
Abstract
Purpose
Both anaemia and allogenic red blood cell transfusion are common and potentially harmful in patients admitted to the intensive care unit. Whilst intravenous iron may decrease anaemia and RBC transfusion requirement, the safety and efficacy of administering iron intravenously to critically ill patients is uncertain.
Methods
The multicentre, randomized, placebo-controlled, blinded Intravenous Iron or Placebo for Anaemia in Intensive Care (IRONMAN) study was designed to test the hypothesis that, in anaemic critically ill patients admitted to the intensive care unit, early administration of intravenous iron, compared with placebo, reduces allogeneic red blood cell transfusion during hospital stay and increases the haemoglobin level at the time of hospital discharge.
Results
Of 140 patients enrolled, 70 were assigned to intravenous iron and 70 to placebo. The iron group received 97 red blood cell units versus 136 red blood cell units in the placebo group, yielding an incidence rate ratio of 0.71 [95 % confidence interval (0.43–1.18), P = 0.19]. Overall, median haemoglobin at hospital discharge was significantly higher in the intravenous iron group than in the placebo group [107 (interquartile ratio IQR 97–115) vs. 100 g/L (IQR 89–111), P = 0.02]. There was no significant difference between the groups in any safety outcome.
Conclusions
In patients admitted to the intensive care unit who were anaemic, intravenous iron, compared with placebo, did not result in a significant lowering of red blood cell transfusion requirement during hospital stay. Patients who received intravenous iron had a significantly higher haemoglobin concentration at hospital discharge.
The trial was registered at http://www.anzctr.org.au as # ACTRN12612001249842.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Anaemia is extremely common in patients admitted to the intensive care unit (ICU) and is the most common indication for allogenic red blood cell (RBC) transfusion even when adherence to transfusion guidelines is high [1, 2]. Both anaemia and RBC transfusion may be harmful to critically ill patients. Anaemia is an independent risk factor for mortality and major morbidity in patients undergoing major surgery and in general ICU patients; RBC transfusion is associated with mortality, nosocomial infection, multi-organ dysfunction syndrome and the acute respiratory distress syndrome (ARDS) in patients treated in an ICU [3–6].
Progressive anaemia and subsequent RBC transfusion are predictable at the time of ICU admission [7]. In selected patients, novel interventions implemented shortly after ICU admission could reduce the incidence and severity of anaemia, the need for RBC transfusion, and therefore the burden of associated morbidity and mortality. Intravenous (IV) iron decreases both the severity of anaemia and incidence of RBC transfusion in non-critically ill patients [8]. However, there is a theoretical risk of causing or worsening infection, and older preparations are associated with anaphylactic reactions [8–10]. High-quality safety and efficacy data for IV iron in the critical care setting are lacking.
We designed the multicentre Intravenous Iron or Placebo for Anaemia in Intensive Care (IRONMAN) randomized controlled trial (RCT) to test the hypothesis that, in critically ill patients admitted to the ICU who are anaemic, early administration of IV ferric carboxymaltose, compared with placebo, reduces the mean number of RBC units transfused between randomization and hospital discharge.
Patients, materials and methods
Study design and oversight
Between 20 June 2013 and 6 June 2015, we conducted a randomized, placebo-controlled, blinded trial in four ICUs in Perth, Western Australia. The study protocol was registered prospectively on the Australian New Zealand Clinical Trials Registry (ACTRN12612001249842), was approved by the ethics committee at each participating site and has been published previously [11]. Prospective consent was obtained from all participants or their legal surrogates. The trial was overseen by an independent data safety monitoring committee. Study drug was supplied by Vifor Pharma which had no other role in the design or conduct of the study or analysis and reporting of the results.
Study population
Patients were eligible to participate if they were 18 years of age or older, within 48 h of admission to ICU, anticipated to require ICU care beyond the next calendar day and had a haemoglobin (Hb) less than 100 g/L at any time in the preceding 24 h. Exclusion criteria included suspected or confirmed severe sepsis, a ferritin greater than 1200 ng/ml or transferrin saturation greater than 50 %. A complete list of the exclusion criteria are provided in the electronic supplementary material.
Randomization and blinding
Eligible patients were randomly assigned in a 1:1 ratio to receive either IV iron or placebo. The randomization sequence was generated by an online resource and was stratified according to study centre [12]. Allocation concealment was maintained by using permuted block randomization and sealed, opaque, consecutively numbered envelopes at each study site that had been generated centrally by staff unrelated to the study or ICU. Randomization was to a study number. Study medication was then prepared by a clinical nurse or pharmacist not involved in the care of the patient. An opaque sleeve covering the study drug infusion syringe and giving set was used to maintain blinding of the participants, treating, site researchers and data collectors [13]. The adequacy of blinding was assessed by conducting a blinding substudy measuring interrater agreement between the study intervention actually delivered and the opinion of the intervention arm according to the attending clinician using a McNemar test.
Study treatments
Patients randomized to the IV iron group received 500 mg of ferric carboxymaltose in 100 ml of 0.9 % saline delivered in two consecutive 50-ml syringes. Details of the study treatment including a photo of the blinding set-up have been published previously [11]. Patients in the placebo group received 100 ml of 0.9 % saline alone. Four days after receiving the initial or subsequent dose of study drug, patients remaining in the ICU were assessed for repeat dosing. Participants were eligible for redosing if they continued to fulfil the study eligibility criteria, including repeated ferritin and transferrin saturation parameters and an Hb less than 100 g/L. Assessment for suitability for redosing continued daily until the patient was discharged from the ICU, received four doses of study drug or died, whichever occurred first.
The IV iron formulation was chosen on the basis of data supporting superiority of ferric carboxymaltose at fixed dose compared with an alternate IV iron formulation and low reported side effect profile [14, 15]. The ferritin and transferrin saturation (TSAT) cutoffs were chosen on the basis of the higher end of the effective reported range (ferritin <1200 mn/ml) and lack of interaction between TSAT and IV iron on RBC transfusion [8, 16].
All aspects of patient management, including decision for RBC transfusion and ICU discharge, were administered according to local practice and at the direction of the treating ICU clinician. There were no RBC transfusion policies in any of the participating centres. Open-label IV iron and erythropoiesis-stimulating agents were strongly discouraged and use of these agents was a protocol violation.
Study outcomes
The primary study outcome was number of RBC units transfused per patient between randomization and hospital discharge reported according to an intention-to-treat analysis. Secondary outcomes included Hb at hospital discharge, proportion of patients receiving RBC transfusion, ICU and hospital length of stay and mortality and infection. Infection was defined as the commencement, escalation or change of IV antibiotics for a confirmed or strongly suspected infection and was adjudicated locally by blinded clinical staff. Clinically confirmed deep vein thrombosis (DVT) and pulmonary embolism (PE) were explicitly collected as serious adverse events (SAEs). Bleeding definitions are provided in the electronic supplementary material. Admission diagnoses were based on acute physiology and chronic health evaluation (APACHE) II diagnostic codes. Events were deemed to be part of the natural history of the primary disease process or expected complications of critical illness were not reported as SAEs unless thought to be causally related to the study intervention.
Statistical analysis
All analyses were conducted on an intention-to-treat basis. No imputation was made for missing data. Continuous variables were reported as mean (±SD) or median and interquartile range (IQR), with between-group differences analysed using Student’s t test or the Wilcoxon rank-sum test for apparently normal and non-normally distributed data respectively. Categorical variables were reported as proportion and analysed using the χ 2 test or Fischer exact test as appropriate. Data was censored at 60 days after enrolment for Hb level, RBC transfusion and vital status. A two-sided P value of 0.05 or less was considered to be statistically significant. All analyses were conducted with Stata version 14 (StataCorp College Station, TX77845, USA). No interim analyses were planned or conducted.
Although the analyses were conducted according to a previously reported statistical analysis plan [13], the number of RBC units was not normally distributed and, in conjunction with advice from an independent statistician (Centre for Applied Statistics, University of Western Australia), the primary outcome has been reported as median and IQR instead of the prespecified mean and standard deviation (SD). The data was then analysed using negative binomial regression with incidence-rate ratios reported. This analysis satisfied the assumptions as count data with overdispersion (variance greater than the mean). The sample size of 140 participants was based on a baseline mean of four RBC transfusions in eligible patients, determined from an observational study conducted in one of the participating study sites, with an SD in the intervention and control groups of 2 and a loss to follow-up of 10 % [7]. This provided 80 % power to detect a decrease in the mean number of RBC transfusions of 1 unit at a significance level of 5 %.
Additional sensitivity analyses of the primary outcome variable adjusted for predefined covariates (enrolment Hb, RBC transfusion prior to enrolment, transferrin saturation, ferritin, soluble transferrin receptor and renal replacement therapy) were performed using negative binomial regression for count data. The effect of IV iron on incidence-rate ratio of RBC transfusion was performed for predefined subgroups including transferrin saturation (<20 or ≥20 %) and ferritin (<200 or ≥200 ng/ml).
Results
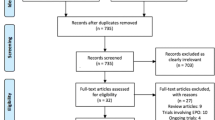
We enrolled 140 patients, with 70 assigned to IV iron and 70 to placebo. All participants received the intervention to which they were randomly allocated and all patients were followed up to discharge from index hospitalisation. One patient declined consent to ongoing participation at time of ICU discharge but consented to data use. Repeat dosing of study drug occurred in 17 patients in the IV iron group (15 patients received two doses, two patients received three doses) and 26 patients in the placebo group (23 patients received two doses, three patients received three doses). Seven participants in the IV iron group and three participants in the placebo group received non-study-drug IV iron either in ICU (n = 1) or post-ICU discharge (n = 9). There was no missing data for the primary or prespecified secondary outcomes (Fig. 1). Demographic and clinical characteristics at baseline were similar between the groups (Table 1), and there was no significant association between perceived and actual study group allocation (McNemar’s test χ 2 = 2.37, p = 0.12).

Participant flow
Primary outcome
The IV iron group was transfused 97 RBC units versus 136 RBC units in the placebo group. The number of RBC units transfused in the ICU was 79 (81 %) and 121 (89 %) for the IV iron and placebo groups respectively. The median (IQR) RBC transfusion in the IV iron and placebo groups [1 unit (0–2) vs. 1 unit (0–3) P = 0.53], incidence rate ratio (IRR) [0.71 (95 % confidence interval (CI) 0.43–1.18) P = 0.19] (Table 2). There was no significant between-group difference in RBC transfusion with the use of multivariable binomial regression adjusting for predefined baseline covariates (P = 0.77), or according to a per protocol analysis (P = 0.15). Between-group RBC transfusion was also similar in the predefined subgroups (Table 3). RBC transfusion (Fig. 3) and median Hb (Fig. 4) by day whilst in ICU are provided in the supplementary appendix.
Secondary outcomes
Overall, the median Hb at hospital discharge was significantly higher in the IV iron group compared with the placebo group (107 g/L (IQR 97–115) vs. 100 g/L (IQR 89–111), P = 0.02). The histograms for the Hb on hospital discharge for the two groups are provided in the electronic supplementary material (Fig. 2). In a post hoc analysis, the proportion of patients discharged from hospital with an Hb less than 100 g/L was significantly lower in the IV iron compared with placebo groups (21/70 (30 %) vs 33/70 (47 %), p = 0.04). The IV iron and placebo groups had similar median lengths of stay in ICU and hospital, and no significant differences in ICU and hospital mortality were observed (Table 2).
Safety
There was no statistical difference between the iron and placebo groups in infection, infection associated with organ failure, or bacteraemia. The number of SAEs did not differ significantly between groups. There were no immediate study-drug-related adverse events in the IV iron group and one in the placebo group where shivering post study drug administration was thought to be possibly related to study drug (Table 4).
Discussion
In this multicentre randomized trial of patients admitted to the ICU who were anaemic, we found that IV iron, compared with placebo, did not result in a significant difference in number of RBC units transfused. IV iron did, however, result in a significantly higher Hb concentration at hospital discharge. Safety outcomes, specifically mortality, infection, clinically diagnosed venous thrombosis and immediate infusion-related adverse events, were not significantly different in those receiving IV iron compared with placebo.
Outside of the critical care setting, trials enrolling patients with similar baseline Hb and haematinics have shown a significant decrease in RBC transfusion associated with IV iron therapy [8]. Although the point estimate for the primary outcome in our study favoured IV iron, the difference was not significant. One possible reason is that IV iron is simply ineffective in patients admitted to the ICU as a result of the modulating effects of severe inflammation on the erythropoietic response to IV iron [17, 18]. Given that the point estimate of the primary outcome favors IV iron with a clinically meaningful decrease in incidence rate ratio of 0.71, and the statistically significant increase in Hb at hospital discharge associated with IV iron, this would appear unlikely. Perhaps more likely is the effect of the mean number of RBC units transfused being substantially lower (1.9 units in the placebo group) than anticipated. Our study was powered to detect a 1-unit reduction from a baseline of 4 units transfused; the observed reduction was 0.5 units. The study was underpowered to detect such a difference leading to the possibility of a type II error (see electronic supplementary material for a power calculation for a future trial of IV iron).
Whilst our study attempted to identify a cohort of patients at high risk of progressive anaemia and subsequent RBC transfusion, characteristics associated with an erythropoiesis response to IV iron in the critical care setting are poorly understood and require further consideration. For example, the relative efficacy of IV iron in patients with anaemia at least partly due to absolute iron deficiency, compared with anaemia of inflammation alone, remains uncertain, and measurement of hepcidin may be of value in this regard [17]. Future trials of IV iron in critical illness should consider adopting a lower Hb threshold for enrolment, only enrolling patients with a longer predicted length of stay, and targeting the intervention at those most likely to mount an erythropoeitic response. This would have the simultaneous effect of identifying a population at higher risk of RBC transfusion and prolonged ICU stay and greater risk of adverse outcomes.
Pieracci et al. conducted an RCT of IV iron sucrose in trauma patients admitted to the ICU and found no difference in Hb concentration [19]. In contrast, our study found that IV iron resulted in a statistically significant increase in Hb at hospital discharge, although the clinical significance of these findings are uncertain.
Compared with Pieracci et al., our study used a higher dose of iron, and an alternative preparation previously shown to be associated with greater erythropoietic response [14]. Our study also enrolled patients at higher risk of RBC transfusion (Hb threshold for enrolment 100 vs 120 g/l) and included a broader range of critically ill patients, potentially at greater risk of pre-existing iron deficiency.
It is plausible that a higher Hb during recovery from critical illness may be of clinical benefit, including more rapid functional recovery and decreased length of stay (LOS). Although our study did not find a significant decrease in hospital LOS associated with IV iron, the median duration from initiation of IV iron to hospital discharge was 11 days, whereas maximal therapeutic effect may not occur for 3–4 weeks. The observed difference may have been greater post-discharge, and the clinical benefits of a higher Hb in a cohort of patients with a longer estimated LOS require further consideration.
Bateman et al. found that moderately severe anaemia at the time of ICU discharge was associated with a markedly reduced health-related quality of life score at 3 and 6 months compared with a non-selected ICU cohort, and that over half remained anaemic at 6 months [20]. Postoperative rehabilitation studies suggest that anaemia is associated with fatigue, reduced exercise capacity, muscle strength and performance in activities of daily living and may impair recovery [21]. Furthermore, Froessler et al. found that IV iron prior to major abdominal surgery was associated with a significant decrease in hospital LOS and a significant increase in Hb at 4 weeks, suggesting a role for IV iron in enhancing recovery [22].
Our study found no association between IV iron and infection. We defined new infection in terms of the commencement, escalation or change of antibiotics. This definition was pragmatic, reflective of clinical practice and assessed by blinded clinicians. Future studies may consider blinded adjudication by independent experts and powering the study to exclude a clinically important difference in infection.
The formulation and dosing of IV iron in our study resulted in no immediate adverse events. Given the lack of data for IV iron use in ICU, we chose a cautious approach to dosing and it is plausible that in future studies, a higher, weight-based dosing and/or continued dosing after ICU discharge may result in a greater response to IV iron. The comparative efficacy of other IV iron preparations in this context remains uncertain.
Strengths
Our study has a number of strengths including a pragmatic design, effective blinding, administration of the study drug to all participants according to the assigned study group, complete follow-up to discharge from index hospitalisation and the use of a restrictive RBC transfusion approach.
Limitations
The data distribution for the primary outcome required a change to the planned statistical analysis, adding to the possibility of a type II error. Baseline transfusion was lower than planned, reducing the power of our study to detect a difference in RBC units. A small proportion of patients received non-study IV iron; however, the numbers were not significantly different between groups and did not change the findings when the groups were analysed per protocol. The significant increase in Hb at discharge was a secondary outcome and there is a risk that this is a chance finding due to multiple testing. However, the point estimate for RBC transfusion also favors IV iron, so a false positive result is considered less likely. Fewer patients required transfusion for major haemorrhage in the IV iron compared with placebo groups, although the difference was not statistically significant. Although a differential effect of mortality or hospital LOS may affect interpretation of the primary end-point, neither was significantly different between groups and so this is considered unlikely. Finally, threshold for RBC transfusion was at the discretion of the treating clinician and not specified as part of the study. Treating clinicians were, however, blinded to the study allocation, and median Hb prior to transfusion was within published guidelines and not significantly different between groups [23].
Conclusion
In patients admitted to the ICU who were anaemic, IV iron compared with placebo did not result in a significant difference in RBC transfusion at hospital discharge. Patients who received IV iron had a significantly higher Hb at hospital discharge.
References
Vincent JL, Baron JF, Reinhart K, Gattinoni L, Thijs L, Webb A, Meier-Hellmann A, Nollet G, Peres-Bota D, ABC (Anemia and Blood Transfusion in Critical Care) Investigators (2002) Anemia and blood transfusion in critically ill patients. JAMA 288:1499–1507
Westbrook A, Pettila V, Nichol A, Bailey MJ, Syres G, Murray L, Bellomo R, Wood E, Phillips LE, Street A, French C, Orford N, Santamaria J, Cooper DJ (2010) Transfusion practice and guidelines in Australian and New Zealand intensive care units. Intensive Care Med 36:1138–1146
Musallam KM, Tamim HM, Richards T, Spahn DR, Rosendaal FR, Habbal A, Khreiss M, Dahdaleh FS, Khavandi K, Sfeir PM, Soweid A, Hoballah JJ, Taher AT, Jamali FR (2011) Preoperative anaemia and postoperative outcomes in non-cardiac surgery: a retrospective cohort study. Lancet 378:1396–1407
Engoren M, Schwann TA, Habib RH, Neill SN, Vance JL, Likosky DS (2014) The independent effects of anemia and transfusion on mortality after coronary artery bypass. Ann Thorac Surg 97:514–520
Corwin HL, Gettinger A, Pearl RG, Fink MP, Levy MM, Abraham E, MacIntyre NR, Shabot MM, Duh MS, Shapiro MJ (2004) The CRIT Study: anemia and blood transfusion in the critically ill–current clinical practice in the United States. Crit Care Med 32:39–52
Holst LB, Petersen MW, Haase N, Perner A, Wetterslev J (2015) Restrictive versus liberal transfusion strategy for red blood cell transfusion: systematic review of randomised trials with meta-analysis and trial sequential analysis. BMJ 350:h1354
Litton E, Xiao J, Allen CT, Ho KM (2015) Iron-restricted erythropoiesis and risk of red blood cell transfusion in the intensive care unit: a prospective observational study. Anaesth Intensive Care 43:612–616
Litton E, Xiao J, Ho KM (2013) Safety and efficacy of intravenous iron therapy in reducing requirement for allogeneic blood transfusion: systematic review and meta-analysis of randomised clinical trials. BMJ 347:f4822
Oppenheimer SJ (2001) Iron and its relation to immunity and infectious disease. J Nutr 131:616S–633S (discussion 633S–635S)
Drakesmith H, Prentice AM (2012) Hepcidin and the iron-infection axis. Science 338:768–772
Litton E, Baker S, Erber W, French C, Ferrier J, Hawkins D, Higgins AM, Hofmann A, Keulenaer BL, Farmer S, McMorrow J, Olynyk J, Richards T, Towler S, Webb S, Australian, New Zealand Intensive Care Society Clinical Trials Group (2014) The IRONMAN trial: a protocol for a multicentre randomised placebo-controlled trial of intravenous iron in intensive care unit patients with anaemia. Crit Care Resusc 16:285–290
Urbaniak GC, Plous S (2013) Research Randomizer (version 4.0) [Computer software]. http://www.randomizer.org/.. Accessed 1 Jan 2016
Litton E (2015) The IRONMAN trial: a protocol for a multicentre randomised placebo-controlled trial of intravenous iron in intensive care unit patients with anaemia. Crit Care Resusc 17:144–145
Evstatiev R, Marteau P, Iqbal T, Khalif IL, Stein J, Bokemeyer B, Chopey IV, Gutzwiller FS, Riopel L, Gasche C, Group FS (2011) FERGIcor, a randomized controlled trial on ferric carboxymaltose for iron deficiency anemia in inflammatory bowel disease. Gastroenterology 141: 846–853, e841–842
MIMS (2012) Ferinject product information. MIMS Australia Pty Ltd & CMPMedica Australia Pty Ltd. New South Wales Australia. http://www.mimsonline-com-au. Accessed 7 Apr 2012
Coyne DW, Kapoian T, Suki W, Singh AK, Moran JE, Dahl NV, Rizkala AR, DRIVE Study Group (2007) Ferric gluconate is highly efficacious in anemic hemodialysis patients with high serum ferritin and low transferrin saturation: results of the Dialysis Patients’ Response to IV Iron with Elevated Ferritin (DRIVE) Study. JASN 18:975–984
Lasocki S, Longrois D, Montravers P, Beaumont C (2011) Hepcidin and anemia of the critically ill patient: bench to bedside. Anesthesiology 114:688–694
Piagnerelli M, Cotton F, Herpain A, Rapotec A, Chatti R, Gulbis B, Vincent JL (2013) Time course of iron metabolism in critically ill patients. Acta Clin Belg 68:22–27
Pieracci FM, Stovall RT, Jaouen B, Rodil M, Cappa A, Burlew CC, Holena DN, Maier R, Berry S, Jurkovich J, Moore EE (2014) A multicenter, randomized clinical trial of IV iron supplementation for anemia of traumatic critical illness. Crit Care Med 42:2048–2057
Bateman AP, McArdle F, Walsh TS (2009) Time course of anemia during six months follow up following intensive care discharge and factors associated with impaired recovery of erythropoiesis. Crit Care Med 37:1906–1912
Carson JL, Terrin ML, Jay M (2003) Anemia and postoperative rehabilitation. Can J Anaesth 50:S60–S64
Froessler B, Palm P, Weber I, Hodyl NA, Singh R, Murphy EM (2016) The important role for intravenous iron in perioperative patient blood management in major abdominal surgery: a randomized controlled trial. Ann Surg 264:41–46
National Blood Authority (2012) Patient blood management guidelines: module 4 critical care. ISBN: 978-0-0873687-0-6. http://www.nba.gov.au. Accessed 1 Jan 2016
Acknowledgments
E.L. is a PhD candidate at the University of Western Australia and this work is submitted in partial fulfilment of the requirement for the Ph.D. The IRONMAN RCT is part of the Blood – CRE, Centre of Research Excellence for Patient Blood Management in Critical Illness and Trauma
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflicts of interest
Dr. Richards reports grants from the UK National Institute for Health Research (NIHR) Health Technology Assessment (HTA), the Australian National Health and Medical Research Council (NHMRC) and the UK National Institute of Academic Anaesthesia (NIAA)/British Journal of Anaesthesia (BJA)/Association of Cardiothoracic Anaesthetists (ACTA)/Vascular Anaesthesia Society of Great Britain and Ireland (VASGBI); grants, personal fees and non-financial support from Pharmocosmos, Vifor Pharma, Acelity and the Stroke Association; grants from Mason Medical Research Foundation and University College Hospital (UCH) League of Friends; grants and non-financial support from Libresse/Bodyform, outside the submitted work. TR is a regular speaker at national and international conferences on anaemia, blood transfusion, wound healing and vascular diseases for which he has received expenses for travel, accommodation and sundries. TR is a director of The Iron Clinic Ltd and director of Veincare London Ltd. TR is also the vascular lead for 18-Week Wait Ltd. Shannon Farmer reports personal fees from Thieme Stuttgart, Germany, and Elsevier Science USA, non-financial support from the National Blood Authority (Australia), the Medical Society for Blood Management and non-financial support from The Health Round Table, outside the submitted work. Dr. Hofmann reports personal fees from Vifor Pharma AG and TEM International GmbH, outside the submitted work. The other authors declare no conflict of interest.
Additional information
A list of additional IRONMAN site investigators and research coordinators is provided in the electronic supplementary material.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
The IRONMAN Investigators., Litton, E., Baker, S. et al. Intravenous iron or placebo for anaemia in intensive care: the IRONMAN multicentre randomized blinded trial. Intensive Care Med 42, 1715–1722 (2016). https://doi.org/10.1007/s00134-016-4465-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-016-4465-6