
Abstract
The receptor for advanced glycation end products (RAGE) is a pattern-recognition receptor and evolutionary member of the immunoglobulin superfamily that is involved in the host response to infection, injury, and inflammation. It exists in two forms: membrane-bound and soluble forms (sRAGE). RAGE recognizes a variety of ligands and, via a receptor-driven signaling cascade, activates the transcription factor NF-κB, leading to the expression of proinflammatory cytokines. The soluble form, sRAGE, is a decoy receptor and competitively inhibits membrane RAGE activation. RAGE is constitutively expressed abundantly in the lung under basal conditions. This expression is enhanced during inflammatory states such as with acute lung injury (ALI) and acute respiratory distress syndrome (ARDS). This review summarizes the characteristics of RAGE, RAGE isoforms, RAGE ligands, and signaling pathways in the pathogenesis of ALI and ARDS. Additionally, the review explores the potential of RAGE as an important therapeutic target in ALI/ARDS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acute lung injury (ALI), and its more severe form, acute respiratory distress syndrome (ARDS), are syndromes of acute respiratory failure associated with severe inflammation and diffuse alveolar damage. Clinically, they are characterized by rapid onset of respiratory failure. ALI/ARDS may occur as a consequence of diverse risk factors including direct injury to lung, such as bacterial or viral pneumonia, gastric aspiration, lung contusion, toxic inhalation, or near drowning. Additionally, there are indirect systemic insults such as sepsis, massive blood transfusion, burn, pancreatitis, or, less frequently, gynecological insults that can predispose to lung injury. The American-European Consensus Committee in 1994 defined ALI/ARDS as radiologically bilateral lung field infiltrates, and physiologically P aO2/F iO2 ≤ 300 mmHg for ALI and ≤200 mmHg for ARDS, with exclusion of left atrial hypertension (pulmonary capillary wedge pressure <18 mmHg) [1]. During the last decade, despite improvements in intensive care, ALI/ARDS still carries a high mortality rate of 29–42 % [2].
The pathophysiology of ALI/ARDS is related to the endothelial injury and increased vascular permeability [3]. Airway epithelium provides a physical and anatomic border between host and environment and protects the host from injurious and infectious stimuli that gain access to the respiratory tract through inspiration or aspiration. Histologically, type I epithelial cells comprise 95 % of the surface area of the alveolus and play a pivotal role in epithelial integrity and alveolar fluid clearance [4]. Given these important functions, impaired integrity of alveolar type I epithelial cells due to a pathological insult could result in significant functional disruption, leading to ALI/ARDS. Newman et al. [5] first described the use of the alveolar type I cell-selective proteins as markers to investigate injury and repair of the alveolar epithelium.
Recent studies have demonstrated that the receptor for advanced glycation end products (RAGE) plays an important role in the pathogenesis of ALI/ARDS, due to the fact that it is abundant in the lung and its expression is primarily located on the basal membranes of alveolar type I epithelial cells [6]. The RAGE–ligand interaction leads to intracellular signaling, with activation of the proinflammatory transcription of NF-κB. This review will focus on recent advances in understanding of the role of RAGE in the pathogenesis of ALI/ARDS.
RAGE and its interaction with ligands
The RAGE is a multi-ligand, immunoglobulin-type transmembrane protein, which serves as one of the pattern-recognition receptors (PRRs) of the innate immune system. RAGE was first characterized almost 20 years ago [7] and has since been actively studied in diverse fields of biomedical research. It was initially identified as a receptor for predominantly advanced glycation end products (AGEs), a heterogeneous group of compounds either formed spontaneously in the body via non-enzymatic glycol-oxidative reactions between reducing sugars, proteins, and lipids [8], or obtained directly from intake of AGE-rich food. Later, it was noted that RAGE also interacts with diverse non-glycated endogenous peptide ligands such as high mobility group box 1 protein (HMGB-1), S100/calgranulin family, amyloid fibrils [9–12], LPS [13], and phosphatidylserine [14]. As shown in Fig. 1, RAGE–ligand interaction results in cellular activation via signaling cascades, which include the proinflammatory transcription factor nuclear factor-κB (NF-κB), the mitogen activated protein (MAP) kinases, and phosphoinositide 3-kinase (PI3K). There is a unique feature of RAGE-induced NF-κB signaling cascade, which is de novo synthesis of NF-κBp65 [15]. These intracellular signalings lead to induction of inflammatory cytokines, proteases, and oxidative stress [16, 17]. Different RAGE–ligand interactions can initiate and stimulate chronic stress pathways and repair, depending on the ligand and environment.

Schematic representation of the cell membrane receptors (TLR, and RAGE) and their ligands. The ligation of the receptors with ligands (DAMPs, PAMPs, and AGEs) leads to the activation of the NF-κB pathway and the release of adhesion molecules and proinflammatory cytokines
RAGE and the danger signals
The host immune system has complete signaling pathways for the detection, containment, and repair of damage caused to cells. The emerging concept of pattern recognition involves RAGE and Toll-like receptors (TLRs) in sensing the danger signals, including pathogen associated molecular patterns (PAMPs) [18] and endogenous damage associated molecular patterns (DAMPs), or Alarmins [19]. PAMPs are a diverse set of microbial molecules which share a number of different recognizable biochemical features and alert the host immune system to invading pathogens [18]. These exogenous PAMPs are recognized by cells of the innate and acquired immune system through the pattern recognition receptors (PRRs). On the other hand, DAMPs, the endogenous equivalents of PAMPs, are a pleiotropic group of intracellular proteins released actively or passively upon the non-programmed cell death, or necrosis, into the extracellular compartment [20–22]. Recent study suggests that DAMPs, including formyl peptides and mitochondrial DNA, are released during injury [23]. DAMPs become “danger signals” by activating PRRs. The best-known DAMPs are high mobility group box-1 (HMGB-1), S100A8 (MRP8, calgranulin A), S100A9 (MRP14, calgranulin B), and serum amyloid A (SAA) [24] (Table 1). As mentioned previously, these molecules are ligands of RAGE.
Although TLRs are the best-studied PRRs for PAMPs and DAMPs [18], RAGE is regarded as a prototypic DAMP receptor. Studies have shown that RAGE plays a key role in the PRR-dependent mechanisms of ALI. RAGE is involved in the recognition of HMGB1- and DNA-containing immune complexes which stimulates TLR9 [25]. The ligation of PAMPs and DAMPs with PRRs leads to oxidative stress and sustained activation of nuclear factor-kappa B (NF-κB). Interestingly, interactions of TLR and RAGE with ligands all lead to activation of NF-κB and the mitogen-activation protein kinase pathway [16], suggesting that both receptor usage and signaling pathways evoke similar responses, when activated by PAMPs and DAMPs. The interaction of RAGE and TLR with their ligands is depicted in Fig. 1. It is possible that RAGE functions as a “non-canonical Toll” that binds with AGEs and other DAMPs, and triggers the host inflammatory response.
The lung is uniquely situated to become a target organ for injury, constantly bombarded by a wide range of infectious pathogens, foreign antigens, and host-derived danger signals. The innate immune mechanism expresses a complete repertoire of PRRs such as RAGE and TLRs, among others, to recognize both PAMPs and DAMPs [26]. Schmidt et al. [10, 27] has posited a ‘two-hit’ model for the inflammatory change and tissue injury mediated by RAGE and its ligands. The engagement of RAGE by its ligands can be considered the ‘first hit’, as discussed above. The ‘second hit’ of cellular perturbation is mediated by superimposed accumulation of invading bacterial pathogens, immune/inflammatory stimuli, modified molecules, ischemic stress, and other factors. During infection, innate immunity is activated by PAMPs expressed on invading microorganisms.
As a result of the expression of proinflammatory cytokines and chemokines, this cellular activation leads to inflammatory processes or tissue injury. This is frequently seen in diabetic complications, neurodegenerative disorders, atherosclerosis, amyloidosis, and immune–inflammatory processes [10, 28, 29]. In ALI, host defense systems encounter PAMPs from exogenous pathogens, and DAMPs after cell death or following immune cell activation and matrix degradation products [21, 22].
RAGE in the lung
It is noteworthy that RAGE is constitutively expressed at low levels in all cells but ubiquitously high in the lung [30–33], even in the physiologic state. In the lung, although primarily located on the basal membranes of alveolar type I epithelial cells [6, 34], RAGE is also expressed in the bronchiolar epithelium, alveolar type II epithelial cells, alveolar macrophages, and vascular endothelial cells [35, 36]. We do not know why RAGE, one of the PRRs to sense environmental and endogenous cues, would be on the basal membrane and not the apical surface. RAGE may have lung-specific functions distinct from the role of RAGE in other organ systems. One study demonstrated that RAGE enhances the adherence of epithelial cells to collagen-coated surfaces and has a striking capacity for inducing cell spreading, and suggested that RAGE might assist AT I cells to acquire a spreading morphology, thereby ensuring effective gas exchange and alveolar stability [37].
Because of its ability to interact with a wide range of endogenous ligands, RAGE may function as a sensor for the environmental cues. Two recent genome-wide association studies for lung function identified multiple gene loci for RAGE acting as one of the important determinants of the pulmonary function [38, 39]. The study also suggested that RAGE coupled with ICAM-1 as a new set of functionally linked adhesion molecules in mediating leukocyte recruitment and adhesion during the acute trauma-induced inflammatory response in vivo [40]. These findings might imply that the lung is highly responsive to RAGE ligands that are associated with potential dangers, so that the lung can rapidly respond to and defend the host from the insults. However, the RAGE–ligand interaction may also act as a double-edged sword. Persistent inflammation from RAGE activation can tilt the protective innate immunity towards harmful effects, resulting in ALI.
Soluble RAGE as a biomarker in ALI/ARDS
In addition to its full-length membrane-bound form of RAGE (mRAGE), soluble RAGE (sRAGE) is a secreted isoform of RAGE that lacks the transmembrane domain and cytoplasmic tail while containing the same V-type and C-type regions found in mRAGE [32]. Consequently, sRAGE is found in the extracellular space. Circulating sRAGE either is produced by receptor ectodomain shedding [41] or represents a splice variant of RAGE, such as endogenous secreted RAGE (esRAGE) [35]. Like mRAGE, sRAGE has a very high level of expression in the lung under normal conditions [30–33]. Indeed, sRAGE has been shown to be both a biomarker for type I alveolar epithelial cell injury, and a key mediator of inflammation. This is important because epithelial injury and inflammation are all involved in the mechanism of ALI, and RAGE is involved in both pathways in the mechanism of ALI.
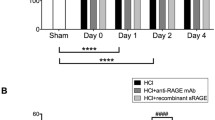
In 2006, Uchida et al. [42] first conducted animal and human studies on the role of RAGE in lung injury. In their animal study, RAGE was released into the BAL and serum as a single soluble isoform sized approximately 48 kDa. The RAGE levels in the blood circulation and BAL increased rapidly following lung injury induced by intratracheal administration of hydrochloric acid. In a murine model of lung injury induced by intratracheal lipopolysaccharide (LPS) administration, sRAGE was detected in the BAL [43]. In the study by Su et al. [44], ALI in mice was induced after exposure to >95 % oxygen at 72 and 96 h. The authors found that the elevations of excess lung water (ELW) and extravascular plasma equivalents (EVPE; an index of lung vascular permeability) correspond to the BAL RAGE levels (Fig. 2). All these animal studies demonstrate that sRAGE is a useful biomarker for type I alveolar epithelial cell injury, and sRAGE correlates with the severity of ALI.

Mice developed ALI after exposure to >95 % oxygen at 72 and 96 h as indicated by an increase in excess lung water (ELW) and extravascular plasma equivalents (EVPE; an index of lung vascular permeability). Corresponding to the severity of lung injury, BAL RAGE levels were increased at 72 and 96 h. *P < 0.01. (The figure is adapted from Su et al. [44] and used with permission from the American Physiological Society)
Similarly, observations in human subjects suggest that sRAGE is a biomarker of severity of ALI/ARDS (Table 2). Uchida and colleagues [42] first illustrated that the RAGE levels in the pulmonary edema fluid from patients with ALI were higher than the levels from patients with hydrostatic pulmonary edema, and the plasma RAGE level in patients with ALI were significantly higher than the healthy volunteers or patients with hydrostatic pulmonary edema. These findings suggest that the vast majority of RAGE in these samples was predominantly from the lung, and that RAGE is a biomarker of acute pulmonary inflammatory response. Jabaudon et al. [45] specifically designed their study to determine whether RAGE was associated with patients who had severe sepsis without ALI or only those with ALI. They found that RAGE levels were higher in those who had ALI independent of their sepsis status. This would imply more of an association with epithelial injury than with inflammation. Elevated plasma levels of sRAGE were also associated with primary graft dysfunction after lung transplantation [46].
Additionally, the levels of sRAGE were found to be of prognostic value in patients with ALI. In the study by Cohen et al. [47], plasma levels of sRAGE were found to increase within 30 min of severe trauma and correlated with the severity of injury, early post-traumatic coagulation, hyper-fibrinolysis, and endothelial cell activation. In a large, randomized, controlled trial of lower tidal volume ventilation in ARDS, increases in the baseline plasma sRAGE levels were associated with worse clinical outcomes in patients randomized to higher tidal volumes (12 mL/kg predicted body weight) [48]. In other words, patients with high RAGE were those who benefited especially from low tidal volume and lung protection. In addition, the higher RAGE levels are associated with higher radiographic and physiological indices of ALI severity as well as the non-pulmonary Acute Physiology and Chronic Health Evaluation (APACHE III) scores [48]. These data suggest that baseline plasma RAGE levels are strongly associated with clinical outcomes in patients with ALI.
Although RAGE may be a biomarker of disease severity, the potential predictive value of RAGE measurement needs to be tested in patients at risk of developing ALI. Further animal and clinical investigation is needed to study the sensitivity and specificity of RAGE in the diagnosis and prognosis of ALI. Conceivably, it would identify subgroups of patients who would benefit from specific therapies targeted at relevant pathogenetic pathways.
Interestingly, Gefter et al. [49] reported that lung exclusively expressed four predominant RAGE protein isoforms which were not found elsewhere, suggesting that those lung-specific RAGE isoforms may play an important role in the pulmonary homeostasis, i.e., to maintain internal equilibrium by adjusting its physiological processes including alveolar inflammatory response. Further investigation to elucidate the expression of RAGE isoforms unique to the lung is warranted.
On the other hand, sRAGE is fully capable of ligand (i.e., AGE) binding, but is devoid of signaling function, giving it a potential protective role in the pulmonary inflammatory response. More details will be discussed in the “sRAGE” section.
RAGE ligands in ALI/ARDS
The ligand–RAGE ligation in the lung has been suggested to play a significant role in the pulmonary pathophysiology [50]. On the other hand, an increased RAGE expression leads to the ligand accumulation [10]. RAGE is able to bind to a variety of endogenous molecules including DAMPs that alert the host immune system and trigger a defensive inflammatory response [51]. Major ligands of RAGE comprise heterogeneous groups of molecules, such as proinflammatory cytokine-like mediators of the S100/calgranulin family (S100A12 and S100B), amphoterin, and β-sheet fibrils of amyloid. RAGE expression is upregulated when there is an elaboration of endogenous host-derived ligands (proteins, lipids, and other products of oxidative stress) [52].
AGEs
AGEs are the prototypal ligand of RAGE. AGEs form spontaneously by the Maillard reaction via non-enzymatic glycol-oxidative reactions between reducing sugars, proteins, and lipids [8]. AGEs have been implicated in various chronic diseases characterized by sustained oxidant stress and multi-organ low-level inflammatory injury [53]. The production and accumulation of AGEs in the body could be due to physiological or pathological conditions (such as diabetes mellitus, renal failure, or aging process), lifestyle habits (such as smoking and unhealthy diets), and environmental pollution [54]. Therefore, by definition, AGEs can be classified as PAMPS if they are from exogenous sources, or DAMPs if are from endogenous sources. Studies have shown that dietary AGEs play a significant role in the initiation and pathogenesis of a variety of diseases, such as diabetes, chronic renal failure, atherosclerosis, rheumatic arthritis, the aging process, and neurodegenerative disorders [55, 56]. A recent study by Tikellis [57] showed that the “Western diet” was associated with cardiac hypertrophy, inflammation, mitochondrial-dependent superoxide production, and cardiac AGE accumulation. The interaction of AGEs with structural and cellular components has emerged as a potential mechanism of environmental toxicity for the general adult population.
In our laboratory, with a mouse model of ALI induced with combined acid and small food particle aspiration, we were the first to demonstrate that a diet high in AGEs exacerbates ALI as evidenced by a significant increase in BAL albumin concentration, the pulmonary PMN counts, and lung parenchymal MPO activity [58]. High AGEs also impaired the pulmonary mechanical compliance. With the most prominent transcription of RAGE being in the lung and the wide range of AGE sources in the body and in the environment, it is speculated that the RAGE–AGE axis plays an important role in mediating pulmonary inflammation and determining the extent of lung injury.
S100/calgranulins
S100 proteins, or calgranulins, are a family of over 20 related calcium-binding proteins. Many of the S100 proteins have been found to bind to RAGE [59]. In particular, S100A8 (calgranulin A), S100A9 (calgranulin B), and S100A12 (EN-RAGE) are expressed by phagocytes and secreted at the sites of inflammation [60]. These proteins induce a specific inflammatory pattern in endothelial cells with increased vascular permeability and pro-thrombotic effects [59].
On the other hand, S100A12 is expressed in the cytoplasm of neutrophils, monocytes, and lymphocytes [61], and solely binds to RAGE. Therefore, the expression may reflect neutrophil activation during lung inflammation and contribute to pulmonary inflammation and endothelial activation via binding to RAGE. As an example, patients with ARDS had significantly enhanced pulmonary S100A12 expression and higher S100A12 protein and RAGE concentrations in bronchoalveolar lavage fluid when compared with controls [62]. Even in healthy individuals, inhalation of LPS increased the BAL levels of S100A12 [62]. In patients who developed ALI after emergency surgery for lower gastrointestinal tract perforation, the blood S100A12 levels were significantly higher in the immediate postoperative period in the group that developed ALI [63].
High mobility group box-1 (HMGB-1)
High mobility group (HMG) proteins are nuclear, non-histone chromosomal proteins. Among them, HMGB-1 is the only one that has been shown to activate RAGE, as well as TLR 4 and 9 [64]. HMGB-1 can be either passively released from necrotic cells or actively secreted by activated immune cells. HMGB-1 is a potent innate “danger signal” for triggering sterile inflammation and the initiation of host defense or tissue repair [64, 65]. HMGB-1 release is associated with both cellular necrosis and apoptosis as well as via a non-classical pathway in immune and non-immune cells [65]. HMBG-1 has been found to be a later mediator of endotoxin-induced ALI in mice [66], and serum levels of HMGB1 were increased in septic shock patients and positively associated with sepsis-related organ failure assessment score [67]. HMGB-1 administered intratracheally in mice produces an acute inflammatory lung injury, with increased production of IL-1β, TNF-α, macrophage-inflammatory protein-2, neutrophil accumulation, and the development of lung edema [66]. Additionally, in endotoxin-induced lung injury, administration of anti-HMG-1 antibodies either before or after endotoxin exposure decreased the migration of neutrophils to the lungs, as well as lung edema [66]. In human subjects, the plasma levels of HMGB-1 were increased within 30 min following severe trauma. These levels correlated with the severity of injury, tissue hypoperfusion, early post-traumatic coagulopathy, and hyperfibrinolysis. Non-survivors had significantly higher plasma levels of HMGB-1 than survivors [68]. Among mechanically ventilated patients, those with long-term ventilator support due to respiratory failure demonstrated higher levels of HMGB-1 in BAL as compared to those with short term ventilation for less than 5 h (for an elective surgical procedure) [69]. These results suggest that HMGB-1 is the archetypal mediator of cellular DAMPs after stress and ALI.
β-sheet fibrils of amyloid and other ligands
RAGE also binds to ligands such as leukocyte β2 integrin Mac-1, β-amyloid peptide, and serum amyloid A [70]. In an animal model of thioglycollate-induced acute peritonitis, RAGE−/− mice showed that leukocyte recruitment was significantly impaired when compared to wild-type animals [70]. An in vitro study by the same authors demonstrated that the ligation of RAGE with β2 integrins mediated recruitment of leukocytes [70]. RAGE-dependent leukocyte adhesion to endothelial cells was initiated by a direct interaction of RAGE with the β2 integrin Mac-1. Interestingly, the RAGE–Mac-1 interaction was augmented by another RAGE ligand, S100 protein. Mac-1 ligation of RAGE probably defines a novel pathway of leukocyte recruitment relevant in the host inflammatory response. However, to date, there is no information regarding the role of the interaction of RAGE with these ligands in ALI/ARDS. Further research on the role of these ligands will be useful in understanding the role of RAGE in the pathogenesis of ALI/ARDS, as well as providing a basis for the development of novel therapeutic applications.
RAGE as a therapeutic target
ALI/ARDS remain an important source of morbidity, mortality, and healthcare costs. Ventilation strategies, fluid management strategies, a wide range of anti-inflammatory, antioxidant, anticoagulant, and other drug therapies, and extracorporeal assist devices are among many interventions for ALI/ARDS. However, only reducing mechanical ventilation-induced lung injury (VILI) has been shown to be beneficial in improving morbidity and mortality. Clinically, before the full-blown ALI/ARDS, the patients are usually not intubated, which makes the low tidal volume ventilation strategy impossible in this subset of patients. Therefore, the urgent need to identify novel approaches to treatment has significant potential for clinical benefit.
Since there is significant upregulation of RAGE expression in the pulmonary epithelial cells during ALI/ARDS, decreasing RAGE activation could be a therapeutic strategy in decreasing the severity of the lung injury secondary to ALI/ARDS. To this end, there are three possible approaches: (1) Decrease the ligand concentration in the blood or BAL. (2) Block RAGE to avoid RAGE–ligand interaction, by administration of anti-RAGE antibody. (3) Competitive binding of RAGE ligands by administration of decoy receptors, i.e., sRAGE.
RAGE ligand interference
The AGE cross-linkage breaker, alagebrium, or ALT-711, has been shown to reduce RAGE expression, improve endothelial function in patients with hypertension, and have an antioxidative stress (OS) effect [71, 72]. Other compounds for preventing AGEs binding with RAGE are aminoguanidine and pyridoxamine, inhibitors of AGE formation. In CD1 mice with end stage renal disease, aminoguanidine and pyridoxamine significantly inhibit glomerular lesions [73]. In a study of obese patients with hepatic steatosis, aminoguanidine and pyridoxamine inhibited the formation of endogenous N-ε-carboxymethyllysine (CML) and increased the concentrations of RAGE, PAI-1, IL-8, IL-6, and CRP expression [74]. As mentioned above, RAGE is a receptor with multiple ligands. Thus, minimizing one potential ligand (AGEs) will probably not be very efficient in limiting the overwhelming inflammatory response in the lung. Interference of other ligand formation has not yet been reported. Future focus on development of pharmacologic compounds targeting DAMPs also deserves special attention.
Anti-RAGE antibody
In a sepsis model, genetically deficient (RAGE−/−) mice that do not express RAGE had a survival advantage following cecal ligation and puncture (CLP) or colon ascendens catheter placement compared with wild-type mice [75]. Subsequently, a neutralizing antibody to the receptor for the advanced glycation end products (anti-RAGE antibody) has been developed as a potential treatment of acute and chronic inflammatory conditions. Using a similar model, Lutterloh et al. [75] concluded that the rat anti-RAGE monoclonal antibody effectively decreased mortality compared with control animals, even when given 24 h after cecal ligation and puncture (CLP). A recent study also demonstrated that a humanized anti-RAGE monoclonal antibody significantly protected mice from pneumococcal pneumonia-induced mortality, even if the treatment was given 6 h after intratracheal infection with Streptococcus pneumonia [76]. The conclusions from the study strongly suggest that inhibition of RAGE ligation and its subsequent activation of inflammatory signaling pathways by anti-RAGE antibody may be a promising therapeutic target in the management of pneumonia and ALI.
sRAGE
Yet another promising approach to neutralize RAGE–ligand interaction is the application of C-terminal truncated RAGE (sRAGE). sRAGE acts as a decoy receptor and fully retains the capacity to bind RAGE ligands which would otherwise interact with mRAGE. Generally, mRAGE is thought to promote disease pathogenesis and injury by activating the NF-κB pathway. In contrast, sRAGE is thought to be protective by preventing membrane RAGE signaling in the systemic diseases, such as tumor growth, metastasis, and diabetic wound healing [11, 12, 77–79]. Indeed, sRAGE administration in a number of cell culture and animal models of RAGE-mediated disorders has been found to successfully prevent or reverse RAGE-associated pathology [77–79]. Specifically in mice with LPS-induced lung injury, treatment with sRAGE significantly attenuated the increases in neutrophil infiltration, lung permeability, proinflammatory cytokine production, NF-κB activation, and number of apoptotic cells [43].
Conclusions
Recent published data strongly suggest a prominent role for RAGE in the pathogenesis of ALI and ARDS. The ligation of RAGE is a double-edged sword, which plays an important role in resolving the pathogenesis of an offending insult but also leads to tissue destruction. During this process, RAGE expression in the pulmonary epithelial cell is upregulated. sRAGE is secreted into the BAL and is detectable in the serum, serving as a biomarker for the degree of lung injury and acting as a decoy receptor to downregulate the injurious pulmonary inflammatory response.
Current measures of pharmacotherapy in ALI/ARDS have been limited in lowering morbidity and mortality. A complete understanding of the relationship between RAGE and other receptors such as TLRs will be beneficial in formulating potential therapeutic approaches. As of now, modification of the RAGE–ligand activation pathway seems to be a promising target for therapeutic intervention. Limiting the RAGE ligation (i.e., by anti-RAGE antibody of sRAGE) may modulate the intensity of an overwhelming pulmonary inflammatory response.
References
Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, LeGall JR, Morris A, Spragg R (1994) Report of the American-European Consensus conference on acute respiratory distress syndrome: definitions, mechanisms, relevant outcomes, and clinical trial coordination. Consensus Committee. J Crit Care 9:72–81
Johnson ER, Matthay MA (2010) Acute lung injury: epidemiology, pathogenesis, and treatment. J Aerosol Med Pulm Drug Deliv 23:243–252
Ware LB, Matthay MA (2001) Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med 163:1376–1383
Johnson MD, Widdicombe JH, Allen L, Barbry P, Dobbs LG (2002) Alveolar epithelial type I cells contain transport proteins and transport sodium, supporting an active role for type I cells in regulation of lung liquid homeostasis. Proc Natl Acad Sci USA 99:1966–1971
Newman V, Gonzalez RF, Matthay MA, Dobbs LG (2000) A novel alveolar type I cell-specific biochemical marker of human acute lung injury. Am J Respir Crit Care Med 161:990–995
Shirasawa M, Fujiwara N, Hirabayashi S, Ohno H, Iida J, Makita K, Hata Y (2004) Receptor for advanced glycation end-products is a marker of type I lung alveolar cells. Genes Cells 9:165–174
Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, Elliston K, Stern D, Shaw A (1992) Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem 267:14998–15004
Thornalley PJ (2003) The enzymatic defence against glycation in health, disease and therapeutics: a symposium to examine the concept. Biochem Soc Trans 31:1341–1342
Yan SF, Barile GR, D’Agati V, Du Yan S, Ramasamy R, Schmidt AM (2007) The biology of RAGE and its ligands: uncovering mechanisms at the heart of diabetes and its complications. Curr DiabRep 7:146–153
Schmidt AM, Yan SD, Yan SF, Stern DM (2001) The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest 108:949–955
Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, Tanji N, Lu Y, Lalla E, Fu C, Hofmann MA, Kislinger T, Ingram M, Lu A, Tanaka H, Hori O, Ogawa S, Stern DM, Schmidt AM (2000) Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature 405:354–360
Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM (1999) RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97:889–901
Yamamoto Y, Harashima A, Saito H, Tsuneyama K, Munesue S, Motoyoshi S, Han D, Watanabe T, Asano M, Takasawa S, Okamoto H, Shimura S, Karasawa T, Yonekura H, Yamamoto H (2011) Septic shock is associated with receptor for advanced glycation end products ligation of LPS. J Immunol 186:3248–3257
He M, Kubo H, Morimoto K, Fujino N, Suzuki T, Takahasi T, Yamada M, Yamaya M, Maekawa T, Yamamoto Y, Yamamoto H (2011) Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep 12:358–364
Bierhaus A, Schiekofer S, Schwaninger M, Andrassy M, Humpert PM, Chen J, Hong M, Luther T, Henle T, Kloting I, Morcos M, Hofmann M, Tritschler H, Weigle B, Kasper M, Smith M, Perry G, Schmidt AM, Stern DM, Haring HU, Schleicher E, Nawroth PP (2001) Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 50:2792–2808
Aleshin A, Ananthakrishnan R, Li Q, Rosario R, Lu Y, Qu W, Song F, Bakr S, Szabolcs M, D’Agati V, Liu R, Homma S, Schmidt AM, Yan SF, Ramasamy R (2008) RAGE modulates myocardial injury consequent to LAD infarction via impact on JNK and STAT signaling in a murine model. Am J Physiol Heart Circ Physiol 294:H1823–H1832
van Beijnum JR, Buurman WA, Griffioen AW (2008) Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis 11:91–99
Janeway CA Jr, Medzhitov R (2002) Innate immune recognition. Annu Rev Immunol 20:197–216
Natarajan B, Gupta PK, Cemaj S, Sorensen M, Hatzoudis GI, Forse RA (2010) FAST scan: is it worth doing in hemodynamically stable blunt trauma patients? Surgery 148:695–700 (discussion 700–691)
Scaffidi P, Misteli T, Bianchi ME (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418:191–195
Bianchi R, Adami C, Giambanco I, Donato R (2007) S100B binding to RAGE in microglia stimulates COX-2 expression. J Leukoc Biol 81:108–118
Kuipers MT, van der Poll T, Schultz MJ, Wieland CW (2011) Bench-to-bedside review: damage-associated molecular patterns in the onset of ventilator-induced lung injury. Crit Care 15:235
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464:U104–U115
Harris HE, Raucci A (2006) Alarmin(g) news about danger: workshop on innate danger signals and HMGB1. EMBO Rep 7:774–778
Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La Rosa G, Bierhaus A, Naworth P, Marshak-Rothstein A, Crow MK, Fitzgerald KA, Latz E, Kiener PA, Coyle AJ (2007) Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol 8:487–496
Xiang M, Fan J (2010) Pattern recognition receptor-dependent mechanisms of acute lung injury. Mol Med (Cambridge, Mass) 16:69–82
Schmidt AM, Yan SD, Yan SF, Stern DM (2000) The biology of the receptor for advanced glycation end products and its ligands. Biochim Biophys Acta 1498:99–111
Chavakis T, Bierhaus A, Nawroth PP (2004) RAGE (receptor for advanced glycation end products): a central player in the inflammatory response. Microbes Infect 6:1219–1225
Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, Stern DM, Nawroth PP (2005) Understanding RAGE, the receptor for advanced glycation end products. J Mol Med (Berlin, Germany) 83:876–886
Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, Nowygrod R, Neeper M, Przysiecki C, Shaw A et al (1993) Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol 143:1699–1712
Hanford LE, Fattman CL, Shaefer LM, Enghild JJ, Valnickova Z, Oury TD (2003) Regulation of receptor for advanced glycation end products during bleomycin-induced lung injury. Am J Respir Cell Mol Biol 29:S77–S81
Hanford LE, Enghild JJ, Valnickova Z, Petersen SV, Schaefer LM, Schaefer TM, Reinhart TA, Oury TD (2004) Purification and characterization of mouse soluble receptor for advanced glycation end products (sRAGE). J Biol Chem 279:50019–50024
Englert JM, Hanford LE, Kaminski N, Tobolewski JM, Tan RJ, Fattman CL, Ramsgaard L, Richards TJ, Loutaev I, Nawroth PP, Kasper M, Bierhaus A, Oury TD (2008) A role for the receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Am J Pathol 172:583–591
McElroy MC, Kasper M (2004) The use of alveolar epithelial type I cell-selective markers to investigate lung injury and repair. Eur Respir J 24:664–673
Cheng C, Tsuneyama K, Kominami R, Shinohara H, Sakurai S, Yonekura H, Watanabe T, Takano Y, Yamamoto H, Yamamoto Y (2005) Expression profiling of endogenous secretory receptor for advanced glycation end products in human organs. Mod Pathol 18:1385–1396
Morbini P, Villa C, Campo I, Zorzetto M, Inghilleri S, Luisetti M (2006) The receptor for advanced glycation end products and its ligands: a new inflammatory pathway in lung disease? Mod Pathol 19:1437–1445
Demling N, Ehrhardt C, Kasper M, Laue M, Knels L, Rieber EP (2006) Promotion of cell adherence and spreading: a novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type I cells. Cell Tissue Res 323:475–488
Repapi E, Sayers I, Wain LV, Burton PR, Johnson T, Obeidat M, Zhao JH, Ramasamy A, Zhai G, Vitart V, Huffman JE, Igl W, Albrecht E, Deloukas P, Henderson J, Granell R, McArdle WL, Rudnicka AR, Barroso I, Loos RJ, Wareham NJ, Mustelin L, Rantanen T, Surakka I, Imboden M, Wichmann HE, Grkovic I, Jankovic S, Zgaga L, Hartikainen AL, Peltonen L, Gyllensten U, Johansson A, Zaboli G, Campbell H, Wild SH, Wilson JF, Glaser S, Homuth G, Volzke H, Mangino M, Soranzo N, Spector TD, Polasek O, Rudan I, Wright AF, Heliovaara M, Ripatti S, Pouta A, Naluai AT, Olin AC, Toren K, Cooper MN, James AL, Palmer LJ, Hingorani AD, Wannamethee SG, Whincup PH, Smith GD, Ebrahim S, McKeever TM, Pavord ID, MacLeod AK, Morris AD, Porteous DJ, Cooper C, Dennison E, Shaheen S, Karrasch S, Schnabel E, Schulz H, Grallert H, Bouatia-Naji N, Delplanque J, Froguel P, Blakey JD, Britton JR, Morris RW, Holloway JW, Lawlor DA, Hui J, Nyberg F, Jarvelin MR, Jackson C, Kahonen M, Kaprio J, Probst-Hensch NM, Koch B, Hayward C, Evans DM, Elliott P, Strachan DP, Hall IP, Tobin MD (2010) Genome-wide association study identifies five loci associated with lung function. Nat Genet 42:36–44
Hancock DB, Eijgelsheim M, Wilk JB, Gharib SA, Loehr LR, Marciante KD, Franceschini N, van Durme YM, Chen TH, Barr RG, Schabath MB, Couper DJ, Brusselle GG, Psaty BM, van Duijn CM, Rotter JI, Uitterlinden AG, Hofman A, Punjabi NM, Rivadeneira F, Morrison AC, Enright PL, North KE, Heckbert SR, Lumley T, Stricker BH, O’Connor GT, London SJ (2010) Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet 42:45–52
Frommhold D, Kamphues A, Hepper I, Pruenster M, Lukic IK, Socher I, Zablotskaya V, Buschmann K, Lange-Sperandio B, Schymeinsky J, Ryschich E, Poeschl J, Kupatt C, Nawroth PP, Moser M, Walzog B, Bierhaus A, Sperandio M (2010) RAGE and ICAM-1 cooperate in mediating leukocyte recruitment during acute inflammation in vivo. Blood 116:841–849
Raucci A, Cugusi S, Antonelli A, Barabino SM, Monti L, Bierhaus A, Reiss K, Saftig P, Bianchi ME (2008) A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J 22:3716–3727
Uchida T, Shirasawa M, Ware LB, Kojima K, Hata Y, Makita K, Mednick G, Matthay ZA, Matthay MA (2006) Receptor for advanced glycation end-products is a marker of type I cell injury in acute lung injury. Am J Respir Crit Care Med 173:1008–1015
Zhang H, Tasaka S, Shiraishi Y, Fukunaga K, Yamada W, Seki H, Ogawa Y, Miyamoto K, Nakano Y, Hasegawa N, Miyasho T, Maruyama I, Ishizaka A (2008) Role of soluble receptor for advanced glycation end products on endotoxin-induced lung injury. Am J Respir Crit Care Med 178:356–362
Su X, Looney MR, Gupta N, Matthay MA (2009) Receptor for advanced glycation end-products (RAGE) is an indicator of direct lung injury in models of experimental lung injury. Am J Physiol Lung Cell Mol Physiol 297:L1–L5
Jabaudon M, Futier E, Roszyk L, Chalus E, Guerin R, Petit A, Mrozek S, Perbet S, Cayot-Constantin S, Chartier C, Sapin V, Bazin JE, Constantin JM (2011) Soluble form of the receptor for advanced glycation end products is a marker of acute lung injury but not of severe sepsis in critically ill patients. Crit Care Med 39:480–488
Christie JD, Shah CV, Kawut SM, Mangalmurti N, Lederer DJ, Sonett JR, Ahya VN, Palmer SM, Wille K, Lama V, Shah PD, Shah A, Weinacker A, Deutschman CS, Kohl BA, Demissie E, Bellamy S, Ware LB (2009) Plasma levels of receptor for advanced glycation end products, blood transfusion, and risk of primary graft dysfunction. Am J Respir Crit Care Med 180:1010–1015
Cohen MJ, Carles M, Brohi K, Calfee CS, Rahn P, Call MS, Chesebro BB, West MA, Pittet JF (2010) Early release of soluble receptor for advanced glycation endproducts after severe trauma in humans. J Trauma 68:1273–1278
Calfee CS, Ware LB, Eisner MD, Parsons PE, Thompson BT, Wickersham N, Matthay MA (2008) Plasma receptor for advanced glycation end products and clinical outcomes in acute lung injury. Thorax 63:1083–1089
Gefter JV, Shaufl AL, Fink MP, Delude RL (2009) Comparison of distinct protein isoforms of the receptor for advanced glycation end-products expressed in murine tissues and cell lines. Cell Tissue Res 337:79–89
Mukherjee TK, Mukhopadhyay S, Hoidal JR (2008) Implication of receptor for advanced glycation end product (RAGE) in pulmonary health and pathophysiology. Respir Physiol Neurobiol 162:210–215
Foell D, Wittkowski H, Roth J (2007) Mechanisms of disease: a ‘DAMP’ view of inflammatory arthritis. Nat Clin Pract Rheumatol 3:382–390
Bucciarelli LG, Wendt T, Rong L, Lalla E, Hofmann MA, Goova MT, Taguchi A, Yan SF, Yan SD, Stern DM, Schmidt AM (2002) RAGE is a multiligand receptor of the immunoglobulin superfamily: implications for homeostasis and chronic disease. Cell Mol Life Sci 59:1117–1128
Ramasamy R, Vannucci SJ, Yan SS, Herold K, Yan SF, Schmidt AM (2005) Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 15:16R–28R
Ulrich P, Cerami A (2001) Protein glycation, diabetes, and aging. Recent Prog Horm Res 56:1–21
Vlassara H (2005) Advanced glycation in health and disease: role of the modern environment. Ann N Y Acad Sci 1043:452–460
Wang H, Vishnubhakat JM, Bloom O, Zhang M, Ombrellino M, Sama A, Tracey KJ (1999) Proinflammatory cytokines (tumor necrosis factor and interleukin 1) stimulate release of high mobility group protein-1 by pituicytes. Surgery 126:389–392
Tikellis C, Thomas MC, Harcourt BE, Coughlan MT, Pete J, Bialkowski K, Tan A, Bierhaus A, Cooper ME, Forbes JM (2008) Cardiac inflammation associated with a Western diet is mediated via activation of RAGE by AGEs. Am J Physiol Endocrinol Metab 295:E323–E330
Ottosen JM, Mullan B, Ohtake PJ, Davidson BA, Knight PR, Guo WA (2010) Diet high in advanced glycation end products exacerbates pulmonary inflammatory response and impairs lung compliance in mice following gastric aspiration. J Surg Res 158:214
Leclerc E, Fritz G, Vetter SW, Heizmann CW (2009) Binding of S100 proteins to RAGE: an update. Biochim Biophys Acta 1793:993–1007
Foell D, Wittkowski H, Vogl T, Roth J (2007) S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol 81:28–37
Vogl T, Propper C, Hartmann M, Strey A, Strupat K, van den Bos C, Sorg C, Roth J (1999) S100A12 is expressed exclusively by granulocytes and acts independently from MRP8 and MRP14. J Biol Chem 274:25291–25296
Wittkowski H, Sturrock A, van Zoelen MA, Viemann D, van der Poll T, Hoidal JR, Roth J, Foell D (2007) Neutrophil-derived S100A12 in acute lung injury and respiratory distress syndrome. Crit Care Med 35:1369–1375
Kikkawa T, Sato N, Kojika M, Takahashi G, Aoki K, Hoshikawa K, Akitomi S, Shozushima T, Suzuki K, Wakabayashi G, Endo S (2010) Significance of measuring S100A12 and sRAGE in the serum of sepsis patients with postoperative acute lung injury. Dig Surg 27:307–312
Dumitriu IE, Baruah P, Manfredi AA, Bianchi ME, Rovere-Querini P (2005) HMGB1: guiding immunity from within. Trends Immunol 26:381–387
Fink MP (2007) Bench-to-bedside review: high-mobility group box 1 and critical illness. Crit Care 11:229
Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ (2000) HMG-1 as a mediator of acute lung inflammation. J Immunol 165:2950–2954
Gibot S, Massin F, Cravoisy A, Barraud D, Nace L, Levy B, Bollaert PE (2007) High-mobility group box 1 protein plasma concentrations during septic shock. Intensive Care Med 33:1347–1353
Cohen MJ, Brohi K, Calfee CS, Rahn P, Chesebro BB, Christiaans SC, Carles M, Howard M, Pittet JF (2009) Early release of high mobility group box nuclear protein 1 after severe trauma in humans: role of injury severity and tissue hypoperfusion. Crit Care 13:R174
van Zoelen MA, Ishizaka A, Wolthuls EK, Choi G, van der Poll T, Schultz MJ (2008) Pulmonary levels of high-mobility group box 1 during mechanical ventilation and ventilator-associated pneumonia. Shock 29:441–445
Chavakis T, Bierhaus A, Al-Fakhri N, Schneider D, Witte S, Linn T, Nagashima M, Morser J, Arnold B, Preissner KT, Nawroth PP (2003) The pattern recognition receptor (RAGE) is a counter receptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med 198:1507–1515
Guo Y, Lu M, Qian J, Cheng YL (2009) Alagebrium chloride protects the heart against oxidative stress in aging rats. J Gerontol A Biol Sci Med Sci 64:629–635
Kim JB, Song BW, Park S, Hwang KC, Cha BS, Jang Y, Lee HC, Lee MH (2010) Alagebrium chloride, a novel advanced glycation end-product cross linkage breaker, inhibits neointimal proliferation in a diabetic rat carotid balloon injury model. Korean Circ J 40:520–526
Sugimoto H, Grahovac G, Zeisberg M, Kalluri R (2007) Renal fibrosis and glomerulosclerosis in a new mouse model of diabetic nephropathy and its regression by bone morphogenic protein-7 and advanced glycation end product inhibitors. Diabetes 56:1825–1833
Gaens KH, Niessen PM, Rensen SS, Buurman WA, Greve JW, Driessen A, Wolfs MG, Hofker MH, Bloemen JG, Dejong CH, Stehouwer CD, Schalkwijk CG (2012) Endogenous formation of Nε-(carboxymethyl)lysine is increased in fatty livers and induces inflammatory markers in an in vitro model of hepatic steatosis. J Hepatol 56:647–655
Lutterloh EC, Opal SM, Pittman DD, Keith JC Jr, Tan XY, Clancy BM, Palmer H, Milarski K, Sun Y, Palardy JE, Parejo NA, Kessimian N (2007) Inhibition of the RAGE products increases survival in experimental models of severe sepsis and systemic infection. Crit Care 11:R122
Christaki E, Opal SM, Keith JC Jr, Kessimian N, Palardy JE, Parejo NA, Tan XY, Piche-Nicholas N, Tchistiakova L, Vlasuk GP, Shields KM, Feldman JL, Lavallie ER, Arai M, Mounts W, Pittman DD (2011) A monoclonal antibody against RAGE alters gene expression and is protective in experimental models of sepsis and pneumococcal pneumonia. Shock 35:492–498
Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ Jr, Chow WS, Stern D, Schmidt AM (1998) Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med 4:1025–1031
Wear-Maggitti K, Lee J, Conejero A, Schmidt AM, Grant R, Breitbart A (2004) Use of topical sRAGE in diabetic wounds increases neovascularization and granulation tissue formation. Ann Plast Surg 52:519–521 (discussion 522)
Goova MT, Li J, Kislinger T, Qu W, Lu Y, Bucciarelli LG, Nowygrod S, Wolf BM, Caliste X, Yan SF, Stern DM, Schmidt AM (2001) Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am J Pathol 159:513–525
Mauri T, Masson S, Pradella A, Bellani G, Coppadoro A, Bombino M, Valentino S, Patroniti N, Mantovani A, Pesenti A, Latini R (2010) Elevated plasma and alveolar levels of soluble receptor for advanced glycation endproducts are associated with severity of lung dysfunction in ARDS patients. Tohoku J Exp Med 222:105–112
Determann RM, Royakkers AA, Haitsma JJ, Zhang H, Slutsky AS, Ranieri VM, Schultz MJ (2010) Plasma levels of surfactant protein D and KL-6 for evaluation of lung injury in critically ill mechanically ventilated patients. BMC Pulm Med 10:6
Briot R, Frank JA, Uchida T, Lee JW, Calfee CS, Matthay MA (2009) Elevated levels of the receptor for advanced glycation end products, a marker of alveolar epithelial type I cell injury, predict impaired alveolar fluid clearance in isolated perfused human lungs. Chest 135:269–275
Acknowledgments
The authors thank Ms. Elizabeth Tona for her assistance with the preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Guo, W.A., Knight, P.R. & Raghavendran, K. The receptor for advanced glycation end products and acute lung injury/acute respiratory distress syndrome. Intensive Care Med 38, 1588–1598 (2012). https://doi.org/10.1007/s00134-012-2624-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-012-2624-y