
Abstract
Summary
This study aimed to delineate the mechanism involved in type 1 diabetes-induced bone loss. The results revealed the alteration of vitamin D metabolic enzyme expression and the downregulation of renal calcium transporter abundance in type 1 diabetic mice.
Introduction
The purpose of this study was to investigate the changes of the expression of vitamin D metabolic enzymes and transcellular calcium-transporting proteins in kidneys from mice with experimentally induced diabetes.
Methods
Male DBA/2J mice were injected with either vehicle (control) or streptozotocin (STZ) daily for five consecutive days. Bone mineral density was measured by peripheral quantitative computerized tomography, and bone histomorphology was analyzed by Safranin O staining. Real-time PCR and Western blotting were applied to determine the expression of target genes and proteins.
Results
Type 1 diabetes produced high urinary calcium excretion and loss of trabecular bone measured at the proximal metaphysis of the tibia and the distal femur. Bone loss was associated with deterioration of trabecular bone microstructure. Quantified PCR results showed that mRNA expression level in the kidney of diabetic mice for 25-hydroxyvitamin D-24-hydroxylase was downregulated at week 10, while those for 25-hydroxyvitamin D-1α-hydroxylase were upregulated at week 20. In addition, mRNA expression levels for renal transient receptor potential V6, plasma membrane Ca-ATPase (PMCA)1b, and vitamin D receptor (VDR) genes were decreased in STZ-treated mice. Western blot analysis showed that protein expression of PMCA1b and VDR was significantly decreased in kidneys from STZ-treated mice compared to that of controls.
Conclusions
The limitation in this study is the lack of vitamin D, parathyroid hormone, and phosphorus levels in serum. However, the present study supports the conclusion that the underlying mechanism contributing to type 1 diabetes-associated bone loss may be alterations of vitamin D metabolic enzyme expression and associated decreases in expression of renal calcium transporters.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The coexistence of diabetes mellitus and altered bone and mineral metabolism has been established by a number of investigations in diabetic patients [1, 2]. Patients with type 1 diabetes, also called insulin-dependent diabetes mellitus, are consistently observed to have low bone mineral density [3–6], and emerging evidence suggests that patients with type 1 diabetes are at high risk for osteoporotic fractures due to low bone mass and bone fragility [7, 8]. The relationship between diabetes and osteoporosis is complex, and the fundamental mechanisms contributing to the development of osteoporosis in diabetes are unclear despite extensive investigation [3].
An experimental animal model, where streptozotocin (STZ) is used to induce diabetes in rodents, has been well accepted as a model for type 1 diabetes in humans [9]. Using this model, pharmacological studies have been performed to investigate the preventive and therapeutic effects of different treatments on various bone phenotypes [10–17]. However, these studies have only evaluated the direct effects of different therapies on bone phenotypes, such as bone mass, biomechanical behavior, and bone structure, and have revealed little about the mechanisms contributing to these altered phenotypes. A better understanding of the fundamental pathogenic mechanisms contributing to type 1 diabetes-associated bone defects may help identify additional potential therapeutic targets and provide a rationale for developing individualized treatments for diabetic patients with osteoporosis.
The exact mechanisms accounting for type 1 diabetes-associated bone defects are not known. Some existing data suggest that the bone defect in type 1 diabetes is attributable to deficient bone formation because of an absence of the anabolic effects of insulin and amylin [4, 6]. Other published data, however, suggest that diabetes-associated bone defects are caused by increased bone resorption. This has been demonstrated by enhanced expression of cathepsin K and tartrate-resistant acid phosphatase, which have the capacity to break down bone matrix [18]. The present study was performed to examine the mechanisms by which other factors contribute to the development and progression of type 1 diabetes-associated bone defects.
Calcium homeostasis plays a major role in regulating bone metabolism in vivo. Calcium mobilization and deposition in skeleton, through the dynamic processes of bone resorption and bone formation, respectively, are actively involved in maintaining calcium homeostasis [19, 20]. Therefore, we hypothesize that systemic factors, which are capable of regulating calcium metabolism and balance, may influence the development of type 1 diabetes-associated bone defects. Transient receptor potential V6 (TRPV6), calcium-binding protein-9k (CaBP-9k), and calcium-binding protein-28k (CaBP-28k) in kidney are responsible for calcium reabsorption in the renal tubules by apical Ca influx and intracellular diffusion [21, 22]. A calcium pump (plasma membrane Ca-ATPase, PMCA), located on the basolateral membrane, has a role in the extrusion of calcium [23]. The active and hormonal form of vitamin D, 1,25(OH)2D3, contributes to the stimulation of intestinal Ca absorption, the maintenance of Ca balance by binding to the vitamin D receptor (VDR) and directly stimulating bone metabolism, and also by altering the rate of transcription of target genes. Renal 25-hydroxyvitamin D-1α-hydroxylase (1-OHase) and 25-hydroxyvitamin D-24-hydroxylase (24-OHase) are enzymes that catalyze the production and inactivation of circulating 1,25(OH)2D3, respectively [24, 25].
In this study, our aim was to investigate the role of the renal handling on calcium reabsorption and the renal vitamin D metabolic enzymes in type 1 diabetes-associated bone defects. Mice rendered diabetic after either 10 or 20 weeks of STZ injection were used in the present study. Blood glucose and urine albumin were measured to determine diabetic status during the experimental period, and the expression of calcium-transporting protein and vitamin D metabolic enzymes in kidney was analyzed by real-time PCR and Western blotting.
Materials and methods
Mouse model and treatment
The animal study protocol was approved by the Institutional Animal Care and Use Committee at the University of Shanghai for Science and Technology. Ten-week-old male DBA/2J mice (Jackson Laboratory, Bar Harbor, ME, USA), which are susceptible to hyperglycemia-induced renal injury [26], were allowed to acclimate to their environment for 1 week before injection. Mice were fasted for 4 h and then given an intraperitoneal injection of freshly prepared STZ (40 mg/kg dissolved in 10 mM citrate buffer, pH 4.2, n = 20) or vehicle (control mice, n = 20) daily for five consecutive days. Ten mice in each group were sacrificed after week 10, and the remainders were sacrificed after 20 weeks. Blood glucose levels were monitored with CONTOUR blood glucose monitoring system (Bayer) at 0, 10, and 20 weeks post-STZ treatment. Spot urine was collected at 0, 5, 10, 15, and 20 weeks after completion of STZ injection. After sacrifice, serum and kidneys were immediately harvested for a variety of biochemical and molecular analyses.
Serum and urine chemistries
Urinary albumin and creatinine levels were determined using a mouse albumin ELISA quantitation kit (BETHYL, Montgomery, TX, USA) and creatinine assay kit (Hayward, CA, USA), respectively. Total calcium in serum and urine was measured by standard colorimetric method according to the manual of the commercial kit purchased from Stanbio Laboratory (Boerne, TX, USA).
Bone histology
The tibias were fixed in 4% formaldehyde/phosphate-buffered saline (pH 7.2), decalcified in 0.5 M EDTA (pH 8.0), and embedded in paraffin by standard histological procedures. Sections of 4 μm were cut and stained with Safranin O (Sigma), followed by counter-staining with hematoxylin, to visualize proteoglycans.
Peripheral quantitative computerized tomography
Trabecular bone mineral density of femoral end and tibial head was measured by peripheral quantitative computerized tomography (pQCT) with an XCT2000 machine (Norland Stratec Medizintechnik GmbH, Birkenfeld, Germany) as described previously [24].
Real-time quantitative PCR
Total RNA from kidneys were isolated using TRIzol reagents (Invitrogen, Carlsbad, CA, USA). First-strand cDNAs were synthesized from 4 μg of total RNA in 20 μl reaction using SuperScript First-Strand Synthesis System (Invitrogen). The first-strand cDNAs served as the template for the regular PCR performed using a Bio-Rad DNA Engine (Bio-Rad). Real-time PCR was performed using SYBR Green Supermix (Bio-Rad Laboratory, USA) as described previously [24]. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal control was used to normalize the data to determine the relative expression of the target genes. The PCR primers used in this study were as described in Table 1.
Western blot
Kidneys were homogenized in Laemmli buffer (Boston Bioproducts, Worcester, MA, USA), followed by 5 min boiling and centrifugation to obtain the supernatant. Protein concentrations were determined using a Bio-Rad Protein Assay kit (Bio-Rad, Hercules, CA, USA). Proteins were separated by SDS-PAGE and electrotransferred onto polyvinylidene difluoride membranes (Immobilon-P, Millipore, MA, USA). Immunoblotting was performed after blocking nonspecific binding on the membrane with 5% nonfat milk in TBST. The membranes were incubated with the following dilutions of primary antibodies: 1:1,000 rabbit anti-calbindin D-9k or anti-plasma membrane Ca2+-ATPase 1b (Swant, Bellinzona, Switzerland), 1:500 rabbit anti-rat VDR (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and 1:2,000 mouse anti-β-actin (Sigma-Aldrich). Membranes were then incubated with peroxidase-conjugated anti-rabbit (1:3,000, Santa Cruz Biotechnology) or mouse IgG (1:3,000, Sigma-Aldrich), which were detected with the ECL Western Blotting Detection Reagents (GE Healthcare, UK).
Statistical analysis
Data values were presented as means ± SEM. Statistical comparisons were made using Student’s t test, with P < 0.05 being considered significant.
Results
Blood glucose level and urinary albumin content
As shown in Table 2, blood glucose levels in the diabetic group rose to >600 mg/dl after 10 weeks of STZ treatment, and the same results were observed after 20 weeks of STZ treatment. As expected, STZ-treated diabetic mice developed time-dependent progressive albuminuria (Table 2), and the urinary albumin-to-creatinine ratio increased more than fourfold (P < 0.001) and sixfold (P < 0.001) over normal controls at weeks 10 and 20, respectively.
Calcium content in serum and urine
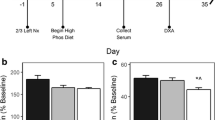
Serum Ca levels did not differ significantly between the normal control and diabetes groups at either week 10 or week 20 (Table 2). In contrast, urinary Ca excretion was significantly higher in the diabetes group than in the control group at all points tested between weeks 5 and 20 (P < 0.05; Fig. 1) with the maximal difference at week 10 (P < 0.01).

Urinary calcium excretion of control mice and diabetic mice at weeks 0, 5, 10, 15, and 20 after STZ treatment. The level of urinary Ca is corrected by the level of urine creatinine (Cr). Data are expressed as mean ± SEM, n = 10. *P < 0.05; **P < 0.01 vs. control group
Bone properties
Histological analyses revealed bone abnormalities in diabetic mice at week 20 post-STZ treatment (Fig. 2a, b). These abnormalities were characterized by a marked expansion of the hypertrophic chondrocyte zone (arrow with solid line) in the proximal metaphysis of tibias. Immature and poorly mineralized woven bones (arrow with dash line) were present in the trabecular bone of STZ-treated mice, indicating impaired mineralization of newly formed bones. pQCT analysis (Fig. 2c) showed that compared to control mice, diabetic mice had a significant decrease in bone mineral density of trabecular bone measured at the proximal metaphysis of the tibia (P < 0.01) and the distal femur (P < 0.05).

Properties of bone in control (a) and diabetic mice (b) 20 weeks after STZ injection. a, b Safranin O staining of the proximal metaphysis of the tibia. c Bone mineral density of the distal metaphysis of the femur and the proximal metaphysis of the tibia. Data are expressed as mean ± SEM, n = 10. *P < 0.05; **P < 0.01 vs. control group
Gene expression of 1α- and 24-vitamin D hydroxylase in kidney
The expression of renal 1-OHase and 24-OHase by regular PCR analysis is shown in Fig. 3a. Quantitative data generated using real-time PCR showed that, compared to controls, the expression of 24-OHase mRNA in kidney of diabetic mice was significantly and markedly decreased at week 10 (P < 0.01; Fig. 3b), but not at week 20 (Fig. 3c). In contrast, the expression of 1-OHase mRNA in kidney of diabetic mice was significantly and markedly increased, compared to controls, at week 20 (P < 0.01; Fig. 3c), but not at week 10 (Fig. 3b).

Gene expression of vitamin D hydroxylase in kidneys of control and diabetic mice. a RT-PCR determination of 1-OHase and 24-OHase. b, c Real-time PCR quantification of 1-OHase and 24-OHase at week 10 (b) and week 20 (c) after STZ treatment. Data are expressed as mean ± SEM, n = 10. **P < 0.01 vs. control group
Gene expression of CaBP-9k, CaBP-28k, TRPV6, PMCA1b, and VDR in kidney
There was no statistically significant difference between the STZ and control groups in mRNA levels for calcium-binding proteins in kidney, including CaBP-9k and CaBP-28k, at either 10 or 20 weeks. In contrast, STZ-injected mice showed the downregulation of the renal TRPV6 gene at week 20 (P < 0.01; Fig. 4b), but not at week 10 (Fig. 4a). mRNA levels for both PMCA1b and vitamin D receptor in kidney were significantly reduced at week 10 (P < 0.01) and week 20 (P < 0.01) in mice subjected to STZ treatment, compared to controls.

Expression of transcellular calcium channels and vitamin D receptor (VDR) determined by real-time PCR in kidney of control mice and diabetes mice at week 10 (a) and week 20 (b) after STZ treatment. Data are expressed as mean ± SEM, n = 10. **P < 0.01 vs. control group
Protein expression of CaBP-9k, PMCA1b, and VDR in kidney
Consistent with the results of gene expression studies, STZ treatment did not change the level of expression of CaBP-9k protein in the kidney compared to controls. Also consistent with the results of gene expression studies, treatment with STZ dramatically downregulated the level of expression of renal PMCA1b and VDR proteins at both week 10 (P < 0.001; Fig. 5a, c) and week 20 (P < 0.05; Fig. 5b, c), compared to controls.

Expression of transcellular calcium channels and vitamin D receptor (VDR) determined by Western blotting in kidney of control mice and diabetes mice at week 10 (a) and week 20 (b) after STZ treatment. c Densitometric quantification of the target proteins. Data are expressed as mean ± SEM, n = 10. *P < 0.05; **P < 0.01; ***P < 0.001 vs. control group
Discussion
STZ-injected mice showed the typical features of type I diabetes in this study, manifested as high serum glucose levels and increased urinary albumin excretion. The development of proteinuria in this study, which is the pathogenic hallmark of diabetic nephropathy, demonstrated the progression of diabetic renal injury. Like diabetic nephropathy, diabetes-induced bone loss is also a chronic disorder. This long-term study was of sufficient duration (20 weeks) to permit the development of prominent bone disorders so that the potential mechanisms underlying these changes could be better evaluated. The present study demonstrated that type 1 diabetes causes trabecular osteopenia in mice, which is consistent with recent findings for type 1 diabetes in rats [27].
Calcium excretion in urine was consistently much higher in diabetic mice than that in normal controls during the experimental period, consistent with findings in diabetic patients [28] and STZ-induced diabetic rats [29]. The current findings suggest that the increase of urinary calcium loss is one of the potential pathogenesis of osteoporosis in type 1 diabetes. It has been shown that calcium intake has a protective role in bone mineral density in patients with diabetes [30]. Consequently, further study addressing the pharmacological and therapeutic effects of calcium supplementation on type 1 diabetes-induced bone loss is probably warranted.
Although mRNA expression of renal vitamin D metabolizing hydroxylases was altered following sustained hyperglycemia in STZ-induced type 1 diabetic mice, the time response and direction of these alterations differed. Gene expression of 1-OHase, which catalyzes the production of circulating 1,25(OH)2D3, was significantly upregulated 20 weeks after STZ treatment but did not differ from controls 10 weeks after STZ treatment. In contrast, gene expression of 24-OHase, which catalyzes the inactivation of circulating 1,25(OH)2D3, was significantly downregulated 10 weeks after STZ treatment but did not differ from controls 20 weeks after treatment. The time-dependent changes of these two hydroxylases, in response to hyperglycemia, have not previously been reported.
Serum 25-hydroxycalciferol vitamin D levels have been characterized as deficient or insufficient in individuals with diabetes [31, 32], and there is a growing body of evidence in humans [33, 34] and animals [35, 36] for the important protective role of vitamin D in diabetes nephropathy. More recently, important immunomodulatory effects have been attributed to vitamin D [37]. Vitamin D deficiency affects immune system behavior in different animal models of immune-mediated disorders, such as type 1 diabetes, and can predispose individuals to type 1 diabetes [38].
In the current study, we report significant downregulation of the VDR in the kidney of type 1 diabetic mice, both at mRNA and protein expression levels. Importantly, an earlier study reported that the distribution of VDR genotype frequencies differed significantly between type 1 diabetic patients and nondiabetic controls [39]. Collectively, these findings strongly support the conclusion that disturbances in expression of the VDR are associated with type 1 diabetes and that these alterations in expression may be affected by genetic polymorphisms within the population.
TRPV6 has been identified as a calcium channel that finely tunes the final urinary calcium concentration [40] and is expressed in both the distal convoluted tubule (DCT) and the terminal nephron (TN). In our study, long-term diabetes-induced downregulation of the TRPV6 gene, suggesting that the defect of calcium entry into the DCT and TN contributes to diabetes-associated renal calcium loss at the terminal stages of renal failure. Intracellular calcium-binding proteins are expressed in the DCT, connecting tubules, and collecting ducts of the kidney [41]. Our findings that mRNA expression levels for renal CaBP-9k and CaBP-28k were not altered in diabetes are consistent with a previous report in which diabetic rats were studied [42]. Interestingly, we demonstrated that both short-term and long-term hyperglycemia resulted in a significant reduction of mRNA and protein of the basolateral calcium efflux protein PMCA1b. These data suggest that PMCA1b plays a functional role in the pathogenesis of renal calcium loss during the development of type 1 diabetes. Since vitamin D is an important positive regulator for calcium-transporting proteins [29, 43], alterations of vitamin D metabolic enzymes expression would appear to be critical to changes in renal calcium-transporting function.
Recent studies have reported histopathologic changes occurring in renal tubules of kidneys from type 1 diabetic mice including fibrosis, inflammation, and epithelial–mesenchymal transition [35, 36, 44]. The present study revealed alterations of vitamin D metabolic enzymes expression and calcium-transporting proteins expression in kidneys from mice with type 1 diabetes, and it is reasonable to hypothesize that these molecular changes contribute to the morphologic changes described above. Thus, this study provides significant insight into the pathogenesis of osteoporosis and renal damage associated with type 1 diabetes.
The limitation in this study is the lack of vitamin D (25-hydroxyvitamin D and 1,25-dihydroxyvitamin D), parathyroid hormone, and phosphorus levels in serum. Although 24-hydroxylase and 1-hydroxylase mRNA expression were measured in kidney, serum levels of the metabolites and calciotropic hormones would provide adequate supports to confirm the alteration of vitamin D metabolism during the development of bone loss in type 1 diabetes, and these queries would be addressed in our further experiment.
In conclusion, we showed the loss of trabecular bone mass and the destruction of trabecular bone microstructure in STZ-induced type 1 diabetic mice. Our data suggest that the underlying mechanism contributing to type 1 diabetes-associated bone loss might be altered vitamin D metabolic enzymes expression and downregulated expression of renal calcium-transporting proteins, leading to renal calcium wasting. Further studies will be required to investigate the potential prophylactic or therapeutic effect of dietary vitamin D and calcium fortification on type 1 diabetes-induced osteoporosis.
References
Tao B, Liu JM, Zhao HY, Sun LH, Wang WQ, Li XY, Ning G (2008) Differences between measurements of bone mineral densities by quantitative ultrasound and dual-energy X-ray absorptiometry in type 2 diabetic postmenopausal women. J Clin Endocrinol Metab 93:1670–1675
Schwartz AV, Sellmeyer DE (2007) Diabetes, fracture, and bone fragility. Curr Osteoporos Rep 5:105–111
Räkel A, Sheehy O, Rahme E, LeLorier J (2008) Osteoporosis among patients with type 1 and type 2 diabetes. Diab Metab 34:193–205
McCabe LR (2007) Understanding the pathology and mechanisms of type I diabetic bone loss. J Cell Biochem 102:1343–1357
Martin LM, McCabe LR (2007) Type I diabetic bone phenotype is location but not gender dependent. Histochem Cell Biol 128:125–133
Hofbauer LC, Brueck CC, Singh SK, Dobnig H (2007) Osteoporosis in patients with diabetes mellitus. J Bone Miner Res 22:1317–1328
Saller A, Maggi S, Romanato G, Tonin P, Crepaldi G (2008) Diabetes and osteoporosis. Aging Clin Exp Res 20:280–289
Nickolas TL, Leonard MB, Shane E (2008) Chronic kidney disease and bone fracture: a growing concern. Kidney Int 74:721–731
Szkudelski T (2001) The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol Res 50:536–546
Altan MF, Kanter M, Donmez S, Kartal ME, Buyukbas S (2007) Combination therapy of Nigella sativa and human parathyroid hormone on bone mass, biomechanical behavior and structure in streptozotocin-induced diabetic rats. Acta Histochem 109:304–314
Ogasawara A, Nakajima A, Nakajima F, Goto K, Yamazaki M (2008) Molecular basis for affected cartilage formation and bone union in fracture healing of the streptozotocin-induced diabetic rat. Bone 43:832–839
Yamaguchi M, Uchiyama S, Lai YL (2007) Oral administration of phytocomponent p-hydroxycinnamic acid has a preventive effect on bone loss in streptozotocin-induced diabetic rats. Int J Mol Med 19:803–807
Kanter M, Altan MF, Donmez S, Ocakci A, Kartal ME (2007) The effects of quercetin on bone minerals, biomechanical behavior, and structure in streptozotocin-induced diabetic rats. Cell Biochem Funct 25:747–752
Uchiyama S, Yamaguchi M (2005) Oral administration of beta-cryptoxanthin prevents bone loss in streptozotocin-diabetic rats in vivo. Biol Pharm Bull 28:1766–1769
Suzuki K, Miyakoshi N, Tsuchida T, Kasukawa Y, Sato K, Itoi E (2003) Effects of combined treatment of insulin and human parathyroid hormone(1–34) on cancellous bone mass and structure in streptozotocin-induced diabetic rats. Bone 33:108–114
Yamaguchi M, Uchiyama S (2003) Preventive effect of zinc acexamate administration in streptozotocin-diabetic rats: restoration of bone loss. Int J Mol Med 12:755–761
Motyl KJ, McCabe LR (2009) Leptin treatment prevents type I diabetic marrow adiposity but not bone loss in mice. J Cell Physiol 218:376–384
Hie M, Shimono M, Fujii K, Tsukamoto I (2007) Increased cathepsin K and tartrate-resistant acid phosphatase expression in bone of streptozotocin-induced diabetic rats. Bone 41:1045–1050
Quarles LD (2008) Endocrine functions of bone in mineral metabolism regulation. J Clin Investig 118:3820–3828
Zhang Y, Leung PC, Che CT, Chow HK, Wu CF, Wong MS (2008) Improvement of bone properties and enhancement of mineralization by ethanol extract of Fructus Ligustri Lucidi. Br J Nutr 99:494–502
Zhang Y, Dong XL, Leung PC, Che CT, Wong MS (2008) Fructus ligustri lucidi extract improves calcium balance and modulates the calciotropic hormone level and vitamin D-dependent gene expression in aged ovariectomized rats. Menopause 15:558–565
Zhang Y, Lai WP, Leung PC, Che CT, Wong MS (2008) Improvement of Ca balance by Fructus Ligustri Lucidi extract in aged female rats. Osteoporos Int 19:235–242
Strehler EE, Caride AJ, Filoteo AG, Xiong Y, Penniston JT, Enyedi A (2007) Plasma membrane Ca2+ ATPases as dynamic regulators of cellular calcium handling. Ann NY Acad Sci 1099:226–236
Zhang Y, Lai WP, Wu CF, Favus MJ, Leung PC, Wong MS (2007) Ovariectomy worsens secondary hyperparathyroidism in mature rats during low Ca diet. Am J Physiol Endocrinol Metab 292:E723–E731
Hendrix I, Anderson P, May B, Morris H (2004) Regulation of gene expression by the CYP27B1 promoter-study of a transgenic mouse model. J Steroid Biochem Mol Biol 89–90:139–142
Qi Z, Fujita H, Jin J, Davis LS, Wang Y, Fogo AB, Breyer MD (2005) Characterization of susceptibility of inbred mouse strains to diabetic nephropathy. Diabetes 54:2628–2637
Silva MJ, Brodt MD, Lynch MA, McKenzie JA, Tanouye KM, Nyman JS, Wang X (2009) Type 1 diabetes in young rats leads to progressive trabecular bone loss, cessation of cortical bone growth, and diminished whole bone strength and fatigue life. J Bone Miner Res 24:1618–1627
Yamaguchi T, Sugimoto T (2008) Calcium homeostasis and osteoporosis in diabetes mellitus and the metabolic syndrome. Clin Calcium 18:904–911
Lee CT, Lien YH, Lai LW, Chen JB, Lin CR, Chen HC (2006) Increased renal calcium and magnesium transporter abundance in streptozotocin-induced diabetes mellitus. Kidney Int 69:1786–1791
de Luis Román DA, Aller R, Perez Castrillon JL, De Luis J, Gonzalez Sagrado M, Izaola O, Romero E, Martín Escudero JC, Herreros V (2004) Effects of dietary intake and life style on bone density in patients with diabetes mellitus type 2. Ann Nutr Metab 48:141–145
Diaz VA, Mainous AG 3rd, Carek PJ, Wessell AM, Everett CJ (2009) The association of vitamin D deficiency and insufficiency with diabetic nephropathy: implications for health disparities. J Am Board Fam Med 22:521–527
Heaney RP (2008) Vitamin D in health and disease. Clin J Am Soc Nephrol 3:1535–1541
Danescu LG, Levy S, Levy J (2009) Vitamin D and diabetes mellitus. Endocr 35:11–17
Mathieu C, Gysemans C, Giulietti A, Bouillon R (2005) Vitamin D and diabetes. Diabetologia 48:1247–1257
Zhang Y, Deb DK, Kong J, Ning G, Wang Y, Li G, Chen Y, Zhang Z, Strugnell S, Sabbagh Y, Arbeeny CM, Li YC (2009) Long-term therapeutic effect of vitamin D analog doxercalciferol on diabetic nephropathy: strong synergism with AT1 receptor antagonist. Am J Physiol Ren Physiol 297:F791–F801
Zhang Z, Zhang Y, Ning G, Kong J, Deb DK, Li YC (2008) Combination therapy with AT1 receptor blocker and vitamin D analog markedly ameliorates diabetic nephropathy. Proc Natl Acad Sci USA 105:15896–15901
Holick MF (2008) Diabetes and the vitamin D connection. Curr Diab Rep 8:393–398
Baeke F, van Etten E, Gysemans C, Overbergh L, Mathieu C (2008) Vitamin D signaling in immune-mediated disorders: evolving insights and therapeutic opportunities. Mol Aspects Med 29:376–387
Panierakis C, Goulielmos G, Mamoulakis D, Petraki E, Papavasiliou E, Galanakis E (2009) Vitamin D receptor gene polymorphisms and susceptibility to type 1 diabetes in Crete, Greece. Clin Immunol 133:276–281
den Dekker E, Hoenderop JG, Nilius B, Bindels RJ (2003) The epithelial calcium channels, TRPV5 & TRPV6: from identification towards regulation. Cell Calcium 33:497–507
Boros S, Bindels RJ, Hoenderop JG (2009) Active Ca(2+) reabsorption in the connecting tubule. Pflugers Arch 458:99–109
Ward DT, Yau SK, Mee AP, Mawer EB, Miller CA, Garland HO, Riccardi D (2001) Functional, molecular, and biochemical characterization of streptozotocin-induced diabetes. J Am Soc Nephrol 12:779–790
Kip SN, Strehler EE (2004) Vitamin D3 upregulates plasma membrane Ca2+-ATPase expression and potentiates apico-basal Ca2+ flux in MDCK cells. Am J Physiol Ren Physiol 286:F363–F369
Reidy K, Susztak K (2009) Epithelial–mesenchymal transition and podocyte loss in diabetic kidney disease. Am J Kidney Dis 54:590–593
Acknowledgments
This project was sponsored by Shanghai Pujiang Program (10PJ1407700) and Innovation Program of Shanghai Municipal Education Commission (11ZZ137) for Yan Zhang. HWD was partially supported by grants from NIH (P50AR055081, R01AG026564, R01AR050496, RC2DE020756, R01AR057049, and R03TW008221) and Franklin D. Dickson/Missouri Endowment.
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, Y., Papasian, C.J. & Deng, HW. Alteration of vitamin D metabolic enzyme expression and calcium transporter abundance in kidney involved in type 1 diabetes-induced bone loss. Osteoporos Int 22, 1781–1788 (2011). https://doi.org/10.1007/s00198-010-1404-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-010-1404-1