
Abstract
Emerging evidence from the last two decades has shown that vascular calcification (VC) is a regulated, cell-mediated process orchestrated by vascular smooth muscle cells (VSMCs) and that this process bears many similarities to bone mineralization. While many of the mechanisms driving VSMC calcification have been well established, it remains unclear what factors in specific disease states act to promote vascular calcification and in parallel, bone loss. Diabetes is a condition most commonly associated with VC and bone abnormalities. In this review, we describe how factors associated with the diabetic milieu impact on VSMCs, focusing on the role of oxidative stress, inflammation, impairment of the advanced glycation end product (AGE)/receptor for AGE system and, importantly, diabetic neuropathy. We also explore the link between bone and VC in diabetes with a specific emphasis on the receptor activator of nuclear factor κβ ligand/osteoprotegerin system. Finally, we describe what insights can be gleaned from studying Charcot osteoarthropathy, a rare complication of diabetic neuropathy, in which the occurrence of VC is frequent and where bone lysis is extreme.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
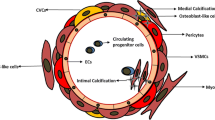
Evidence emerging from the past 20 years has shown that vascular calcification (VC) is a major health problem, associated with increased cardiovascular morbidity and mortality. Calcification or biomineralization of arteries involves the deposition of hydroxyapatite (HA) in the extracellular matrix and occurs at two distinct sites within the vessel wall: the intima and the media [1]. Intimal calcification is commonly found in the aorta and coronary arteries, whereas medial calcification affects large-, medium- and small-sized arteries [2]. Intimal calcification is associated with lipid, macrophages and vascular smooth muscle cells (VSMCs) and occurs in the context of atherosclerosis, whereas medial calcification can exist independently of atherosclerosis and is associated with extracellular matrix components such as elastin and collagen and VSMCs [1]. HA deposition in the intima is often eccentric and is associated with luminal narrowing, plaque formation and rupture [3]. In contrast, medial calcification can occur throughout the vessel media and contributes to vascular stiffness, which in turn increases pulse–wave velocity to decrease diastolic blood pressure and increase systolic blood pressure [1].
It is now well-established that arterial calcification is a regulated, cell-mediated process orchestrated by VSMCs and that this process bears many similarities to bone mineralization [4]. Under normal conditions, VSMCs express potent inhibitors of calcification including matrix Gla protein and pyrophosphate and there are also powerful circulating inhibitors of HA formation such as fetuin A. In calcifying environments, expression or function of these inhibitory proteins is impaired [5]. In addition, VSMCs undergo phenotypic modulation and upregulate expression of a number of bone proteins including the master transcription factors Runx 2 and Msx2, as well as their downstream targets such as bone sialoprotein, osteocalcin and alkaline phosphatase (ALP) which act to further promote mineralization [6]. In particular, ALP has been shown to degrade the potent calcification inhibitor pyrophosphate to promote VSMC calcification [7]. Concomitant with VSMC phenotypic change, cell death by apoptosis as well as matrix vesicle release, provide membranes and protein complexes that act as a nidus to nucleate mineral [8]. Importantly, many of the signals that promote VSMC osteo/chondrocytic change also have profound effects on bone mineralization leading to the suggestion that these processes not only share features in common, but may also be pathologically linked in some disease processes [4].
Whilst many of the mechanisms driving VSMC calcification have been well established, it remains unclear what factors in specific disease states act to promote vascular calcification and in parallel, bone loss. Diabetes is a condition most commonly associated with VC and bone abnormalities and with the global increase in the prevalence of diabetes, the burden of VC and its related adverse outcomes is also expected to rise (http://www.idf.org/diabetesatlas/5e/mortality). Indeed, VC is four times more common in diabetic patients compared with healthy subjects [9] and is associated with significant morbidity and mortality [10, 11]. It is a significant predictor of future fatal and nonfatal myocardial infarction, stroke and amputation [12] and is also a strong independent predictor of cardiovascular mortality in type 2 diabetes [13]. It is associated with the duration of diabetes, poor glycaemic control and nephropathy [10] but is often independent of chronic kidney disease [14], as it also occurs in subjects with preserved renal function [11], suggesting that diabetes itself can modulate calcification. Patients with diabetes are also at greater risk of developing osteoporosis and fragility fractures [15], and a link between VC and impaired bone health is now well established [16].
Many factors lead to the development of VC in diabetes, and drivers that require specific attention include oxidative stress, inflammation and derangement in the advanced glycation end product (AGE)/receptor for AGE (RAGE) system. Although these factors are common players in the pathogenesis of VC per se, in diabetes their importance is further emphasised by their association with the development of diabetic neuropathy, a common complication of diabetes, identified as the harbinger of VC [17]. Importantly, neuropathy not only modulates various pathways and mechanisms that cause calcification in diabetes [17] but also impacts on bone mineralization. The receptor activator of nuclear factor κβ ligand (RANKL)/osteoprotegerin (OPG) signalling pathway is of particular importance as these factors are key regulators of VC and bone loss potentially modulated by neuropathy, via the calcitonin-gene related protein (C-GRP) [18]. Disturbance of this pathway is significantly marked in Charcot osteoarthropathy, a rare complication of diabetes presenting with specific neuropathy, extensive local VC and severe osteolysis. However, the interactions between these pathologies have not been fully investigated.
To address this deficit, in this review, we describe how factors associated with the diabetic milieu impact on VSMCs to promote calcification and we also explore the link between bone and VC in diabetes with a specific emphasis on the RANKL/OPG system. Finally, we describe what lessons can be learnt from Charcot osteoarthropathy, where VC and extreme bone lysis occur concomitantly, in the context of neuropathy and inflammation.
Vascular calcification and neuropathy are linked in diabetes
Both intimal and medial artery calcification correlate with significant cardiovascular morbidity and are important predictors of cardiovascular mortality [1]. In diabetes, VC occurs at both anatomical sites and although either process may arise independently, a combination of intimal and medial calcification is frequently found [19]. Factors commonly associated with driving intimal calcification and atherosclerosis include oxidative stress, inflammation and accumulation of AGEs although these may also contribute to medial calcification. In contrast, neuropathy appears to be exclusively associated with medial calcification suggesting that the neuropathic environment has adverse effects on local VSMCs. However, both processes are likely to be interlinked as inflammation, oxidative stress and AGE/RAGE derangement can also lead to nerve impairment and hence neuropathy. Thus, there may be a vicious cycle of VSMC and nerve damage, which acts synergistically to further promote VC in the context of diabetic neuropathy (Fig. 1). How these factors potentially act to drive both VSMC and nerve damage are described below [17].

Interaction between common risk factors for VC and neuropathy in diabetes. Diabetes is associated with VC and neuropathy and these two pathologies share common risk factors including oxidative stress, inflammation and AGE/RAGE derangement. Neuropathy is also linked with VC. It is possible that in diabetes, there may be a vicious cycle of parallel VSMC and nerve damage, which acts synergistically to further promote VC
Oxidative stress
Oxidative stress plays a critical role in the pathobiology of VC [20] and has been identified as a key event in the pathogenesis of diabetic complications. Chronic hyperglycaemia leads to excessive free radical production and oxidative stress resulting in cellular injury and death. Increased oxidative stress occurs early in the onset of diabetes and is characterised by the presence of a decreased antioxidant capacity and an increased rate of lipoperoxidation [21] with clinical studies suggesting that this environment may be associated with the development of intimal calcification. For example, increased levels of oxidised low-density lipoprotein (LDL) immune complexes at baseline, in patients with type 1 diabetes, were associated with an increased risk of developing clinically significant coronary artery calcification, detected by computed tomography after a period of 11–20 years [22]. Atherosclerotic plaques contain high levels of oxysterols [23] which enhance VSMC calcification in vitro by inducing increased ALP activity and apoptosis [24]. Moreover, numerous studies have implicated reactive oxygen species (ROS) in promoting the phenotypic switch of VSMCs from a contractile to an osteogenic phenotype, characterised by increased expression of the key osteogenic differentiation factor Runx2 [25]. In addition, hyperglycemia-induced oxidative stress is also associated with inflammation, another factor shown to promote VC in diabetes [26].
Inflammation
Inflammatory markers including acute-phase response proteins (C-reactive protein (CRP), haptogobin, fibrinogen, plasminogen activator inhibitor and serum amyloid A) as well as cytokines (tumour necrosis factor α (TNF-α) and interleukin-6) are raised in type 2 diabetes and correlate with calcification [27]. Raised inflammatory markers in patients with type 1 diabetes are also strongly associated with the progression of coronary artery calcification [28], while increased plasma levels of high sensitivity CRP and matrix GLA protein have been noted in patients with type 2 diabetes, suggesting that the process of VC may be paralleled by low-grade inflammation [29].
Raised levels of ROS and inflammatory cytokines in diabetes may link vascular and nerve damage. Chronic hyperglycaemia has been shown to promote endogenous TNF-α production by activated macrophages in microvascular and neural tissues, promoting increased microvascular permeability, hypercoagulation and associated nerve damage [30]. Hyperglycaemia-induced oxidative stress also leads to increased TNF-α expression in response to oxidised LDL, and evidence from pigs with streptozotocin-induced diabetes has shown increased expression of TNF-α and IL-6 in both the coronary media and adventitia, as a result of increased ROS production via augmented NAD(P)H oxidase activity [31]. Importantly, the proinflammatory cytokines TNF-α and IL-6 have been identified as key cytokines involved in promoting VC. These factors have been shown to promote VSMC osteogenic differentiation in vitro as blocking antibodies can inhibit TNF-α-induced ALP activity in human VSMCs. Upregulation of adventitial TNF-α production by activated monocytes and macrophages has also been shown to induce pro-calcific MSX-2 activity via the NF-kappaB pathway and paracrine Wnt signalling cascades leading to mural calcification and fibrosis via activation of mesenchymal progenitors in the arterial wall [32]. IL-6 can induce osteoblastic differentiation of VSMCs by increasing STAT3 phosphorylation and ALP activity [33] and in diabetes, IL-6 also triggers increased expression of heat shock proteins, thus potentiating bone morphogenic protein (BMP)2/4 action to promote VSMC osteogenic differentiation [34].
AGE/RAGE
A further signalling pathway that may be important in the pathogenesis of VC in diabetes is the AGE/RAGE system. Hyperglycaemia increases protein glycation and leads to formation of AGEs and tissue accumulation of fluorescent AGEs, detected by skin autofluorescence, is a significant predictor of cardiac mortality in diabetic patients [35]. Some of the biological effects of AGEs are modulated through the interaction with RAGE which activates signalling pathways leading to cellular dysfunction, organ damage and subsequently to complications. In diabetes, activation of AGEs/RAGE contributes to the acceleration of vascular inflammation as a result of enhanced inflammatory gene expression and proatherogenic responses in VSMCs [36].
Activation of RAGE plays a central role in the development of accelerated atherosclerosis in diabetes. A study in streptozotocin-induced diabetic RAGE (−/−)/apolipoprotein E (apoE−/−) double knockout mice clearly showed that RAGE activation has a central role in the formation and progression of atherosclerotic lesions, as the absence of RAGE was associated with significant attenuation of atherosclerotic plaque. The anti-atherosclerotic effects seen in these mice were associated with reduced expression of ROS and inflammatory cytokines, ultimately leading to reduced leukocyte recruitment [37]. Additional evidence from both animal and cell studies on the role of non-cross-linked and nonfluorescent Nε-carboxymethyl-lysine (CML), a major immunogen of AGEs, on the progression of atherosclerotic calcification, showed that the CML/RAGE axis induces apoptosis of macrophages, followed by osteogenic differentiation of VSMCs [38]. Furthermore, in vitro RAGE activation has been shown to induce osteogenic differentiation of VSMCs through Notch/MSX-2 induction, providing further evidence of its role in VC [39].
Diabetic neuropathy
Neuropathy is a common complication of diabetes and is an important risk factor for diabetic foot complications [40]. The first evidence to suggest the link between neuropathy and calcified arteries came from clinical observations of diabetic foot patients [41] where it was noted that VC and bone loss often have a predominant peripheral distribution, similar to the distal “stocking” distribution of neuropathy. Calcified arteries can be identified on plain foot and ankle radiographs and recognised by the typical “pipe-stem or tramline” appearance of continuous parallel lines of calcification of foot and leg arteries [41]. This distal distribution suggests an interaction with peripheral diabetic neuropathy and indeed impaired vibration sensation, a clinical measure of neuropathy, has long been noted as a risk factor for VC [10]; more recently, a significant association between distal sensorimotor polyneuropathy and VC has been documented [9].
Studies focused on histological examination of the peripheral artery specimens from diabetic patients with neuropathy revealed medial calcification, characterised by the presence of calcified elastic fibres in the mildly affected arteries, to extensive calcium deposits and areas of true bone formation in the advanced lesions [19, 42]. Using immunohistochemisty and in situ hybridization, it was demonstrated that calcified vessels from diabetic patients showed diminished expression of matrix Gla-protein and osteonectin, key inhibitors of vascular calcification. Conversely, they showed increased expression of ALP, bone sialoprotein, bone Gla protein and collagen II—indicators of osteo/chondrogenesis [19].
Importantly, inflammation, oxidative stress and AGEs/RAGE disturbances are common players in the pathogenesis of both VC and neuropathy that impact on VSMCs and nerve fibres [43–45. Excess production of mitochondrial ROS and reactive nitrogen species is a pivotal step in hyperglycemia-induced nerve damage [46]. It is also the main cause of nerve damage in diabetic patients with good overall glucose control, as a result of short episodes of postprandial hyperglycaemia and increased ROS production [43]. Subclinical inflammation is also associated with the development of neuropathy in vivo [47] while in animal models, inactivation of TNF-α via either genetic inactivation or by neutralisation of the TNF-α protein using the monoclonal antibody infliximab, ameliorates diabetic neuropathy in mice [48]. Tissue accumulation of fluorescent AGEs, detected by skin autofluorescence, is associated with the severity of diabetic neuropathy [49] and accumulation of AGEs has been noted in peripheral nerves from diabetic rats and dogs [50]. Moreover, the absence of RAGE attenuates both structural and electrophysiological changes within the peripheral nerves in diabetic RAGE−/− mice [51]; it has been postulated that strategies targeting RAGE activation could be helpful to prevent both diabetic vascular complications and neuropathy.
Thus, the same factors and their complex interactions in “at risk” individuals modulate the occurrence and progression of both VC and nerve damage. However, an association does not always mean causation. While the same factors may drive both vascular and nerve damage concomitantly, it remains unknown whether loss of trophic factors, from damaged nerves in neuropathic tissues in diabetes, directly impacts on the vessel wall to drive VSMC-mediated calcification.
The impact of neuropathy on VC and bone in diabetes—the role of RANKL/OPG
Neuropathy may also provide a link between the vasculature and the skeletal system [18]. A high prevalence and coincidence of VC and cardiovascular disease in patients with osteoporosis has been well documented [52–54] and such an association has also been noted in patients with diabetes. Moreover, patients with both type 1 (due to underlying osteopenia) and type 2 diabetes (due to impaired bone quality and increased risk of falls) have increased osteoporotic fracture risk [15], while osteopenia in the diabetic lower limb is associated with peripheral nerve damage [55, 56].
The interaction between arteries and bone in diabetes is complex. However, one signalling pathway which could account for this association is the RANKL/OPG cytokine system [57, 58], which is also potentially modulated by neuropathy [18]. This cytokine system was initially linked with bone break down as RANKL, a cytokine from the TNF superfamily, has been described as a key regulator of osteoclast maturation and differentiation. RANKL binds to its target RANK, expressed on mononuclear osteoclastic precursors, induces NF-kβ signalling, resulting in NF-kβ translocation to the nucleus and drives osteoclastogenesis [59]. Apart from bone-forming osteoblasts, it has been shown that RANKL is expressed by a variety of cells including endothelial cells, T-cells, dendritic cells and fibroblasts. OPG, a soluble decoy receptor for RANKL, blocks the interaction between RANKL-RANK, thus acting as an antagonist for RANKL [59].
The first evidence to suggest the importance of the RANKL/OPG signalling pathway in the pathogenesis of calcification came from animal knockout studies. Mice lacking OPG develop arterial calcification of the aorta and renal arteries together with severe osteoporosis [60], and conversely transgenic mice overexpressing OPG develop osteopetrosis [61]. These important observations have led to a number of studies exploring this signalling pathway and its link with VC. There is evidence that calcified arteries of OPG−/− mice express RANKL and RANK, proteins not normally present in non-calcified arteries [62]. Similarly, a study in atherosclerotic human arteries has shown the presence of OPG and RANKL in advanced calcified lesions [63]. In vitro studies revealed that VSMCs incubated with RANKL, show a dose-dependent increase in calcification via RANKL/RANK interaction, associated with increased expression of BMP4 [64]. RANK activation leads to nuclear translocation of NF-kβ with involvement only of the alternative pathway with IKK-α activation, but not of the classical pathway of IKK-β. Furthermore, the process of calcification was abolished by co-inhibition with OPG [64]. Immunohistochemistry of atherosclerotic tissue specimens revealed that OPG is deposited at sites of calcification [65], and this may be a protective mechanism to block RANKL-induced calcification. Paradoxically, OPG inhibits calcification of VSMCs induced by high calcium/phosphate treatment only in physiological concentrations, as its inhibitory effect is blunted at high concentrations [65]. This observation of a bi-phasic effect of its action on calcification supports the clinical findings of elevated serum OPG levels in patients with vascular disease and it has been suggested that this glycoprotein could serve as a marker of arterial vascular damage [65].
Similarly, in diabetes, serum OPG levels are raised [66], and significantly correlate with cardiovascular mortality [67]. In a 17-year prospective observational follow-up study in type 2 diabetes, elevated plasma OPG was a strong predictor of all-cause mortality, an effect which was independent of kidney function [68]. Furthermore, high serum OPG levels but not fibroblast growth factor 23 and 25-hydroxyvitamin D3 (modulators of VC in renal disease) were closely associated with the presence of arterial calcification in patients with type 2 diabetes [69]. Thus, in diabetes, there is a shift in classical factors known to modulate VC in chronic kidney disease, providing further evidence to support the notion that VC in diabetes is independent from progression of diabetic nephropathy [69].
Interestingly, it has been recently postulated that upregulated RANKL-mediated effects on arteries and bone could be triggered by the loss of nerve-derived peptides, e.g. calcitonin gene-related peptide (CGRP) [18]. The latter is a neuropeptide that acts as a neurotransmitter in small fibres (C-fibres) [70]. Local innervation plays a modulating role in bone growth, repair and remodelling. The terminal structure of the osseous CGRP-containing nerves directly contact osteoblasts, osteoclasts and the periosteal lining cells, and are a source of local CGRP, which can act as a local modulator of bone metabolism. CGRP increases osteoblastic cyclic adenosine monophosphate via acting on bone specific CGRP receptors [71] thus stimulating osteogenesis [72]. Osseous CGRP-containing fibres are also involved in pathologic events in bone. The density of CGRP fibres is increased near sites of post-fracture osteogenesis (healing callus) and is decreased at the stumps of non-union [73]. Evidence from bone marrow macrophage cultures has shown that CGRP inhibits RANKL-induced NF-κB activation, downregulates osteoclastic genes like tartarate-resistant acid phosphatase (TRAP) and cathepsin K, decreases the number of TRAP+ cells and RANKL-mediated bone resorption [74]. Interestingly, CGRP is also known as a potent vasodilatory peptide and an inhibitor of vascular hypertrophy. Loss of CGRP innervation in large vessels in rats contributes to age-related hypertrophy of VSMCs and intimal thickening [75]. However, whether it has any direct effects on VC has not been explored.
Thus, CGRP deficiency in diabetes may lead to impaired osteogenesis and delayed fracture healing, and also increased RANKL-mediated osteoclastic activity. It may also be responsible for enhanced RANKL-mediated calcification as calcified arteries and bone loss are frequently seen in patients with diabetic neuropathy. Although the full impact of neuropathy on vascular damage and bone loss has not been fully investigated, the magnitude of this interaction may be most dramatically illustrated in Charcot osteoarthropathy.
VC and bone destruction in Charcot osteoarthropathy are mediated via the RANKL/OPG signalling pathway
The association between diabetic neuropathy and VC extends to neuropathy-related complications including foot ulceration, osteomyelitis and Charcot osteoarthropathy. VC in peripheral arteries was reported in 54 % of patients presenting with uncomplicated foot ulcer and in 66 % of patients with osteomyelitis [76]. A condition in which VC is particularly common is Charcot osteoarthropathy (or Charcot foot) [76]; in some groups, VC has been observed in up to 90 % of cases [77]. This rare but devastating complication of diabetes requires specific attention as it links VC, neuropathy and bone destruction in diabetes and could provide novel insights into the interactions between these processes.
Although neuropathic joint disease has been associated with many conditions such as tabes dorsalis, syringomyelia, leprosy, hereditary sensory neuropathy and familial amyloid neuropathy, in the twenty-first century, Charcot foot is most frequently seen in patients with diabetes [77]. It has been reported that a prevalence of 1:680 of people with diabetes develop this condition [78]; however, more recent radiological analysis has shown the presence of Charcot joints on foot and ankle radiographs in up to 10 % of the patients with diabetes and neuropathy [79]. It commonly presents in the mid-foot with severe metatarsal/tarsal fracture/dislocation (Fig. 2) but also occurs in the forefoot and hind foot [77]. Rarely, in diabetes, the knee and the wrist can be affected [77]. Neuropathic joint disease is associated with enormous morbidity and disability [80]. The development of Charcot osteoarthropathy is preceded by trauma which subsequently leads to severe bone and joint destruction, foot deformity, ulceration and then sepsis and often amputation. It is also associated with significant mortality [81, 82], possibly related to the high prevalence of VC and extensive neuropathy, affecting not only the peripheral nervous system but also the autonomic nervous system [17].

Anterior–posterior foot radiograph (a) and lateral ankle radiograph (b) in a patient with left foot Charcot osteoarthropathy. Severe metatarsal-tarsal Lisfranc fracture/dislocation of the left mid-foot (a) with collapse of the medial arch and associated rocker-bottom foot deformity (b). Bilateral VC of the left and right dorsalis pedis arteries (a) and of the left posterior tibial and anterior tibial arteries (b) (white arrows)
The exact mechanisms which link neuropathy, VC and bone loss in Charcot osteoarthropathy are not fully understood but evidence from the last few years has highlighted the importance of RANKL/OPG. This signalling pathway is upregulated in patients with Charcot osteoarthropathy who exhibit elevated serum levels of RANKL, OPG and RANKL/OPG ratio [73], with high serum OPG significantly associated with the presence of VC in foot and leg arteries in neuropathic patients [83].
In the vasculature histological examination of tibial peripheral artery specimens from Charcot patients, undergoing surgery identified positive RANKL staining in both medial and intimal calcified areas, which were not present in non-calcified areas or in specimens from control subjects [84]. Moreover, VSMCs from Charcot patients treated with RANKL show a greater capacity to mineralise and this increased osteoblastic differentiation was inhibited by OPG, confirming the role of RANKL in the process of calcification [84]. In bone, evidence from in vitro studies has shown that newly formed osteoclasts, derived from patients with Charcot osteoarthropathy, exhibit extensive resorbing activity in the presence of macrophage-colony stimulating factor (a survival factor) and RANKL, a response which is attenuated by OPG [85]. Thus, in the vessel wall, progenitor cells differentiate into osteoblast-like cells, depositing mineralised matrix, whereas in bone, the enhanced osteoclastic activation results in osteolysis and severe bone damage [86].
If RANKL and OPG are key players in both VC and bone loss in Charcot osteoarthropathy, what are the factors that activate this pathway? Importantly, evidence suggests that both neuropathy and inflammation may be causal.
Neuropathy and inflammation promote RANKL/OPG
The exact type of nerve damage in the pathogenesis of Charcot foot is not fully understood. However, a selective small fibre neuropathy has been noted [87] with a possible impairment of C-fibres and associated lack of CGRP. Whether CGRP modulates the vessel wall in diabetes is unknown; however, a small pilot immunohistological study has shown a trend towards reduced expression of CGRP in bone specimens from Charcot patients compared with controls [31]. The role of CGRP deficiency in the aetiology of Charcot osteorthropathy has not been fully proven; however, it is thought that deficiency of this neuropeptide may enhance the ratio of RANKL to OPG and thus promote VC and accelerate bone resorption [88]. Further work on the direct effects of CGRP on diabetic vessels and bone is now required.
A red hot inflamed foot is the classical clinical presentation of Charcot osteoarthropathy and uncontrolled inflammation has been shown to have deleterious effect on both vessels and bone. Indeed, VSMCs treated with Charcot serum showed increased capacity to mineralise suggesting that there are secreted mediators including proinflammatory cytokines, which can directly induce osteoblastic differentiation of VSMCs and promote calcification within the Charcot milieu [84]. Indeed, Charcot patients present with raised serum inflammatory markers (hsCRP, TNF-α and IL-6) as well as markers of bone resorption (C-terminal telopeptide of type I collage and alkaline phosphatase) when compared with controls [89]. Calcaneal bone mineral density of the affected foot is decreased when compared with the non-affected contralateral foot [90]. A further reduction in bone mineral density of the affected foot is noted in the natural history of the osteoarthropathy and this fall is closely associated with inflammation [91]. Recent in vitro studies on newly formed osteoclasts in patients with Charcot osteoarthropathy also suggest that inflammation modulates osteoclastic activity with a particular interaction between TNF-α, IL-6 and RANKL [92]. It is possible that in Charcot osteoarthropathy, TNF-α could promote bone resorption, as this pro-inflammatory cytokine not only induces RANKL expression but also enhances osteoclastogenesis in the presence of permissive levels of RANKL (Fig. 4) [93]. This is in agreement with a previous histological study of bone specimens from Charcot patients which has demonstrated that osteoclastic bone resorption is taking place in the presence of TNF-α, and IL-1β [94], cytokines also known to modulate VC [95].
All these observations provide firm evidence to support the recent hypothesis of the role of proinflammatory cytokines and RANKL activation in the pathogenesis of Charcot osteoarthropathy [96] and these could also promote VC. Questions that remain to be answered are why VC is common in diabetic neuropathy, whereas bone destruction leading to Charcot joints affects only a small subset of patients.
One possible explanation is that there may be feedback mechanisms which elaborate RANKL signalling in both the vasculature and bone in the Charcot foot. For example, the osteogenic transcription factor Runx2, which is upregulated in the calcified vessel wall, is known to bind directly to the promoter region of RANKL and to promote its expression [97]. Increased RANKL in the vessel wall results in enhanced migration and differentiation of macrophages into osteoclast-like cells, ultimately leading to calcification and true bone formation in the affected arteries (Fig. 3) [98]. It is thought that these osteoclast-like cells may drive VC. It is plausible that in the context of Charcot foot, where there is great availability/differentiation of osteoclastic precursors [99], that VC is accelerated via this pathway. Moreover, in bone, trauma (with or without fracture), sets off an exaggerated inflammatory response with the release of proinflammatory cytokines and RANKL activation [96]. Fracture itself triggers a coordinated healing cytokine response with enhanced production of IL-1 and TNF-α [100]. Cytokine stimulation of the endothelium results in upregulation of receptors and adhesion molecules which in turn are capable of forming firm attachment with osteoclast precursors [94]. Furthermore, RANKL itself exhibits chemotactic properties towards human monocytes (osteoclastic precursors) [88]. Thus, local inflammation and enhanced RANKL expression may contribute to fracture and bone destruction as well as VC in Charcot osteoarthropathy (Fig. 4).

The potential role of RANKL in the pathogenesis of VC in Charcot osteoarthropathy. In neuropathy, activation of VSMCs via lack of CGRP and inflammation associated with a rise in TNF-α and IL-6 could upregulate RANKL and possibly MSX-2 and Runx2 resulting in enhanced osteoblastic activity and formation of osteoblast-like cells. Upregulated Runx-2 in VSMCs in response to oxidative stress leads to enhanced RANKL expression due to direct promoter binding. RANKL attracts circulating microphages which differentiate into vascular osteoclasts. This results in mineralisation and true bone formation in the vessel wall ultimately leading to VC

The potential role of RANKL in bone destruction in Charcot osteoarthropathy. Nerve damage in diabetes is associated with loss of CGRP and increased RANKL-mediated osteoclastic activity. Trauma itself triggers an inflammatory response with a rise in TNF-α and IL-6, which modulates RANKL activation and osteoclastogenesis. TNF-α itself acts on osteoclast precursors and has a synergistic effect with RANKL on osteoclast activation. The increased local RANKL expression exhibits chemotactic properties towards circulating osteoclastic precursors resulting in increased osteoclastic activity, bone fracture and ultimately bone destruction. Reduced endothelial expression of nitric oxide synthase (eNOS) in diabetic neuropathy also contributes to this increased osteoclastic activity in the diabetic neuropathic foot
Although the interaction between VC, bone lysis and neuropathy in Charcot osteoarthropathy is highly probable, it has yet to be established why the frequently seen VC in diabetic peripheral neuropathy is bilateral, whereas the occurrence of Charcot joints is more commonly unilateral and is associated with the site of foot trauma and inflammation. Furthermore, RANKL itself has contrasting action on vessels and bone. In the vasculature, upregulated RANKL promotes both osteoclast differentiation of macrophages and osteoblast differentiation of progenitor cells (via binding with RANK expressed by VSMCs) [64, 98], whereas in bone, its effects are mainly limited to osteoclast regulation [86]. Furthermore, the temporal association between VC and bone destruction in Charcot osteoarthropaty is unknown. Although VC is frequently seen in diabetic neuropathy, only a subset of patients develops Charcot joints. Furthermore, it is not clear whether the local production of RANKL and cytokines in the vessels could increase osteoclastic activity and lead to destruction of bone through loss of mineral [86]. Alternatively, release of calcium and minerals could modulate the process of VC in Charcot osteoarthopathy, although evidence to support this hypothesis is sparse. A better understanding of the mechanisms and interaction of VC and bone could contribute to better management of this condition, as the current standard of treatment includes only casting immobilisation during the acute active stage of bone destruction [101]. Although recent studies have highlighted the importance of RANKL and inflammation in the pathogenesis of osteoclast activation and VC in Charcot osteoarthropathy, currently, there is no clinical experience with the use of therapies aimed to inhibit RANKL or TNF-α activation [101].
In addition, emerging evidence suggests that other potential vascular/bone pathways, mediated by neuropathy, may also be involved. Endothelial nitric oxide synthase (eNOS) is the enzyme that generates the vasoprotective molecule nitric oxide which acts to suppress osteoclast activity. In neuropathy, and particularly in Charcot osteoarthropathy, eNOS levels are significantly decreased suggesting endothelial dysfunction could also impact on bone loss [31].
In summary, these studies suggest that enhanced RANKL activity (via neuropathy, inflammation and oxidative stress), as well as other mechanisms, could modulate both VSMCs and osteoclasts and lead to VC and bone destruction. More research is now needed to establish whether VC in diabetic neuropathy identifies subjects “at risk” of developing Charcot osteoarthropathy, or alternatively could neuropathy-related bone destruction itself drive calcium deposition in diabetic arteries.
Conclusions
Bone and vessels are frequently damaged in diabetes and this damage could be modulated by neuropathy. Further studies of the complex relationship between VC and bone damage in the presence of neuropathy are now needed for a better understanding of the mechanisms whereby the diabetic milieu impacts on these processes. Charcot osteoarthropathy, a rare neuropathy-related complication in diabetes, is a useful model of VC and bone destruction, processes mediated via the RANKL/OPG signalling pathway and inflammatory modulators that now requires further investigation with the ultimate aim of devising therapeutics to ameliorate these interrelated and morbid conditions.
References
Proudfoot D, Shanahan CM (2001) Biology of calcification in vascular cells: intima versus media. Herz 26:245–251
London GM (2011) Arterial calcification: cardiovascular function and clinical outcome. Nefrologia 31:644–647
Maldonado N, Kelly-Arnold A, Vengrenyuk Y, Laudier D, Fallon JT, Virmani R, Cardoso L, Weinbaum S (2012) A mechanistic analysis of the role of microcalcifications in atherosclerotic plaque stability: potential implications for plaque rupture. Am J Physiol Heart Circ Physiol 303:H619–H628
Iyemere VP, Proudfoot D, Weissberg PL, Shanahan CM (2006) Vascular smooth muscle cell phenotypic plasticity and the regulation of vascular calcification. J Intern Med 260:192–210
Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM (2011) Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res 109:697–711
Tyson KL, Reynolds JL, McNair R, Zhang Q, Weissberg PL, Shanahan CM (2003) Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arterial calcification. Arterioscler Thromb Vasc Biol 23:489–494
Villa-Bellosta R, Wang X, Millan JL, Dubyak GR, O′Neill WC (2011) Extracellular pyrophosphate metabolism and calcification in vascular smooth muscle. Am J Physiol Heart Circ Physiol 301:H61–H68
Kapustin AN, Davies JD, Reynolds JL et al (2011) Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res 109:e1–e12
Moon J-S, Clark VM, Beabout JW, Swee RG, Dyck PJ (2011) A controlled study of medial arterial calcification of legs: implications for diabetic polyneuropathy. Arch Neurol 68:1290–1294
Everhart JE, Pettitt DJ, Knowler WC, Rose FA, Bennett PH (1988) Medial arterial calcification and its association with mortality and complications of diabetes. Diabetologia 31:16–23
Singh DK, Winocour P, Summerhayes B, Kaniyur S, Viljoen A, Sivakumar G, Farrington K (2012) Prevalence and progression of peripheral vascular calcification in type 2 diabetes subjects with preserved kidney function. Diabetes Res Clin Pract 97:158–165
Lehto S, Niskanen L, Suhonen M, Ronnemaa T, Laakso M (1996) Medial artery calcification. A neglected harbinger of cardiovascular complications in non-insulin-dependent diabetes mellitus. Arterioscler Thromb Vasc Biol 16:978–983
Niskanen L, Siitonen O, Suhonen M, Uusitupa MI (1994) Medial artery calcification predicts cardiovascular mortality in patients with NIDDM. Diabetes Care 17:1252–1256
Mehrotra R, Budoff M, Christenson P, Ipp E, Takasu J, Gupta A, Norris K, Adler S (2004) Determinants of coronary artery calcification in diabetics with and without nephropathy. Kidney Int 66:2022–2031
Hamann C, Kirschner S, Gunther K-P, Hofbauer LC (2012) Bone, sweet bone—osteoporotic fractures in diabetes mellitus. Nat Rev Endocrinol 8:297–305
Bandeira E, Neves AP, Costa C, Bandeira F (2012) Association between vascular calcification and osteoporosis in men with type 2 diabetes. J Clin Densitom 15:55–60
Jeffcoate WJ, Rasmussen LM, Hofbauer LC, Game FL (2009) Medial arterial calcification in diabetes and its relationship to neuropathy. Diabetologia 52:2478–2488
Jeffcoate W (2004) Vascular calcification and osteolysis in diabetic neuropathy-is RANK-L the missing link? Diabetologia 47:1488–1492
Shanahan CM, Cary NRB, Salisbury JR, Proudfoot D, Weissberg PL, Edmonds ME (1999) Medial localization of mineralization-regulating proteins in association with Monckeberg’s sclerosis: evidence for smooth muscle cell-mediated vascular calcification. Circulation 100:2168–2176
Liu Y, Shanahan CM (2011) Signalling pathways and vascular calcification. Front Biosci 16:1302–1314
Marra G, Cotroneo P, Pitocco D, Manto A, Di Leo MAS, Ruotolo V, Caputo S, Giardina B, Ghirlanda G, Santini SA (2002) Early increase of oxidative stress and reduced antioxidant defenses in patients with uncomplicated type 1 diabetes: a case for gender difference. Diabetes Care 25:370–375
Lopes-Virella MF, Baker NL, Hunt KJ, Lachin J, Nathan D, Virella G, Group DER (2011) Oxidized LDL immune complexes and coronary artery calcification in type 1 diabetes. Atherosclerosis 214:462–467
Brown AJ, Jessup W (1999) Oxysterols and atherosclerosis. Atherosclerosis 142:1–28
Liu H, Yuan L, Xu S, Zhang T, Wang K (2004) Cholestane-3beta, 5alpha, 6beta-triol promotes vascular smooth muscle cells calcification. Life Sci 76:533–543
Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, McDonald JM, Chen Y (2008) Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor runx2 by AKT signaling. J Biol Chem 283:15319–15327
Node K, Inoue T (2009) Postprandial hyperglycemia as an etiological factor in vascular failure. Cardiovasc 8:23
Donath MY, Shoelson SE (2011) Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 11:98–107
Alman AC, Kinney GL, Tracy RP, Maahs DM, Hokansen JE, Rewere MJ, Snell-Bergeon JK (2013) Prospective association between inflammatory markers and progression of coronary artery calcification in adults with and without type1 diabetes. Diabetes Care 36(7):1967–1973
Thomsen SB, Rathcke CN, Zerahn B, Vestergaard H (2010) Increased levels of the calcification marker matrix gla protein and the inflammatory markers YKL-40 and CRP in patients with type 2 diabetes and ischemic heart disease. Cardiovasc 9:86
Satoh J, Yagihashi S, Toyota T (2003) The possible role of tumor necrosis factor-alpha in diabetic polyneuropathy. Exp Diabesity Res 4:65–71
La Fontaine J, Harkless LB, Sylvia VL, Carnes D, Heim-Hall J, Jude E (2008) Levels of endothelial nitric oxide synthase and calcitonin gene-related peptide in the Charcot foot: a pilot study. J Foot Ankle Surg 47:424–429
Thompson B, Towler DA (2012) Arterial calcification and bone physiology: role of the bone-vascular axis. Nat Rev Endocrinol 8:529–543
Abedin M, Lim J, Tang TB, Park D, Demer LL, Tintut Y (2006) N-3 fatty acids inhibit vascular calcification via the p38-mitogen-activated protein kinase and peroxisome proliferator-activated receptor-gamma pathways. Circ Res 98:727–729
Shao J-S, Cheng S-L, Sadhu J, Towler DA (2010) Inflammation and the osteogenic regulation of vascular calcification: a review and perspective. Hypertension 55:579–592
Lutgers HL, Graaff R, Links TP, Ubink-Veltmaat LJ, Bilo HJ, Gans RO, Smit AJ (2006) Skin autofluorescence as a noninvasive marker of vascular damage in patients with type 2 diabetes. Diabetes Care 29:2654–2659
Li S-l, Reddy MA, Cai Q, Meng L, Yuan H, Lanting L, Natarajan R (2006) Enhanced proatherogenic responses in macrophages and vascular smooth muscle cells derived from diabetic db/db mice. Diabetes 55:2611–2619
Soro-Paavonen A, Watson AMD, Li J et al (2008) Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes 57:2461–2469
Wang Z, Jiang Y, Liu N, Ren L, Zhu Y, An Y, Chen D (2012) Advanced glycation end-product N-carboxymethyl-lysine accelerates progression of atherosclerotic calcification in diabetes. Atherosclerosis 221:387–396
Suga T, Iso T, Shimizu T, Tanaka T, Yamagishi S-i, Takeuchi M, Imaizumi T, Kurabayashi M (2011) Activation of receptor for advanced glycation end products induces osteogenic differentiation of vascular smooth muscle cells. J Atheroscler Thromb 18:670–683
Tesfaye S, Boulton AJM, Dyck PJ et al (2010) Diabetic neuropathies: update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes Care 33:2285–2293
Edmonds ME, Morrison N, Laws JW, Watkins PJ (1982) Medial arterial calcification and diabetic neuropathy. Br Med J (Clin Res Ed) 284:928–930
Shanahan CM (2007) Inflammation ushers in calcification: a cycle of damage and protection? Circulation 116:2782–2785
Vincent AM, Russell JW, Low P, Feldman EL (2004) Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr Rev 25:612–628
Yorek MA (2003) The role of oxidative stress in diabetic vascular and neural disease. Free Radic Res 37:471–480
Huijberts MSP, Schaper NC, Schalkwijk CG (2008) Advanced glycation end products and diabetic foot disease. Diabetes Metab Res Rev 24(Suppl 1):S19–S24
Figueroa-Romero C, Sadidi M, Feldman EL (2008) Mechanisms of disease: the oxidative stress theory of diabetic neuropathy. Rev Endocr Metab Disord 9:301–314
Herder C, Lankisch M, Ziegler D et al (2009) Subclinical inflammation and diabetic polyneuropathy: MONICA/KORA survey F3 (Augsburg, Germany). Diabetes Care 32:680–682
Yamakawa I, Kojima H, Terashima T et al (2011) Inactivation of TNF-α ameliorates diabetic neuropathy in mice. Am J Physiol Endocrinol Metab 301:E844–E852
Meerwaldt R, Links TP, Graaff R, Hoogenberg K, Lefrandt JD, Baynes JW, Gans ROB, Smit AJ (2005) Increased accumulation of skin advanced glycation end-products precedes and correlates with clinical manifestation of diabetic neuropathy. Diabetologia 48:1637–1644
Vlassara H, Brownlee M, Cerami A (1981) Nonenzymatic glycosylation of peripheral nerve protein in diabetes mellitus. Proc Natl Acad Sci U S A 78:5190–5192
Toth C, Rong LL, Yang C et al (2008) Receptor for advanced glycation end products (RAGEs) and experimental diabetic neuropathy. Diabetes 57:1002–1017
Hak AE, Pols HA, van Hemert AM, Hofman A, Witteman JC (2000) Progression of aortic calcification is associated with metacarpal bone loss during menopause: a population-based longitudinal study. Arterioscler Thromb Vasc Biol 20:1926–1931
Kado DM, Browner WS, Blackwell T, Gore R, Cummings SR (2000) Rate of bone loss is associated with mortality in older women: a prospective study. J Bone Miner Res 15:1974–1980
den Uyl D, Nurmohamed MT, van Tuyl LH, Raterman HG, Lems WF (2011) (Sub)clinical cardiovascular disease is associated with increased bone loss and fracture risk; a systematic review of the association between cardiovascular disease and osteoporosis. Arthritis Res Ther 13(1):R5
Young MJ, Marshall A, Adams JE, Selby PL, Boulton AJ (1995) Osteopenia, neurological dysfunction, and the development of Charcot neuroarthropathy. Diabetes Care 18:34–38
Rix M, Andreassen H, Eskildsen P (1999) Impact of peripheral neuropathy on bone density in patients with type 1 diabetes. Diabetes Care 22:827–831
Schoppet M, Preissner KT, Hofbauer LC (2002) RANK ligand and osteoprotegerin: paracrine regulators of bone metabolism and vascular function. Arterioscler Thromb Vasc Biol 22:549–553
Collin-Osdoby P (2004) Regulation of vascular calcification by osteoclast regulatory factors RANKL and osteoprotegerin. Circ Res 95:1046–1057
Boyce BF, Xing L (2008) Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys 473:139–146
Bucay N, Sarosi I, Dunstan CR et al (1998) Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 12:1260–1268
Simonet WS, Lacey DL, Dunstan CR et al (1997) Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89:309–319
Min H, Morony S, Sarosi I et al (2000) Osteoprotegerin reverses osteoporosis by inhibiting endosteal osteoclasts and prevents vascular calcification by blocking a process resembling osteoclastogenesis. J Exp Med 192:463–474
Dhore CR, Cleutjens JP, Lutgens E, Cleutjens KB, Geusens PP, Kitslaar PJ, Tordoir JH, Spronk HM, Vermeer C, Daemen MJ (2001) Differential expression of bone matrix regulatory proteins in human atherosclerotic plaques. Arterioscler Thromb Vasc Biol 21:1998–2003
Panizo S, Cardus A, Encinas M, Parisi E, Valcheva P, Lopez-Ongil S, Coll B, Fernandez E, Valdivielso JM (2009) RANKL increases vascular smooth muscle cell calcification through a RANK-BMP4-dependent pathway. Circ Res 104:1041–1048
Schoppet M, Kavurma MM, Hofbauer LC, Shanahan CM (2011) Crystallizing nanoparticles derived from vascular smooth muscle cells contain the calcification inhibitor osteoprotegerin. Biochem Biophys Res Commun 407:103–107
Browner WS, Lui LY, Cummings SR (2001) Associations of serum osteoprotegerin levels with diabetes, stroke, bone density, fractures, and mortality in elderly women. J Clin Endocrinol Metab 86:631–637
Anand DV, Lahiri A, Lim E, Hopkins D, Corder R (2006) The relationship between plasma osteoprotegerin levels and coronary artery calcification in uncomplicated type 2 diabetic subjects. J Am Coll Cardiol 47:1850–1857
Reinhard H, Lajer M, Gall M-A, Tarnow L, Parving H-H, Rasmussen LM, Rossing P (2010) Osteoprotegerin and mortality in type 2 diabetic patients. Diabetes Care 33:2561–2566
Aoki A, Murata M, Asano T et al (2013) Association of serum osteoprotegerin with vascular calcification in patients with type 2 diabetes. Cardiovasc 12:11
Pittenger G, Vinik A (2003) Nerve growth factor and diabetic neuropathy. Exp Diabesity Res 4:271–285
Bjurholm A, Kreicbergs A, Schultzberg M, Lerner UH (1992) Neuroendocrine regulation of cyclic AMP formation in osteoblastic cell lines (UMR-106-01, ROS 17/2.8, MC3T3-E1, and Saos-2) and primary bone cells. J Bone Miner Res 7:1011–1019
Bernard GW, Shih C (1990) The osteogenic stimulating effect of neuroactive calcitonin gene-related peptide. Peptides 11:625–632
Santavirta S, Konttinen YT, Nordstrom D, Makela A, Sorsa T, Hukkanen M, Rokkanen P (1992) Immunologic studies of nonunited fractures. Acta Orthop Scand 63:579–586
Wang L, Shi X, Zhao R, Halloran BP, Clark DJ, Jacobs CR, Kingery WS (2010) Calcitonin-gene-related peptide stimulates stromal cell osteogenic differentiation and inhibits RANKL induced NF-kappaB activation, osteoclastogenesis and bone resorption. Bone 46:1369–1379
Connat JL, Busseuil D, Gambert S et al (2001) Modification of the rat aortic wall during ageing; possible relation with decrease of peptidergic innervation. Anat Embryol (Berl) 204:455–468
Sharma A, Scammell BE, Fairbairn KJ, Seagrave MJ, Game FL, Jeffcoate WJ (2010) Prevalence of calcification in the pedal arteries in diabetes complicated by foot disease. Diabetes Care 33:e66
Petrova NL, Edmonds ME (2008) Charcot neuro-osteoarthropathy—current standards. Diabetes Metab Res Rev 24(Suppl 1):S58–S61
Sinha S, Munichoodappa CS, Kozak GP (1972) Neuro-arthropathy (Charcot joints) in diabetes mellitus (clinical study of 101 cases). Medicine (Baltimore) 51:191–210
Cavanagh PR, Young MJ, Adams JE, Vickers KL, Boulton AJ (1994) Radiographic abnormalities in the feet of patients with diabetic neuropathy. Diabetes Care 17:201–209
Sinacore DR, Withrington NC (1999) Recognition and management of acute neuropathic (Charcot) arthropathies of the foot and ankle. J Orthop Sports Phys Ther 29:736–746
van Baal J, Hubbard R, Game F, Jeffcoate W (2010) Mortality associated with acute Charcot foot and neuropathic foot ulceration. Diabetes Care 33:1086–1089
Gazis A, Pound N, Macfarlane R, Treece K, Game F, Jeffcoate W (2004) Mortality in patients with diabetic neuropathic osteoarthropathy (Charcot foot). Diabet Med 21:1243–1246
Edmonds MEKBA, Saldana Chaparro R, Dew T, Stock S, Moniz C, Petrova NL (2009) Serum levels of osteoprotegerin are raised in diabetic peripheral neuropathy and are significantly correlated with peripheral arterial calcification. Diabetologia 52:S97
Ndip A, Williams A, Jude EB, Serracino-Inglott F, Richardson S, Smyth JV, Boulton AJM, Alexander MY (2011) The RANKL/RANK/OPG signaling pathway mediates medial arterial calcification in diabetic Charcot neuroarthropathy. Diabetes 60:2187–2196
Mabilleau G, Petrova NL, Edmonds ME, Sabokbar A (2008) Increased osteoclastic activity in acute Charcot’s osteoarthropathy: the role of receptor activator of nuclear factor-kappaB ligand. Diabetologia 51:1035–1040
Alexander MY (2009) RANKL links arterial calcification with osteolysis. Circ Res 104:1032–1034
Stevens MJ, Edmonds ME, Foster AV, Watkins PJ (1992) Selective neuropathy and preserved vascular responses in the diabetic Charcot foot. Diabetologia 35:148–154
Rogers LC, Frykberg RG, Armstrong DG et al (2011) The Charcot foot in diabetes. Diabetes Care 34:2123–2129
Edmonds M, Dew T, Musto R, Thomson S, Sherwood R, Moniz C, Petrova NL (2008) Serum proinflammatory cytokines TNF-alpha and IL-6 are raised in acute Charcot osteoarthropathy and correlate with a marker of bone resorption. Diabetologia 51(Suppl 1):S510
Petrova NL, Foster AVM, Edmonds ME (2005) Calcaneal bone mineral density in patients with Charcot neuropathic osteoarthropathy: differences between type 1 and type 2 diabetes. Diabet Med 22:756–761
Petrova NL, Edmonds ME (2010) A prospective study of calcaneal bone mineral density in acute Charcot osteoarthropathy. Diabetes Care 33:2254–2256
Petrova NL, Shanahan C, Edmonds M (2011) The proinflammatory cytokines TNF-α and IL-6 modulate RANKL-mediated osteoclastic resorption in vitro in patients with acute Charcot osteoarthropathy. Diabetologia 54(Suppl 1):S11
Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL (2000) TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Investig 106:1481–1488
Baumhauer JF, O′Keefe RJ, Schon LC, Pinzur MS (2006) Cytokine-induced osteoclastic bone resorption in charcot arthropathy: an immunohistochemical study. Foot Ankle Int 27:797–800
Al-Aly Z (2007) Medial vascular calcification in diabetes mellitus and chronic kidney disease: the role of inflammation. Cardiovasc Hematol Disord Drug Targets 7:1–6
Jeffcoate WJ, Game F, Cavanagh PR (2005) The role of proinflammatory cytokines in the cause of neuropathic osteoarthropathy (acute Charcot foot) in diabetes. Lancet 366:2058–2061
Sun YBC, Yuan K, Chen J, Mao X, Heath JM, Javed A, Zhang K, Anderson PG, Chen Y (2012) Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ Res 111:543–552
Byon CH, Sun Y, Chen J et al (2011) Runx2-upregulated receptor activator of nuclear factor kB ligand in calcifying smooth muscle cells promotes migration and osteoclastic differentiation of macrophages. Arterioscler Thromb Vasc Biol 31:1387–1396
Mabilleau G, Petrova N, Edmonds ME, Sabokbar A (2011) Number of circulating CD14-positive cells and the serum levels of TNF-α are raised in acute charcot foot. Diabetes Care 34:e33
Kon T, Cho TJ, Aizawa T, Yamazaki M, Nooh N, Graves D, Gerstenfeld LC, Einhorn TA (2001) Expression of osteoprotegerin, receptor activator of NF-kappaB ligand (osteoprotegerin ligand) and related proinflammatory cytokines during fracture healing. J Bone Miner Res 16:1004–1014
Petrova NL, Edmonds M (2013) Medical management of Charcot arthropathy. Diab Obes Met 15:193–197
Acknowledgments
We thank Professor Michael Edmonds for critical reading of the manuscript and useful comments.
Funding support
The British Heart Foundation and Diabetes Research and Wellness Foundation supported this study.
Conflict of interests
None
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Petrova, N.L., Shanahan, C.M. Neuropathy and the vascular-bone axis in diabetes: lessons from Charcot osteoarthropathy. Osteoporos Int 25, 1197–1207 (2014). https://doi.org/10.1007/s00198-013-2511-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-013-2511-6