
Abstract
Purpose
Nicotine has been reported that it has a dose-dependent effect on matrix mineralization by human bone marrow cells. However, there is no relevant research concerning on chondrogenic differentiation potential of bone marrow stromal stem cells (BMSCs) treated with nicotine in vitro. The aims of the study were to examine the effects of nicotine (0, 10−7, 10−6 and 10−5 M) on the proliferation and chondrogenic differentiation of BMSCs from three healthy donors in vitro.
Methods
BMSCs proliferation was analyzed by CCK8 assay and real-time polymerase chain reaction was used to assay the expression of type II collagen, aggrecan, type I collagen and type X collagen. The proteoglycan content was stained by Alcian blue, and the sulfated glycosaminoglycan (sGAG) content of BMSCs was quantified spectrofluorometrically using dimethylmethylene blue.
Results
The cell viability was not significantly impaired until up to a concentration of 10−5 M nicotine. Nicotine promoted the proliferation and enhanced the expression of type II collagen at the level up to 10−6 M (P < 0.05). The expression of aggrecan was reduced at the concentration of 10−5 M nicotine at day 14 (P < 0.05), and there was no significant difference in aggrecan gene expression at 10−7 and 10−6 M nicotine levels compared to control group (n.s.). Also the fibroblastic and hypertrophic gene expressions were down-regulated in the chondrogenic medium with 10−7–10−5 M nicotine (P < 0.05).
Conclusion
It was implied that local application of nicotine at an appropriate concentration may be a promising approach for enhancing chondrogenic differentiation capacity of BMSCs in cell-based cartilage tissue engineering. Also these results indicate that nicotine maybe a potentially useful drug for the treatment of Osteoarthritis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Articular cartilage has limited capacity to repair damage caused by trauma or disease because of its avascularity and low cellular mitotic activity [13]. The chondral lesions often result in progressive deterioration and eventual osteoarthritis [19]. Surgical procedures are quite unsatisfactory in long-term evaluation and often lead to endoprothetic joint replacement. Other strategies for cartilage repair include transplantation of autograft cartilage or culture-expanded autologous chondrocytes [3]. However, the in vitro expanded chondrocyte experiment results in a progressive loss of specific markers that lead to fibrocartilage formation, devoid of the specific properties of the articular cartilage, as well as dedifferentiation. Schulze et al. [27] found that dedifferentiation makes chondrocytes which were expanded in vitro no longer capable of redifferentiation when they were re-implanted. In recent years, the use of bone marrow stromal stem cells (BMSCs) represents a good alternative to all of these techniques. BMSCs are characterized by a multilineage differentiation potential and a high proliferative capacity without losing their genetic stability [1]. Therefore, BMSCs have considerable potential for use in the therapeutic regeneration of tissues due to their ability to differentiate into various cell lineages, including osteoblasts and chondrocytes [1, 29].
Cigarette smoking has been associated with chronic musculoskeletal conditions, such as low-back pain and degenerative disc disease [6, 15]. Jaiswal et al. [14] using a case-controlled study to look at whether smoking has a deleterious effect in the outcome of autologous chondrocyte implantation for the treatment of full thickness chondral defects of the knee, and they found that patients who smoke have worse pre-operative function and obtain less benefit from this procedure than non-smokers. Recently, Meidinger et al. [20] found that smoking was one of the risk factors with a statistically significant influence on the development of a non-union after medial open-wedge high tibial osteotomy (HTO). Of the more than 400 agents found in cigarette smoke, nicotine is one of the most physiologically active components. Gullahorn et al. [8] reported that nicotine could up-regulate collagen synthesis of human articular chondrocytes in vitro. Also we previously showed that nicotine can promote the proliferation of articular chondrocytes isolated from normal human, and osteoarthritis patients enhance the expression of cartilage-specific type II collagen [34]. Recently, autologous culture-expanded BMSCs have been applied in patients with osteoarthritis [32], and its therapeutic potential for osteoarthritis or the repair of cartilage defect holds out new hope to reverse a painful and debilitating condition suffered by millions of people globally. To our knowledge, there was no relevant research concerning on chondrogenic differentiation potential of BMSCs treated with nicotine. The aims of the study were to examine the effects of various concentrations of nicotine (0, 10−7, 10−6 and 10−5 M) on the proliferation and chondrogenic differentiation of human BMSCs in vitro. We hypothesized that nicotine had a significant positive effect on cell proliferation and chondrogenic differentiation of BMSCs.
Materials and methods
BMSC isolation and culture
Bone marrow was aseptically aspirated from the iliac crest of three human donors (aged 35–56 years, 1 man and 2 women) who were undergoing elective orthopedic procedures. All of the three patients were non-smokers. Full ethical consent was obtained from all patients, and the study was granted ethical approval by the Medical Ethical Committee of the Second Affiliated Hospital, Wenzhou Medical College. Upon collection, bone marrow was washed with growth culture medium (DMEM, Gibco) supplemented with 10% (V/V) fetal bovine serum (FBS, Gibco), 1% (V/V) penicillin and streptomycin (Gibco). The mixture of bone marrow and medium was gently added to the 50% Percoll solution (Sigma) and centrifuged at 3,000 rpm for 30 min. The cell suspension was obtained between the layer of Percoll and the supernatant liquid layer. Cells were plated and then incubated in a humidified atmosphere of 5% CO2 at 37°C. The BMSCs were passaged every 3–4 days using 0.25% (w/v) trypsin–EDTA solution (Gibco), and the third or fourth passage was used in our experiments. To induce chondrogenic differentiation, the BMSCs were cultured in 96-well culture plates with chondrogenic medium (growth culture medium supplemented with 50 μg/ml ascorbate-2-phosphate (Fluka), Premix ITSþ (BD Biosciences), 10 ng/ml TGF-β3 (Sigma) and 100 nM dexamethasone (Sigma)) at an initial density of 1 × 104 cells/cm2. After that they were exposed to varying dosages of nicotine (0, 10−7, 10−6 and 10−5 M) (Sigma) which bracketed the average 10−8–10−7 M seen in chronic smokers [12].
Cytotoxicity studies
Cytotoxicity of nicotine was evaluated by the neutral red assay [2, 30]. Cells that cultured in 96-well plates at 1 × 104 cells per well were treated with increasing concentrations of nicotine (0, 10−7, 10−6 and 10−5 M). After 24-h incubation, the medium was replaced by medium containing neutral red (3-amino-m-dimethylamino-2-methylphenazine hydrochloride) (Sigma) and cells were incubated for 3 h. The neutral red dye was extracted by a bleaching solution containing 50% ethanol, 1% acetic acid and 49% H2O. Absorbance was read at 540 nm (ELX 800, Bio-Tek), and cell viability was calculated as percentage of medium treated control cells.
Cell proliferation by cell counting kit-8 (CCK8) assay
For the cell proliferation assays, BMSCs were cultured in 96-well plates at 1 × 104 cells per well with growth culture medium. Twenty-four hours later, cells were switched to the nicotine containing media. The proliferation of BMSCs was determined by the cell counting kit-8 (Kumamoto, Japan) and measured by microplate reader scanning at 450 nm as previously described elsewhere [35].
Alcian blue staining of BMSCs
Alcian blue staining was used to detect proteoglycan synthesis as an indicator of cartilage matrix production. Four groups of cells were stained: control group (BMSC cultured only with chondrogenic medium), chondrogenic medium with 10−7 M nicotine, chondrogenic medium with 10−6 M nicotine and chondrogenic medium with 10−5 M nicotine. The cells at an initial density of 1 × 104 cells were washed with phosphate-buffered saline (PBS) after 7 and 14 days, fixed for 10 min with 4% paraformaldehyde, stained with 1% alcian blue (Fluka, Ronkonkoma, NY) for 30 min and rinsed with distilled water. The cells were assessed by an inverted microscope.
Glycosaminoglycan quantification
For glycosaminoglycan quantification assays, cells were seeded in 24-well plates at 1 × 104 per well. At indicated time intervals (at 7 and 14 day), cells were collected and digested for 18 h at 60°C using 300 mg/ml papain solution (1 ml/sample papain solution). Sulfated glycosaminoglycan (sGAG) accumulation was quantified spectrofluorometrically using dimethylmethylene blue (DMMB) [5]. Standard curves for DMMB assay was generated using an aqueous solution of chondroitin sulfate C (Sigma), with concentrations ranging from 0 to 25 mg/ml. The DNA content in each sample was quantified using a DNA Quant-iT kit (Molecular Probes, Eugene, OR) according to the manufacturer’s instructions. All absolute sGAG quantities were normalized by their DNA content.
RNA isolation and real-time polymerase chain reaction (real-time PCR)
Real-time PCR was used to detect the expression of several chondrogenic differentiation related marker genes (COL II, aggrecan, COL I, COL X) at 7, 14 and 21 days respectively. Total RNA was extracted using TriZol (Invitrogen) according to the manufacturer’s instructions and quantified. Its concentration was determined spectrophotometrically at 260 nm (HP 8452A Diode Array Spectrophotometer). First strand complementary DNAs (cDNAs) were synthesised from 0.3 mg of the isolated RNA by oligo(deoxythymidine) (oligo(dT)) using DyNamoTM cDNA Synthesis Kit (Fermentas) and used as templates for real-time PCR. The expression of mRNAs was determined quantitatively using DyNamo SYBR1 Green qPCR kit (Takara, Japan).The PCR was performed on a final volume of 25 ml containing 2 ml cDNA, 7.5 pmol of each primer, 1 ml ROX reference dye and 12.5 ml of SYBR Green Master mix (TIANGEN), with ABI Prism 7300 (Applied Biosystems, Foster City, CA, USA). The samples underwent 40 cycles consisting of the following steps: initial denaturation at 95°C for 5 min, followed by a set cycle of denaturation at 94°C for 10 s, different annealing temperatures for each pair of primers (ranging between 53 and 62°C) for 10 s, extension at 72°C for 27 s and a final elongation at 72°C for 5 min. Fold increment of any assayed gene was calculated by normalizing its expression level to that of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene, which was used as an internal control. Each gene analysis was performed in triplicate. Primer’s sequences of the targeted genes were listed in Table 1.
Measuring type II collagen secretion by ELISA
BMSCs were plated at 1 × 104 cells/well in a 96-well plate in chondrogenic medium with different concentrations of nicotine. Four days later, the medium was removed and the cells were washed twice with PBS and lysed in 0.5 ml PBS by three freeze–thaw cycles. Finally, 20–200 μl of cell lysate was analyzed for type II collagen with the use of an ELISA kit (United States ADL’s). The results are indicated as μg/l of protein in each sample. The content of type II collagen was measured at 450 nm as OD values using a spectrophotometer after cooling. The amount of type II collagen secreted by the BMSCs under the chondrogenic medium with different concentrations of nicotine was calculated and compared to control group. The results were rounded off to one decimal place.
Statistical analysis
All experiments reported in this study were performed independently at least three times, and data (expressed as mean ± SD) from a representative experiment are shown. Results were expressed as mean ± SD. Analysis of variance (ANOVA) was used to determine statistical significance; values of P < 0.05 were considered significant.
Results
Potential cytotoxic effects of nicotine
To study the effect of nicotine on cell viability, Cells were treated with increasing concentrations of nicotine (0, 10−7, 10−6 and 10−5 M). As shown in Fig 1, cell viability of BMSCs was not significantly impaired until up to a concentration of 10−5 M nicotine.

Effect of nicotine on BMSCs viability was determined by the neutral red assay, and cell viability is expressed as percentage of control (untreated cells). Cells were treated with increasing concentrations of nicotine for 24 h. Nicotine did not impair cell viability until at the concentration of 10−5 M. *P < 0.05
Cell proliferation
Cell proliferation of BMSCs was analyzed at 1, 4, 7 and 14 days respectively. By the CCK8 assay (Fig. 2), the relative cell number (value of OD) was significantly increased at the concentration of 10−6 M compared to the control in a time-dependent manner (P < 0.05). We also found that the cell proliferation was inhibited significantly in the cultures treated with 10−5 M nicotine compared to the control (P < 0.05).

Effect of nicotine on cell proliferation was analyzed at 1, 4, 7 and 14 days. The relative cell number (value of OD) was significantly increased at the concentration of 10−6 M compared to the control in a time-dependent manner, and it was inhibited significantly in the culture treated with 10−5 M compared to the control. *P < 0.05
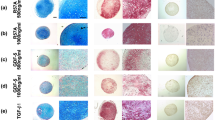
Alcian blue staining
After 7 and 14 days of chondrogenic differentiation, the proteoglycan content was stained by Alcian blue. The BMSCs treated with nicotine (10−5 M) showed slightly reduced alcian blue staining compared to the control group at day 14 (Fig. 3).

Alcian blue staining for proteoglycan accumulation in BMSCs treated with chondrogenic media and nicotine. The BMSCs treated with nicotine (10−5 M) showed slightly reduced alcian blue staining compared to the control group at day 14. All images were taken at 100× magnification
Quantification of sulfated glycosaminoglycans
Because differentiation in Alcian blue staining with or without nicotine treatment could be subtle and non-quantitative, we evaluated total sGAG synthesis, normalized to total DNA content at 7 (Fig. 4a) and 14 (Fig. 4b) days. The sGAG content of the BMSCs that cultured under the chondrogenic medium with 10−7 and 10−6 M nicotine showed a little increased compared with the control group, but no significant differences were observed (n.s).

Quantification of sulfated glycosaminoglycans normalized to total protein content at 7 (a) and 14 days (b) respectively. The sGAG content was reduced in the BMSCs that cultured under the chondrogenic medium with 10−5 M nicotine and were increased at 14 days when compared to the day 7 in all groups except the 10−5 M nicotine group. Values are mean ± SD, *P < 0.05
The mRNA expression of chondrogenic differentiation related marker genes
Real-time PCR was used to detect the expression of several chondrogenic differentiation related marker genes (COL II, aggrecan, COL I, COL X) at 7,14 and 21 days respectively (Fig. 5). Collagen type II was up-regulated in chondrogenic medium with 10−7 and 10−6 M nicotine respectively (Fig. 5a). The expression of aggrecan was reduced at the concentration of 10−5 M nicotine at 14 days (P < 0.05), and there was no significant difference in aggrecan gene expression with other nicotine concentrations (n.s) (Fig. 5b). Both the fibroblastic and hypertrophic gene expressions were down-regulated in the chondrogenic medium with 10−7 to 10−5 M nicotine, as compared to the control group (Fig. 5c, d).

Real-time polymerase chain reaction analysis indicated more chondrogenic markers but less fibroblastic and hypertrophic markers of BMSCs cultured in chondrogenic medium with different concentrations of nicotine than control group (chondrogenic medium without nicotine): a COL II: 10−7 M nicotine (collagen type II: day 7–day 21, P < 0.05) and 10−6 M nicotine (collagen type II: day 7, P < 0.05; day 14–day 21, P < 0.01); b aggrecan; c COL I: from day 7 to 21 (day 7–day 14, P < 0.05; day 21, P < 0.01); d COL X: from day 7 to 21 (day 7, P > 0.05; day 14–21, P < 0.01). Values are mean ± SD, n = 8, *P < 0.05, **P < 0.01
Detecting type II collagen by ELISA
The content of type II collagen produced by BMSCs which were cultured under the chondrogenic medium with 10−7 and 10−6 M nicotine was significantly increased compared with control group (P < 0.05). Also we discovered that nicotine had shown to keep type II collagen expression positive and increasing from 0 to 10−6 M in a time-dependent manner from day 7 to 21 (P < 0.05) (Table 2).
Discussion
The most important finding of the present study was that nicotine could promote the proliferation of human BMSCs from 0 to 10−6 M and enhance the expression of type II collagen at the level up to 10−6 M. Also the study demonstrated that 10−7 and 10−6 M nicotine could decrease the expression of fibrocartilage marker genes (collagen type I) and hypertrophic marker (collagen type X) in human BMSCs.
The concentrations (10−7, 10−6 and 10−5 M) of nicotine that we used in the study were in the range of those (1 μM–10 mM) in previously reported studies [4]. Hukkanen et al. [12] reported that blood concentrations of nicotine obtained from chronic cigarette smokers were in the range of 10−8–10−7 M. However, the concentrations of nicotine in the saliva of chronic snuff users reached from 0.6 to 9.6 mM [10]. Therefore, the effects of high but physiological concentrations of nicotine on the proliferation and chondrogenic differentiation of cultured BMSCs were shown in the present study.
The results of cell proliferation in the study were consistent with the research from Pereira et al. [24], and they found that exposure of human bone marrow (HBM) cells to nicotine resulted in increased cell proliferation at levels up to 0.2 mg/ml, an initial inhibitory effect in cell growth in the presence of 0.3 mg/ml and a dose-dependent deleterious effect at higher levels. The present study also proved that 10−5 M nicotine could decrease the cell viability, and this marked decrease at the concentration of 10−5 M was most likely a result of cytotoxic effects. Schraufstatter et al. [26] also reported that high concentrations of nicotine (10−5 M)-induced hMSC apoptosis, physiological concentrations (10−7–10−6 M) did not interfere with cell survival.
Cartilage extracellular matrix is the place where chondrocytes play the physical effect in vivo. It is the place chondrocytes absorption nutrition and as a carrier to deliver the signal. Metabolic balance of the extracellular matrix is an important part to maintain the normal function of cartilage. Type II collagen and aggrecan were the most widely recognized marker of chondrogenic differentiation [28, 31, 33]. The changes of type II collagen and aggrecan in the quantity, and quality are the direct cause of the loss of the normal articular cartilage biomechanical properties [9]. In this study, we concluded that nicotine has shown to keep type collagen expression positive and increasing from 0 to 10−6 M in a time-dependent manner, and it was reduced at the higher nicotine level of 10−5 M. RNA level for aggrecan was not influenced at the concentration of 10−7 to 10−5 M at 7 days when compared to control group; however, it was decreased at 10−5 M nicotine at 14 days. Additionally, we detected the proteoglycan content by Alcian blue stain after 7 and 14 days. The BMSCs treated with nicotine (10−5 M) showed slightly reduced alcian blue staining compared to the control group at 14 days. Because the differences in Alcian blue staining with or without nicotine treatment could be subtle and non-quantitative, we evaluated total sGAG synthesis, normalized to total DNA quantities. We found that the sGAG content was a little increased at 10−7 and 10−6 M nicotine levels compared with the control group, although there were no significant differences and it was reduced in the BMSCs that cultured under the chondrogenic medium with 10−5 M nicotine. In the literature, there was no study published, related to the interaction of nicotine with chondrogenic differentiation capacity of BMSCs. However, the studies related to the effect of nicotine on chondrocytes had been reported previously. Gullahorn et al. [8] reported that nicotine (2.5, 12.5 and 25 ng/ml) can up-regulate collagen II synthesis of human articular chondrocytes in vitro. Also, it was consistent with what we have reported previously that nicotine can promote the proliferation of articular chondrocytes isolated from normal human and osteoarthritis patients enhance the expression of cartilage specific type II collagen [34]. In contrast, Kawakita et al. [17] discovered that nicotine, from cigarette smoking, acted directly on growth plate chondrocytes to decrease matrix synthesis, suppressed hypertrophic differentiation via alpha7 nAChR, leading to delay skeletal growth. The great variability of results regarding chondrogenic response to nicotine reflected differences in the experimental conditions maybe relate to the differences of cell sources and culture conditions. In Kawakita study, the chondrocytes were harvested from growth plate of human, a major component of endochondral ossification, and cultured in agarose gel culture and alginate bead culture. But in Gullahorn study, the chondrocytes were harvested from human articular and cultured in monolayer in vitro.
In vitro culture of BMSCs eventually leads to hypertrophic chondrocytes, which differentiates toward osteogenic lineages [23]. Gene expression of collagen type X has been reported to be the marker of hypertrophic chondrocytes [22]. In this study, the expression of collagen type I, dominantly expressed in fibrocartilage, was down-regulated in hBMSCs cultured in the chondrogenic medium with 10−7–10−5 M nicotine, as compared to the control group. Moreover, the gene expression level of the hypertrophic marker, collagen type X, was also lower in hBMSCs cultured in the chondrogenic medium with 10−7–10−5 M nicotine at day 7, 14 and 21. Our results indicated that the chondrogenic medium with different concentrations of nicotine inhibited fibrocartilage formation and hypertrophy in hBMSCs.
Ma et al. [18] showed recently that nicotine at low dose had no significant effect on the expression of bone morphogenetic protein-2 and on the radiodensity of bone regeneration. However, the delayed bone healing was detected in the nicotine group by histologic examination, subsequently, there is a potential risk of compromised bone healing in patients taking nicotine medication and maybe also then smoking as well. Nelson et al. [21] discovered that smoking not only reduced body length but also brought ossification retardation in a rat-smoking model. From these results, we speculated that nicotine may suppress the mesenchymal stem cells on osteogenic pathway and guide them instead into a chondrogeneic route. Many studies had proved that elements of the cholinergic system including acetyltransferase, acetylcholinesterase and acetylcholine receptors (AChRs) were expressed in a large array of non-neural cells including epithelial and endothelial cells, mature blood cells, hematopoietic progenitors, osteoblasts and mesenchymal stem cells [7, 11, 25]. And nicotine, as an agonist of the nicotinic acetylcholine receptors (nAChR), maybe play an important part in the process of the proliferation and chondrogenic differentiation of BMSCs. Kalamida et al. [16] reported that a nAChR consisting of a homopentamer of α7 atypically provides gating to calcium ions. Schraufstatter et al. [26] discovered that nicotine evoked transient calcium fluxes and increased intracellular calcium. From these, we speculated that changes in the intracellular levels of Ca2+ ions, following binding of nicotine to its receptor and subsequent activation of voltage-dependent Ca2+ channels, may modulate, at least in part, the effects of nicotine on the proliferation and chondrogenic differentiation of BMSCs.
Articular cartilage has limited capacity to repair damage caused by trauma or disease because of its avascularity and low cellular mitotic activity [13]. The chondral lesions often result in progressive deterioration and eventual osteoarthritis [19]. Articular hyaline cartilage injuries still pose a big challenge to orthopedic surgeons, because these defects have poor capacity for intrinsic repair. In recent years, cell-based articular cartilage tissue engineering studies were focused on using either differentiated chondrocytes or BMSCs for transplantation. However, limited proliferative capacity of BMSCs possesses a major challenge in providing adequate cell numbers for viable transplantations and cartilage repair. Construction of a 3D biomaterial with autologous BMSCs to regenerate defected articular cartilage may become a viable clinical option. In our study, we found nicotine could promote the proliferation of human BMSCs and enhance the expression of type II collagen, these results implied that local application of nicotine at an appropriate concentration with 3D biomaterial such as Poly-lactic-co-gly-colic acid (PLGA) may be a promising approach for enhancing chondrogenic differentiation capacity of BMSCs in cartilage tissue engineering.
The limitation of this study was that we did not test the chondrogenic differentiation capacity of BMSCs with nicotine influence ceases, so further studies upon the influence of nicotine ceases and the molecular mechanism would be performed to bring to light how nicotine affects the process of chondrogenic differentiation of BMSCs. As the results of nicotine on chondrogenic differentiation of BMSCs in the present study were cultured in monolayer and in vitro, a three-dimensional culture needs further validation in vitro and in vivo.
Conclusion
This is the first study to demonstrate that nicotine could promote the proliferation of human BMSCs from 0 to 10−6 M and enhance the expression of type II collagen at the level up to 10−6 M. However, both the cell proliferation and the express of collagen type II were inhibited when treated with 10−5 M nicotine. The expression of aggrecan was reduced at the concentration of 10−5 M nicotine at 14 day, and there was no significant difference in aggrecan gene expression at 10−7 M and 10−6 M nicotine levels compared to control group. Also the reduced expressions of the fibroblastic and hypertrophic gene in the chondrogenic medium with 10−7 to 10−5 M nicotine were demonstrated in the study. These results implied that local application of nicotine at an appropriate concentration may be a promising approach for enhancing chondrogenic differentiation capacity of BMSCs in cartilage tissue engineering.
References
Abdallah BM, Kassem M (2009) The use of mesenchymal (skeletal) stem cells for treatment of degenerative diseases: current status and future perspectives. J Cell Physiol 218(1):9–12
Borenfreund E, Puerner JA (1985) Toxicity determined in vitro by morphological alterations and neutral red absorption. Toxicol Lett 24(2–3):119–124
Brittberg M, Lindahl A, Nilsson A, Ohlsson C, Isaksson O, Peterson L (1994) Treatment of deep cartilage defects in the knee with autologous chondrocyte transplantation. N Engl J Med 331(14):889–895
Fang MA, Frost PJ, Iida-Klein A, Hahn TJ (1991) Effects of nicotine on cellular function in UMR 106–01 osteoblast-like cells. Bone 12(4):283–286
Farndale RW, Buttle DJ, Barrett AJ (1986) Improved quantitation and discrimination of sulphated glycosaminoglycans by use of dimethylmethylene blue. Biochim Biophys Acta 883(2):173–177
Fogelholm RR, Alho AV (2001) Smoking and intervertebral disc degeneration. Med Hypotheses 56(4):537–539
Fujii T, Takada-Takatori Y, Kawashima K (2008) Basic and clinical aspects of non-neuronal acetylcholine: expression of an independent, non-neuronal cholinergic system in lymphocytes and its clinical significance in immunotherapy. J Pharmacol Sci 106(2):186–192
Gullahorn L, Lippiello L, Karpman R (2005) Smoking and osteoarthritis: differential effect of nicotine on human chondrocyte glycosaminoglycan and collagen synthesis. Osteoarthr Cartil 13(10):942–943
Henrotin Y, Addison S, Kraus V, Deberg M (2007) Type II collagen markers in osteoarthritis: what do they indicate? Curr Opin Rheumatol 19(5):444–450
Hoffmann D, Adams JD (1981) Carcinogenic tobacco-specific N-nitrosamines in snuff and in the saliva of snuff dippers. Cancer Res 41(11 Pt 1):4305–4308
Hoogduijn MJ, Cheng A, Genever PG (2009) Functional nicotinic and muscarinic receptors on mesenchymal stem cells. Stem Cells Dev 18(1):103–112
Hukkanen J, Jacob P 3rd, Benowitz NL (2005) Metabolism and disposition kinetics of nicotine. Pharmacol Rev 57(1):79–115
Hunziker EB (2002) Articular cartilage repair: basic science and clinical progress. A review of the current status and prospects. Osteoarthr Cartil 10(6):432–463
Jaiswal PK, Macmull S, Bentley G, Carrington RW, Skinner JA, Briggs TW (2009) Does smoking influence outcome after autologous chondrocyte implantation?A case-controlled study. J Bone Joint Surg Br 91(12):1575–1578
Kaila-Kangas L, Leino-Arjas P, Riihimaki H, Luukkonen R, Kirjonen J (2003) Smoking and overweight as predictors of hospitalization for back disorders. Spine 28(16):1860–1868
Kalamida D, Poulas K, Avramopoulou V, Fostieri E, Lagoumintzis G, Lazaridis K, Sideri A, Zouridakis M, Tzartos SJ (2007) Muscle and neuronal nicotinic acetylcholine receptors. Structure, function and pathogenicity. FEBS J 274(15):3799–3845
Kawakita A, Sato K, Makino H, Ikegami H, Takayama S, Toyama Y, Umezawa A (2008) Nicotine acts on growth plate chondrocytes to delay skeletal growth through the alpha7 neuronal nicotinic acetylcholine receptor. PLoS One 3(12):e3945
Ma L, Sham MH, Zheng LW, Cheung LK (2011) Influence of low-dose nicotine on bone healing. J Trauma 70(6):E117–E121
Mandelbaum BR, Browne JE, Fu F, Micheli L, Mosely JB Jr, Erggelet C, Minas T, Peterson L (1998) Articular cartilage lesions of the knee. Am J Sports Med 26(6):853–861
Meidinger G, Imhoff AB, Paul J, Kirchhoff C, Sauerschnig M, Hinterwimmer S (2011) May smokers and overweight patients be treated with a medial open-wedge HTO? Risk factors for non-union. Knee Surg Sports Traumatol Arthrosc 19(3):333–339
Nelson E, Jodscheit K, Guo Y (1999) Maternal passive smoking during pregnancy and fetal developmental toxicity. Part 1: gross morphological effects. Hum Exp Toxicol 18(4):252–256
Ortega N, Behonick DJ, Werb Z (2004) Matrix remodeling during endochondral ossification. Trends Cell Biol 14(2):86–93
Pelttari K, Winter A, Steck E, Goetzke K, Hennig T, Ochs BG, Aigner T, Richter W (2006) Premature induction of hypertrophy during in vitro chondrogenesis of human mesenchymal stem cells correlates with calcification and vascular invasion after ectopic transplantation in SCID mice. Arthritis Rheum 54(10):3254–3266
Pereira ML, Carvalho JC, Peres F, Fernandes MH (2009) Effect of nicotine in matrix mineralization by human bone marrow and Saos-2 cells cultured on the surface of plasma-sprayed titanium implants. J Biomed Mater Res A 88(1):84–93
Rothem DE, Rothem L, Soudry M, Dahan A, Eliakim R (2009) Nicotine modulates bone metabolism-associated gene expression in osteoblast cells. J Bone Miner Metab 27(5):555–561
Schraufstatter IU, DiScipio RG, Khaldoyanidi SK (2009) Alpha 7 subunit of nAChR regulates migration of human mesenchymal stem cells. J Stem Cells 4(4):203–215
Schulze-Tanzil G, de Souza P, Villegas Castrejon H, John T, Merker HJ, Scheid A, Shakibaei M (2002) Redifferentiation of dedifferentiated human chondrocytes in high-density cultures. Cell Tissue Res 308(3):371–379
Solchaga LA, Penick K, Goldberg VM, Caplan AI, Welter JF (2010) Fibroblast growth factor-2 enhances proliferation and delays loss of chondrogenic potential in human adult bone-marrow-derived mesenchymal stem cells. Tissue Eng Part A 16(3):1009–1019
Tuan RS, Boland G, Tuli R (2003) Adult mesenchymal stem cells and cell-based tissue engineering. Arthritis Res Ther 5(1):32–45
Valacchi G, Rimbach G, Saliou C, Weber SU, Packer L (2001) Effect of benzoyl peroxide on antioxidant status, NF-kappa B activity and interleukin-1 alpha gene expression in human keratinocytes. Toxicology 165(2–3):225–234
Vasan NS, Lash JW (1979) Monomeric and aggregate proteoglycans in the chondrogenic differentiation of embryonic chick limb buds. J Embryol Exp Morphol 49:47–59
Wakitani S, Goto T, Pineda SJ, Young RG, Mansour JM, Caplan AI, Goldberg VM (1994) Mesenchymal cell-based repair of large, full-thickness defects of articular cartilage. J Bone Joint Surg Am 76(4):579–592
Wu SC, Chang JK, Wang CK, Wang GJ, Ho ML (2010) Enhancement of chondrogenesis of human adipose derived stem cells in a hyaluronan-enriched microenvironment. Biomaterials 31(4):631–640
Ying X, Cheng S, Shen Y, Cheng X, An Rompis F, Wang W, Lin Z, Chen Q, Zhang W, Kou D, Peng L, Tian XQ, Lu CZ (2012) Nicotine promotes proliferation and collagen synthesis of chondrocytes isolated from normal human and osteoarthritis patients. Mol Cell Biochem 359(1–2):263–269
Ying X, Cheng S, Wang W, Lin Z, Chen Q, Zhang W, Kou D, Shen Y, Cheng X, Peng L, Zi Xu H, Zhu Lu C (2011) Effect of lactoferrin on osteogenic differentiation of human adipose stem cells. Int Orthop. doi:10.1007/s00264-011-1303-x
Acknowledgments
The authors thank all the staff in the Laboratory of Orthopaedic Research Institute and Scientific Research Center of Second Affiliated Hospital of Wenzhou Medical College. This work was supported by grants from the National Natural Science Foundation of China (31010121), Hainan Provincial Health Department (2011-34), Medicines and Health Research fund of Zhejiang (2007B147), Zhejiang Science and Technology Project (2010C34G2090013), Wenzhou Technology Project (Y20090288), Wenzhou Technology Project (Y20070133).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Ying, X., Zhang, W., Cheng, S. et al. Nicotine-induced chondrogenic differentiation of human bone marrow stromal cells in vitro. Knee Surg Sports Traumatol Arthrosc 20, 2329–2336 (2012). https://doi.org/10.1007/s00167-012-1890-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00167-012-1890-0