGarry et al. [44] have shown that the NO response of osteocytes to mechanical loading by pulsating fluid flow is dependent on the actin cytoskeleton but not the microtubules. From the field of physics, it is known that the effects of stresses applied at different rates at an object are largely determined by its material properties. For bone cells, it was shown that the production of signaling molecules such as NO in response to an in vitro fluid shear stress (at 5 and 9 Hz) and vibration stress (at 5–100 Hz) correlated with the applied stress rate [32]. The faster the stress was applied, the stronger the observed response of the cells [34]. This suggests that bone cellular response to loading and mechanical properties of the cell are related, which implies that the response of bone cells to loading is related to cytoskeletal properties.
Bacabac and colleagues [38] developed a novel application of two-particle microrheology, for which they devised a 3D in vitro system, using optical traps to quantify cell traction on attached fibronectin-coated probes. Using the optical trap device, cells loaded with an NO-sensitive fluorescent dye [45] were mechanically stimulated, and the NO response of the cells to forces applied on the cell using the attached probes was studied. Osteocytes under round-suspended morphology required lower force stimulation in order to show an increase in NO production, even though they were an order-of-magnitude more elastic compared to flat-adherent cells [46]. Apparently elastic osteocytes seem to require less mechanical forces in order to respond than stiffer cells. Differences in mechanosensitivity between cells might not be directly related to the elasticity of the cell but might be more related to other cell-specific properties, i.e., the presence of receptors or ion channels in the membrane.
Simultaneous with the increased NO release in response to mechanical stimulation, osteocytes showed increased force traction on the attached beads. In other words, the cells started to “pull harder” on the beads and generated a force up to nearly 30 pN, which interestingly is within the order of forces necessary for activating integrins. Whether there was a causal link between loading-induced NO production by the cells and force generation is currently unknown. Since force generation and cell elasticity are (indirectly) related, these results might indicate that osteocytes adapt their elasticity in response to a mechanical stimulus. Indeed, experiments with an atomic force microscope and optical tweezers have shown that osteocytes become “stiffer” after mechanical loading [38]. This stiffening response was related to actual changes in material properties of the cell, suggesting that the cells actively change their cytoskeleton in response to a mechanical load.
Taken together, mechanical stimuli induce NO signaling via strain-induced changes in fluid flow that affect mechanosensory molecules on the osteocyte, which are connected to the cytoskeleton. The activation of mechanosensory molecules sets in motion a cascade of events that result in NO production. Such signals are communicated through the network to the bone cells on the surface (bone lining cells, osteoblasts, and osteoclasts), which allows the adaptation of bone mass and structure to its environment. We will now first discuss how NO is produced by osteocytes in response to mechanical loading, and then we will discuss which other signaling molecules are produced by osteocytes in response to mechanical loading, and how NO is related to the production of these molecules.
NOS
The importance of NOS activity for the regulation of bone mass in response to mechanical stimuli is demonstrated by the inhibition of NO production with pharmacological agents in organ cultures or in intact animals, which inhibits the anabolic response of bone to mechanical stimulation [33, 47]. Which NOS isoform is specifically important for mediating the NO response to mechanical loading in osteoblasts and osteocytes remains to be decided. Because in vitro fluid flow stimulation of osteoblasts and osteocytes results in rapid NO production [48, 49], similar to the NO response in endothelial cells [4], attention has originally focused on eNOS. There are strong analogies between the modes of activation of endothelial cells and osteocytes to produce NO after mechanical stimulation. Fluid shear stress produces drag on the cell membrane of endothelial cells, which leads to eNOS activation via a series of phosphorylation and dephosphorylation events, largely within the caveoli in the plasma membrane where eNOS resides [50]. In bone, a similar drag of interstitial fluid over the processes of osteocytes has been proposed [48], and it seemed logical to assume that eNOS was responsible for increased NO production after mechanical stimulation. Additional weight to this argument was provided by the fact that eNOS null mice are mildly osteoporotic and show a blunted response to estrogen, leading to the view that NO acts as an anabolic factor in bone [16, 17]. However, recent work has questioned the involvement of eNOS in osteocyte/osteoblast-derived NO production in response to mechanical stimulation in vitro [51, 52], while others have reported that the low bone mass phenotype in eNOS−/− mice is only transient. This reopens the discussion on the importance of eNOS for bone cell autonomous NO production. Fluid shear stress leads to S-nitrosylation of at least 100 proteins in endothelial cells [53], raising the possibility that redox regulation via S-nitrosylation and denitrosylation may also be important in mechanically challenged bone cells.
NO signaling
NO signaling activates various signal transduction pathways in different cell types. However, there is a paucity of information of the detailed signaling mechanisms of NO in bone cells. The most significant biologically relevant targets of NO have been shown to be metalloproteins (i.e., iron heme proteins), thiol proteins, and free radicals, with the primary target of NO being soluble guanylate cyclase via formation of a ferrous–NO heme protein. The guanylate cyclase–cGMP pathway is a classical target for NO, and cGMP is well known to be crucial to normal bone formation [22]. The signaling function of NO starts with its interaction with guanylate cyclase on the cell membrane or following diffusion into cells. The binding triggers an allosteric change in the protein that in turn triggers formation of a second messenger within the cell. The most common protein target for NO is guanylate cyclase that generates the second messenger cGMP [54]. Type II cGMP-dependent protein kinase has been shown to mediate osteoblast mechanotransduction [55]. Due to its reactive nature, NO readily reacts with cGMP in an autocrine manner.
NO plays both an autocrine and a paracrine role in bone cell metabolism. It modulates the activity of both osteoblasts and osteoclasts in vitro. Dependent on the concentration, NO has an anabolic effect on bone tissue and stimulates osteoblast-induced mineralization in vitro [22]. The release of large amounts of NO by cytokine-stimulated cells inhibits osteoblast proliferation [16] and increases osteoblast apoptosis as well as osteoclast-mediated bone resorption [56, 57]. Thus NO has a biphasic effect on osteoblasts: low NO concentrations stimulate, while high concentrations inhibit bone formation [58]. In mature osteoclasts, NO both stimulates and inhibits activity: a bidirectional effect [58] related to changes in the local concentration of NO. Exposure of mature osteoclasts to NO stimulates contraction and causes the cells to become detached from the underlying bone surface [59].
NO and growth factor release
NO might play a role in hepatocyte growth factor (HGF)-mediated bone remodeling. Juffer and colleagues [60] found that NO modulates HGF protein secretion by osteocytes in response to mechanical loading in vitro. HGF plays a role in bone formation, as it stimulates osteoblast differentiation and inhibits mineralization [61]. HGF is also involved in bone resorption, as it stimulates osteoclast formation [62].
NO might also play a role in vascular endothelial growth factor (VEGF)-mediated angiogenesis during bone remodeling. Recently, it has been reported that mechanical loading stimulates VEGF mRNA and protein release in osteocytes in vitro [60, 63]. VEGF expression is also upregulated in mechanically loaded tibia of ovariectomized mice [64]. Inhibition of NO synthesis decreases mechanical loading-increased VEGF expression, suggesting that VEGF expression by osteocytes in response to mechanical loading is modulated by NO [60]. During bone remodeling, resorption and formation of the bone matrix are generally accompanied by angiogenesis in the cutting cone of the osteon. Since bone remodeling is induced mechanically and since mechanical loading stimulates NO production resulting in upregulation of VEGF mRNA and protein in osteocytes, this might implicate that the NO-induced VEGF production contributes to the vascularization within the cutting cone of the osteon. VEGF not only stimulates blood vessel formation but also increases bone formation by stimulating osteoblastogenesis [65]. Therefore, the mechanical loading-induced VEGF production in osteocytes is likely not only contributing to vascularization within the bone matrix but is also involved in mechanically induced bone remodeling.
Wnts are involved in bone mechanical adaptation. Santos et al. [66] has shown that osteocytes respond to mechanical stimulation by fluid shear stress in vitro by modulating expression of Wnts and activation of Wnt responsive genes through activation of the canonical Wnt signaling pathway (Fig. 3). NO is important for activation of the canonical Wnt signaling pathway in osteocytes [66]. Mechanical loading results in increased NO production as well as in activation of focal adhesion kinase (FAK) and the Akt signaling pathway, which results in β-catenin stabilization, followed by translocation of β-catenin to the nucleus and expression of β-catenin target genes. After induction of Wnt production by mechanical loading, the Wnt signal is propagated, resulting in reactivation of the Wnt/β-catenin signaling pathway. Wnts and the Wnt/β-catenin signaling pathway are key elements for the adaptive response of bone to mechanical loading [66–68]. Wnts not only regulate osteoblast maturation and activity [67, 69] but also control osteoblast and osteocyte apoptosis [70] and suppress the osteoclastogenesis-stimulating factor RANKL [71]. Cellular levels of the Wnt inhibitor sclerostin, a protein product of the SOST gene and a mature osteocyte-specific marker [72], are reduced in osteocytes after mechanical stimulation of bone in vivo [73]. Sclerostin regulates osteoclast activity by promoting osteoblast-mediated inhibition of osteoclast differentiation via increased osteoprotegerin (OPG), a decoy receptor of RANKL [73]. Thus mechanically induced NO release followed by Wnt production and regulation of the Wnt antagonist sclerostin seem to be involved in bone remodeling.

Mechanical loading results in increased production of NO as well as in activation of the Wnt/β-catenin pathway via a concerted mechanism in osteocytes. Mechanical loading activates focal adhesion kinase (FAK) and the Akt signaling pathway, which results in β-catenin stabilization, followed by translocation of β-catenin to the nucleus and expression of β-catenin target genes. After induction of Wnt production by mechanical loading, the Wnt signal is propagated resulting in reactivation of the Wnt/β-catenin signaling pathway
Mechanical adaptation
Mechanical adaptation is facilitated by the continuous remodeling of bone, which is an integral part of the protection of bone tissue against fatigue fractures. The microscopic damage that occurs due to tissue fatigue is assumed to be the actual signal for activation of the process of bone remodeling [74]. Fatigue damage produces osteocyte apoptosis and RANKL production, which attracts osteoclasts, thereby activating bone remodeling [74–77]. After activation, bone remodeling involves groups of osteoblasts and osteoclasts, which collaborate in so-called basic multicellular units (BMUs). A “cutting cone” of osteoclasts excavates a tunnel in compact bone or a trench along the surface of trabeculae, followed by “closing cone” of osteoblasts that refills the tunnel or trench [78]. The osteoblasts do not completely fill the tunnel, but a space is left in the middle for blood vessels, providing the osteocytes with nutrients and oxygen. Many osteoblasts are buried during bone formation, thereby becoming osteocytes. The entombed cells assume a stellate shape, with cell bodies positioned in lacunae in the matrix, from which slender cell processes (about 60 per cell) radiate in all directions. The cell processes pass through the bone matrix via small canals, the canaliculi, to keep in contact with other osteocytes and the cells on the surface. Osteocytes also extend their cell processes into bone marrow spaces where osteoclast precursors are present [79]. This network allows rapid transmission of biochemical signals.
Burger and colleagues have developed a theory that links patterns of strain and strain-derived fluid flow around a BMU to the coordinated activity of osteoblasts and osteoclasts [80, 82]. Volumetric strain in the bone around a BMU cutting cone has been related to canalicular fluid flow [82] and with low canalicular flow in front of the cutting cone and high flow at the sides, the so-called “reversal zone.” The low flow in front of the cutting cone was proposed to induce local osteocyte apoptosis. Mechanical loading by fluid shear stress has been shown to promote osteocyte survival, while unloading is associated with osteocyte apoptosis [83, 84]. NO production by osteocytes plays a major role in preventing apoptosis [85, 86]. Osteocyte apoptosis at the tip of the cutting cone would attract osteoclasts, leading to further excavation of bone in the direction of the loading [87]. Alternatively, the low flow at the tip of the cutting cone could result in RANKL production by osteocytes, thereby actively attracting osteoclasts [88, 89]. It has been shown by Tatsumi et al. [90] that osteocytes are essential for inducing bone loss after unloading. Recently, it has been shown that osteocytes regulate bone mass by producing RANKL in vivo [87, 88]. Osteocytes support osteoclastogenesis under normal (unloaded) culture conditions, but not after being subjected to mechanical load [91–93]. The model by Smit and colleagues [82] further predicts that at the sides of the cutting cone and further down the reversal zone, osteocytes receive enhanced fluid shear stress during loading. This could prevent osteocyte apoptosis but may also stimulate the release of signaling molecules that promote the retraction and detachment of osteoclasts from the bone surface. NO has been proposed as a likely candidate [81, 93]. It is released by osteocytes after fluid flow stimulation [32] and causes osteoclasts to detach form the bone surface [94]. The two mechanisms, attraction of osteoclasts to the cutting cone tip and induction of osteoclast detachment from the cutting cone base, together could explain the mechanically meaningful behavior of osteoclasts during remodeling (Fig. 4).

Drawing of the postulated events in the cutting cone of a basic multicellular unit (BMU). Osteocyte apoptosis (indicated with black X) is caused by lack of fluid flow at the tip of the cutting cone, where the matrix strains are relatively low during normal loading. Multinucleated osteoclasts are attracted by apoptotic and RANKL producing osteocytes, and as a result, the cutting cone follows the loading direction. Osteoclasts are repelled at the sides of the cutting cone as a result of high amounts of NO produced by well-stressed osteocytes. As NO production remains high further down the reversal zone, osteoclasts remain within the cutting cone and may even reenter the resorption cycle, leading to a “treadmill” of active and inactive osteoclasts that together dig the resorption tunner or trench. Vertical arrow heads indicate release of NO by well-stressed osteocytes. Light colors denote places in the matrix that experience low strains during loading in the normal direction, while dark areas denote areas of high strain. NO nitric oxide
Building on earlier bone adaptation models of Huiskes [95], Van Oers et al. [96] developed a computer model of bone remodeling at the BMU level (Fig. 5). In this model, osteocytes sense a mechanical stimulus (represented by strain energy density) and respond with the release of signals to repel osteoclasts and recruit osteoblasts. Since mechanical forces must travel around the BMU cavity, the strains concentrate at the sides, while the region in front (along the loading direction) is unloaded. Strain-induced osteocyte signals from the sides of the cavity constrain the osteoclasts toward the loading direction and recruit osteoblasts at the tunnel wall.

Mechanical regulation of osteon development. A volume of bone with a cavity is loaded in vertical direction, causing strain concentrations at the sides, while the regions above and below the cavity are unloaded. In other words, the strains around the cavity are low in the loading direction (vertical) and high in transverse direction (horizontal). Osteocytes around the cavity translate these strains into a signal (right) which guides osteoclasts (red) in the loading direction and recruits osteoblasts (black) to form new bone (yellow) on the tunnel wall
Crucial in this model is the range of the osteocyte signal. The signal must diffuse far enough to reach the osteoclasts and osteoblasts on the bone surface but not too far to lose its meaning (“this region of bone is strained”). Huiskes and coworkers [95] assumed that the signal exponentially decreases in strength with increasing distance d from the osteocyte, according to:
where D dd is a diffusion–decay constant. It is also the distance where the signal has decreased to 37 % of its initial strength. Mullender and Huiskes [97] found that the trabecular thickness in their simulations was related to the value of D dd . They estimated D dd at 100 μm, in line with the experimental finding that the trabecular thickness of the iliac cancellous bone in normal humans is 100–200 μm. The exponential function represents the steady-state distribution of a signal molecule, where synthesis and decay are in balance. Once a cell starts synthesizing a signal molecule, its concentration rises in the cell and in its immediate vicinity and spreads out in a decreasing fashion. Soon the concentration at all points reaches a steady state, where the rates of synthesis, diffusion, and decay are in balance. This steady state can be reached very quickly, depending on the diffusion and decay rates of the signal molecule. In the case of a single endothelial cell synthesizing NO, this steady state is achieved in 15 to 20 s [98]. For a 1D-point source S [mol s−1] at d = 0, secreting a signal molecule with diffusion coefficient D [m2 s−1] and decay rate λ [s−1], the steady-state concentration C follows [99]:
As this function resembles the previous one, it can be seen that the diffusion–decay constant D dd of Huiskes’ model equals \( \sqrt{D/\lambda } \). For NO, a diffusion coefficient D of 3,300 μm2/s has been reported and a half-life t 1/2 in the range of 5–15 s [98]. Since λ = ln(2)/t 1/2, the decay rate λ of NO ranges from 0.05–0.14 s−1. Then the diffusion–decay constant D dd for NO would be in the range of 150–250 μm, which is similar in magnitude to the value of Huiskes’ hypothetical signal.
Osteocyte signaling may be more than just the passive diffusion of signal molecules from the secreting osteocyte to the bone surface. Osteocytes could, like neurons, actively transmit signals to one another. Guo et al. [100] cultured osteocytes on a micropatterned network, where the cells could connect via cell processes. They then poked one cell with a nano-indenter until it gave a calcium response, which was picked up by neighbor cells and passed along. The response of the indented cell was highest, but later responses did not differ in strength, indicating that this was more than diffusion from the poked osteocyte. Vatsa et al. [101] found similar active transmission via soluble NO signaling, i.e., mechanical stimulation of a single MLO-Y4 osteocyte-like cell resulted in the upregulation of NO production not only in the stimulated cell but also in the surrounding osteocytes. Such active transmission would substantially alter the range and perhaps also the timeframe, of NO signaling.
Conclusions
Important progress has been made over the last few years regarding the understanding of the role of osteocytes in bone metabolism and turnover. The critical role of intercellular communication via chemical signals like NO in the dynamic process of remodeling has been recognized. The collaboration between experimental investigation and theoretical analysis of the response of bone cells to stress has proven effective for advanced understanding of the underlying processes in bone mechanotransduction. The studies agree that the network of osteocytes provides the cellular structure that allows bone organs to determine local needs for bone augmentation or reduction in response to mechanical loads.
The role of the osteocytes as the mechanosensors of bone has been elucidated in the past years with more detail. The NO response of osteocytes to dynamic stress has indicated that the rate rather than magnitude of loading is important for the effect of mechanical loading on bone. The osteocytic release of NO as well as force traction and morphology change are all related in similar pathways in response to environmental stress conditions. Since NO can inhibit bone loss, there are multiple opportunities for therapeutic interventions using the NO-cGMP pathway. The most attractive novel indication for NO donor therapy is the prevention and treatment of osteoporosis in men and women.
Given the crucial importance of osteocytes for maintaining a proper resistance against bone fracture, it seems obvious that a much greater knowledge of the molecular mechanisms that govern the adaptive response of osteocytes is needed. Since NO is an early signaling molecule that mediates the anabolic response of bone to mechanical loading in vivo, recognition of the missing links in the mechanistic model that explains the involvement of NO in the activation of important signaling pathways in bone adaptation deserves to be the focus of future work. Key to proper research on this subject is the development of tools that allows studying osteocytes in their natural 3D environment.
References
Wei XQ, Charles IG, Smith A, Ure J, Feng GJ, Huang FP, Xu D, Muller W, Moncada S, Liew FY (1995) Altered immune responses in mice lacking inducible nitric oxide synthase. Nature 375:408–411
Fox SW, Chambers TJ, Chow JW (1996) Nitric oxide is an early mediator of the increase in bone formation by mechanical stimulation. Am J Physiol 270:E955–E960
Van ‘t Hof RJ, Ralston SH (2001) Nitric oxide and bone. Immunology 103:255–261
Moncada S, Higgs EA (2006) The discovery of nitric oxide and its role in vascular biology. Br J Pharmacol 147(Suppl 1):S193–S201
Zhou L, Zhu DY (2009) Neuronal nitric oxide synthase: structure, subcellular localization, regulation, and clinical implications. Nitric Oxide 20:223–230
Zaman G, Pitsillides AA, Rawlinson SC, Suswillo RF, Mosley JR, Cheng MZ, Platts LA, Hukkanen M, Polak JM, Lanyon LE (1999) Mechanical strain stimulates nitric oxide production by rapid activation of endothelial nitric oxide synthase in osteocytes. J Bone Miner Res 14:1123–1131
Hukkanen MV, Platts LA, Fernandez DM, O’Shaughnessy M, MacIntyre I, Polak JM (1999) Developmental regulation of nitric oxide synthase expression in rat skeletal bone. J Bone Miner Res 14:868–877
Helfrich MH, Evans DE, Grabowski PS, Pollock JS, Ohshima H, Ralston SH (1997) Expression of nitric oxide synthase isoforms in bone and bone cell cultures. J Bone Miner Res 12:1108–1115
Loveridge N, Fletcher S, Power J, Caballero-Alias AM, Das-Gupta V, Rushton N, Parker M, Reeve J, Pitsillides AA (2002) Patterns of osteocytic endothelial nitric oxide synthase expression in the femoral neck cortex: differences between cases of intracapsular hip fracture and controls. Bone 30:866–871
Lirani-Galvão AP, Lazaretti-Castro M, Portero-Muzy N, Bergamaschi CT, Silva OL, Carvalho AB, Delmas PD, Chavassieux P (2010) Is nitric oxide a mediator of the effects of low-intensity electrical stimulation of bone in ovariectomized rats? Calcif Tissue Int 87:52–59
Zweier JL, Wang P, Samouilov A, Kuppusamy P (1995) Enzyme-independent formation of nitric oxide in biological tissues. Nat Med 1:804–809
Godber BL, Doel JJ, Sapkota GP, Blake DR, Stevens CR, Eisenthal R, Harrison R (2000) Reduction of nitrite to nitric oxide catalyzed by xanthine oxidoreductase. J Biol Chem 275:7757–7763
Foster MW, Hess DT, Stamler JS (2009) Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med 15:391–404
Moncada S, Higgs A (1993) The l-arginine-nitric oxide pathway. New Engl J Med 329:2002–2012
Van ‘t Hof RJ, Armour KJ, Smith LM, Armour KE, Wei XQ, Liew FY, Ralston SH (2000) Requirement of the inducible nitrix oxide synthase pathway for IL-1-induced osteoclastic bone resorption. Proc Natl Acad Sci U S A 97:7993–7998
Armour KE, Armour KJ, Gallagher ME, Gödecke A, Helfrich MH, Reid DM, Ralston SH (2001) Defective bone formation and anabolic response to exogenous estrogen in mice with targeted disruption of endothelial nitric oxide synthase. Endocrinology 142:760–766
Aguirre J, Buttery L, O’Shaughnessy M, Afzal F, Fernandez DM, Hukkanen M, Huang P, MacIntyre I, Polak J (2001) Endothelial nitric oxide synthase gene-deficient mice demonstrate marked retardation in postnatal bone formation, reduced bone volume, and defects in osteoblast maturation and activity. Am J Pathol 158:247–257
Van ‘t Hof RJ, MacPhee J, Libouban H, Helfrich MH, Ralston SH (2004) Regulation of bone mass and bone turnover by neuronal nitric oxide synthase. Endocrinology 145:5068–5074
Cuzzocrea S, Mazzon E, Dugo L, Genovese T, Di PR, Ruggeri Z, Vegeto E, Caputi AP, Van De Loo FA, Puzzolo D, Maggi A (2003) Inducible nitric oxide synthase mediates bone loss in ovariectomized mice. Endocrinology 144:1098–1107
Watanuki M, Sakai A, Sakata T, Tsurukami H, Miwa M, Uchida Y, Watanabe K, Ikeda K, Nakamura T (2002) Role of inducible nitric oxide synthase in skeletal adaptation to acute increases in mechanical loading. J Bone Miner Res 17:1015–1025
Sabanai K, Tsusui M, Sakai A, Hirasawa H, Tanaka S, Nakamura E, Tanimoto A, Sasaguri Y, Ito M, Shimokawa H, Nakamura T, Yanagihara N (2008) Genetic disruption of all NO synthase isoforms enhances BMD and bone turnover in mice in vivo: involvement of the renin-angiotensin system. J Bone Miner Res 23:633–643
Wimalawansa SJ (2007) Rationale for using nitric oxide donor therapy for prevention of bone loss and treatment of osteoporosis in humans. Ann NY Acad Sci 1117:283–297
Skerry TM (2008) The response of bone to mechanical loading and disuse: fundamental principles and influences on osteoblast/osteocyte homeostasis. Arch Biochem Biophys 473:117–123
Kamioka H, Kameo Y, Imai Y, Bakker AD, Bacabac RG, Yamada N, Takaoka A, Yamashiro T, Adachi T, Klein-Nulend J (2012) Microscale fluid flow analysis in a human osteocyte canaliculus using a realistic high-resolution image-based three-dimensional model. Integr Biol 4:1198–1206
Burger EH, Klein-Nulend J (1999) Mechanotransduction in bone—role of the lacuno-canalicular network. FASEB J 13:S101–S112
Klein-Nulend J, van der Plas A, Semeins CM, Ajubi NE, Frangos JA, Nijweide PJ, Burger EH (1995) Sensitivity of osteocytes to biomechanical stress in vitro. FASEB J 9:441–445
Scheiner S, Pivonka P, Hellmich C (2013) Poromechanical stimulation of bone remodeling: a continuum micromechanics-based mathematical model and experimental validation. Poromechanics V:1867–1876
Turner CH, Owan I, Takano Y (1995) Mechanotransduction in bone: role of strain rate. Am J Physiol 269:E438–E442
Fritton SP, McLeod KJ, Rubin CT (2000) Quantifying the strain history of bone: spatial uniformity and self-similarity of low-magnitude strains. J Biomech 33:317–325
De Jong WC, Korfage JA, Langenbach GE (2010) Variations in habitual bone strains in vivo: long bone versus mandible. J Struct Biol 172:311–318
Nordstrom P, Pettersson U, Lorentzon R (1998) Type of physical activity, muscle strength, and pubertal stage as determinants of bone mineral density and bone area in adolescent boys. J Bone Miner Res 13:1141–1148
Bacabac RG, Smit TH, Mullender MG, Dijcks SJ, van Loon JJWA, Klein-Nulend J (2004) Nitric oxide production by bone cells is fluid shear stress rate dependent. Biochem Biophys Res Commun 315:823–829
Turner CH, Owan I, Jacob DS, McClintock R, Peacock M (1997) Effects of nitric oxide synthase inhibitors on bone formation in rats. Bone 21:487–490
Rubin CT, Lanyon LE (1984) Regulation of bone formation by applied dynamic loads. J Bone Joint Surg Am 66:397–402
Bacabac RG, Smit TH, Mullender MG, van Loon JJWA, Klein-Nulend J (2005) Initial stress-kick is required for fluid shear stress-induced rate dependent activation of bone cells. Ann Biomed Eng 33:104–110
You L, Cowin SC, Schaffler MB, Weinbaum S (2001) A model for strain amplification in the actin cytoskeleton of osteocytes due to fluid drag on pericellular matrix. J Biomech 34:1375–1386
Han Y, Cowin SC, Schaffler MB, Weinbaum S (2004) Mechanotransduction and strain amplification in osteocyte cell processes. Proc Natl Acad Sci U S A 101:16689–16694
Bacabac RG, Smit TH, van Loon JJWA, Zandieh-Doulabi B, Helder MN, Klein-Nulend J (2006) Bone cell responses to high-frequency vibration stress: does the nucleus oscillate within the cytoplasm? FASEB J 20:858–864
McGarry JG, Klein-Nulend J, Mullender MG, Prendergast PJ (2005) A comparison of strain and fluid shear stress in stimulating bone cell responses—a computational and experimental study. FASEB J 19:482–484
Klein-Nulend J, Helfrich MH, Sterck JG, MacPherson H, Joldersma M, Ralston SH, Semeins CM, Burger EH (1998) Nitric oxide response to shear stress by human bone cell cultures is endothelial nitric oxide synthase dependent. Biochem Biophys Res Commun 250:108–114
Rath AL, Bonewald LF, Ling J, Jiang JX, Van Dyke ME, Nicolella DP (2010) Correlation of cell strain in single osteocytes with intracellular calcium, but not intracellular nitric oxide, in response to fluid flow. J Biomech 43:1560–1564
Bakker AD, da Silva VC, Krishnan R, Bacabac RG, Blaauboer ME, Lin YC, Marcantonio RA, Cirelli JA, Klein-Nulend J (2009) Tumor necrosis factor alpha and interleukin-1beta modulate calcium and nitric oxide signaling in mechanically stimulated osteocytes. Arthr Rheum 60:3336–3345
Sugawara Y, Ando R, Kamioka H, Ishihara Y, Murshid SA, Hashimoto K, Kataoka N, Tsujioka K, Kajiya F, Yamashiro T, Takano-Yamamoto T (2008) The alteration of a mechanical property of bone cells during the process of changing from osteoblasts to osteocytes. Bone 43:19–24
McGarry JG, Klein-Nulend J, Prendergast PJ (2005) The effect of cytoskeletal disruption on pulsatile fluid flow-induced nitric oxide and prostaglandin E2 release in osteocytes and osteoblasts. Biochem Biophys Res Commun 330:341–348
Vatsa A, Mizuno D, Smit TH, Schmidt CF, MacKintosh FC, Klein-Nulend J (2006) Bio imaging of intracellular NO production in single bone cells after mechanical stimulation. J Bone Miner Res 21:1722–1728
Bacabac RG, Mizuno D, Schmidt CF, MacKintosh FC, Van Loon JJWA, Klein-Nulend J, Smit TH (2008) Round versus flat: bone cell morphology, elasticity, and mechanosensing. J Biomech 41:1590–1598, Erratum in: J Biomech 41:2786
Chow JW, Fox SW, Lean JM, Chambers TJ (1998) Role of nitric oxide and prostaglandins in mechanically induced bone formation. J Bone Miner Res 13:1039–1044
Klein-Nulend J, Semeins CM, Ajubi NE, Nijweide PJ, Burger EH (1995) Pulsating fluid flow increases nitric oxide (NO) synthesis by osteocytes but not periosteal fibroblasts—correlation with prostaglandin upregulation. Biochem Biophys Res Commun 217:640–648
Johnson DL, McAllister TN, Frangos JA (1996) Fluid flow stimulates rapid and continuous release of nitric oxide in osteoblasts. Am J Physiol Endocrinol Metab 271:E205–E208
Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM (1999) Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399:601–605
Das-Gupta V, Williamson RA, Pitsillides AA (2012) Expression of endothelial nitric oxide synthase protein is not necessary for mechanical strain-induced nitric oxide production by cultured osteoblasts. Osteoporos Int 23:2635–2647
Bakker AD, Huesa C, Hughes A, Aspden RM, van’t Hof RJ, Klein-Nulend J, Helfrich MH (2013) Endothelial nitric oxide synthase is not essential for nitric oxide production by osteoblasts subjected to fluid shear stress in vitro. Calcif Tissue Int 92:228–239
Huang B, Chen SC, Wang DL (2009) Shear flow increases S-nitrosylation of proteins in endothelial cells. Cardiovasc Res 83:536–546
Murad F, Mittal CK, Arnold WP, Katsuki S, Kimura H (1978) Guanylate cyclase: activation by azide, nitro compounds, nitric oxide, and hydroxyl radical and inhibition by hemoglobin and myoglobin. Adv Cyclic Nucleotide Res 9:145–158
Rangaswami H, Marathe N, Zhuang S, Chen Y, Yeh JC, Frangos JA, Boss GR, Pilz RB (2009) Type II cGMP-dependent protein kinase mediates osteoblast mechanotransduction. J Biol Chem 284:14796–14808
Löwik CW, Nibbering PH, van de Ruit M, Papapoulos SE (1994) Inducible production of nitric oxide in osteoblast-like cells and in fetal mouse bone explants is associated with suppression of osteoclastic bone resorption. J Clin Invest 93:1465–1472
Armour KE, Van ‘t Hoff RJ, Grabowski PS, Reid DM, Ralston SH (1999) Evidence for a pathogenic role of nitric oxide in inflammation-induced osteoporosis. J Bone Miner Res 14:2137–2142
Brandi ML, Hukkanen M, Umeda T, Moradi-Bidhendi N, Bianchi S, Gross SS, Polak JM, MacIntyre I (1995) Bidirectional regulation of osteoclast function by nitric oxide synthase isoforms. Proc Natl Acad Sci U S A 92:2954–2958
MacIntyre I, Zaidi M, Alam AS, Datta HK, Moonga BS, Lidbury PS, Hecker M, Vane JR (1991) Osteoclastic inhibition: an action of nitrix oxide not mediated by cyclic GMP. Proc Natl Acad Sci U S A 88:2936–2940
Juffer P, Jaspers RT, Lips P, Bakker AD, Klein-Nulend J (2012) Expression of muscle anabolic and metabolic factors in mechanically loaded MLO-Y4 osteocytes. Am J Physiol Endocrinol Metab 302:E389–E395
Standal T, Abildgaard N, Fagerli U-M, Stordal B, Hjertner O, Borset M, Sundan A (2007) HGF inhibits BMP-induced osteoblastogenesis: possible implications for the bone disease of multiple myeloma. Blood 109:3024–3030
Adamopoulos IE, Xia Z, Lau YS, Athanasou NA (2006) Hepatocyte growth factor can substitute for M-CSF to support osteoclastogenesis. Biochem Biophys Res Commun 350:478–483
Cheung WY, Liu C, Tonelli-Zasarsky RM, Simmons CA, You L (2011) Osteocyte apoptosis is mechanically regulated and induces angiogenesis in vitro. J Orthop Res 29:523–530
Zaman G, Saxon LK, Sunters A, Hilton H, Underhill P, Williams D, Price JS, Lanyon LE (2010) Loading-related regulation of gene expression in bone in the contexts of estrogen deficiency, lack of estrogen receptor alpha and disuse. Bone 46:628–642
Furumatsu T, Shen ZN, Kawai A, Nishida K, Manabe H, Oohashi T, Inoue H, Ninomiya Y (2003) Vascular endothelial growth factor principally acts as the main angiogenic factor in the early stage of human osteoblastogenesis. J Biochem 133:633–639
Santos A, Bakker AD, Zandieh-Doulabi B, Semeins CM, Klein-Nulend J (2009) Pulsating fluid flow modulates gene expression of proteins involved in Wnt signaling pathways in osteocytes. J Orthop Res 27:1280–1287
Robinson JA, Chatterjee-Kishore M, Yaworsky PJ, Cullen DM, Zhao W, Li C, Kharode Y, Sauter L, Babij P, Brown EL, Hill AA, Akhter MP, Johnson ML, Recker RR, Komm BS, Bex FJ (2006) Wnt/β-catenin signaling is a normal physiological response to mechanical loading in bone. J Biol Chem 281:31720–31728
Javaheri B, Stern A, Lara N, Dallas M, Zhao H, Liu Y, Bonewald LF, Johnon ML (2013) Deletion of a single β-catenin allele in osteocytes abolishes the bone anabolic response to loading. J Bone Miner Res. doi:10.1002/jbmr.2064, Epub ahead of print
Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PV, Komm BS, Javed A, van Wijnen AJ, Stein JL, Stein GS, Lian JB (2005) Canonical Wnt signaling promotes osteogenesis by directly stimulating RUNX2 gene expression. J Biol Chem 280:33132–33140
Bodine PV, Billiard J, Moran RA, Ponce-de-Leon H, Mangine A, Scrimo MJ, Bhat RA, Stauffer B, Green J, Stein GS, Lian JB, Komm BS (2005) The wnt antagonist secreted-related protein-1 controls osteoblast and osteocyte apoptosis. J Cell Biochem 96:1212–1230
Glass DA 2nd, Bialek P, Ahn JD, Starbuch M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, Karsenty G (2005) Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell 8:751–764
Delgado-Calle J, Saňudo C, Bolado A, Fernández AF, Arozamena J, Pascual-Carra MA, Rodriguez-Rey JC, Fraga MF, Bonewald L, Riancho JA (2012) DNA methylation contributes to the regulation of sclerostin expression in human osteocytes. J Bone Miner Res 27:926–937
Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, Mantila SM, Gluhak-Heinrich J, Bellido TM, Harris SE, Turner CH (2008) Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem 283:5866–5875
Bentolila V, Boyce TM, Fyhrie DP, Drumb R, Skerry TM, Schaffler MB (1998) Intracortical remodeling in adult rat long bones after fatigue loading. Bone 23:275–281
Bronckers ALJJ, Goei SW, Luo G, Karsenty G, D’souza RN, Lyaruu DM, Burger EH (1996) DNA fragmentation during bone formation in neonatal rodents assessed by transferase-mediated end labeling. J Bone Miner Res 11:1281–1291
Verborgt O, Gibson GJ, Schaffler MB (2000) Loss of osteocyte integrity in association with microdamage and bone remodeling after fatigue in vivo. J Bone Miner Res 15:60–67
Kennedy OD, Herman BC, Laudier DM, Majeska RJ, Sun HB, Schaffler MB (2012) Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone 50:1115–1122
Parfitt AM (1994) Osteonal and hemi-osteonal remodeling: the spatial and temporal framework for signal traffic in adult human bone. J Cell Biochem 55:273–286
Kamioka H, Honjo T, Takano-Yamamoto T (2001) A three-dimensional distribution of osteocyte processes revealed by the combination of confocal laser scanning microscopy and differential interference contrast microscopy. Bone 28:145–149
Smit TH, Burger EH (2000) Is BMU-coupling a strain-regulated phenomenon? A finite element analysis. J Bone Miner Res 15:301–307
Burger EH, Klein-Nulend J, Smit TH (2003) Strain-derived canalicular fluid flow regulates osteoclast activity in a remodeling osteon—a proposal. J Biomech 36:1453–1459
Smit TH, Burger EH, Huyghe JM (2002) A case for strain-induced fluid flow as a regulator of BMU-coupling and osteonal alignment. J Bone Miner Res 18:2021–2029
Tan SD, Kuijpers-Jagtman AM, Semeins CM, Bronckers ALJJ, Maltha JC, Von den Hoff JW, Everts V, Klein-Nulend J (2006) Fluid shear stress inhibits TNFalpha-induced osteocyte apoptosis. J Dent Res 85:905–909
Bakker AD, Klein-Nulend J, Burger EH (2004) Shear stress inhibits while disuse promotes osteocyte apoptosis. Biochem Biophys Res Commun 320:1163–1168
Tan SD, Bakker AD, Semeins CM, Kuijpers-Jagtman AM, Klein-Nulend J (2008) Inhibition of osteocyte apoptosis by fluid flow is mediated by nitric oxide. Biochem Biophys Res Commun 369:1150–1154
Marathe N, Rangaswami H, Zhuang S, Boss GR, Pilz RB (2012) Pro-survival effects of 17β-estradiol on osteocytes are mediated by nitric oxide/cGMP via differential actions of cGMP-dependent protein kinases I and II. J Biol Chem 287:978–988
Kogianni G, Mann V, Noble BS (2008) Apoptotic bodies convey activity capable of initiating osteoclastogenesis and localized bone destruction. J Bone Miner Res 23:915–927
Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, Bonewald LF, Kodama T, Wutz A, Wagner EF, Penninger JM, Takayanagi H (2011) Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med 17:1231–1234
Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA (2011) Matrix-embedded cells control osteoclast formation. Nat Med 17:1235–1241
Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, Ito M, Takeshita S, Ikeda K (2007) Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab 5:464–475
You L, Temiyasathit S, Lee P, Kim CH, Tummala P, Yao W, Kingery W, Malone AM, Kwon RY, Jacobs CR (2008) Osteocytes as mechanosensors in the inhibition of bone resorption due to mechanical loading. Bone 42:172–179
Kulkarni RN, Bakker AD, Everts V, Klein-Nulend J (2010) Inhibition of osteoclastogenesis by mechanically loaded osteocytes: involvement of MEPE. Calcif Tissue Int 87:461–468
Tan SD, de Vries TJ, Kuijpers-Jagtman AM, Semeins CM, Everts V, Klein-Nulend J (2007) Osteocytes subjected to fluid flow inhibit osteoclast formation and bone resorption. Bone 41:745–751
Mancini L, Moradi-Bidhendi N, Brandi ML, MacIntyre I (1998) Nitric oxide superoxide and peroxynitrite modulate osteoclast activity. Biochem Biophys Res Commun 243:785–790
Huiskes R, Ruimerman R, van Lenthe GH, Janssen JD (2000) Effects of mechanical forces on maintenance and adaptation of form in trabecular bone. Nature 405:704–706
Van Oers RFM, Ruimerman R, Tanck E, Hilbers PAJ, Huiskes R (2008) A unified theory for osteonal and hemi-osteonal remodeling. Bone 42:250–259
Mullender MG, Huiskes R (1995) Proposal for the regulatory mechanism of Wolff’s law. J Orthop Res 13:503–512
Lancaster JR Jr (1997) A tutorial on the diffusibility and reactivity of free nitric oxide. Nitric Oxide 1:18–30
Lancaster JR Jr (1996) Diffusion of free nitric oxide. Methods Enzymol 268:31–50
Guo XE, Takai E, Jiang X, Xu Q, Whitesides GM, Yardley JT, Hung CT, Chow EM, Hantschel T, Costa KD (2006) Intracellular calcium waves in bone cell networks under single cell nanoindentation. Mol Cell Biomech 3:95–107
Vatsa A, Smit TH, Klein-Nulend J (2007) Extracellular NO signalling from a mechanically stimulated osteocyte. J Biomech 40(Suppl 1):S89–95
Acknowledgments
R.F.M. van Oers was supported by a grant of the University of Amsterdam.
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Klein-Nulend, J., van Oers, R.F.M., Bakker, A.D. et al. Nitric oxide signaling in mechanical adaptation of bone. Osteoporos Int 25, 1427–1437 (2014). https://doi.org/10.1007/s00198-013-2590-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-013-2590-4