
Abstract
Purpose
Hypercapnic acidosis often occurs in critically ill patients and during protective mechanical ventilation; however, the effect of hypercapnic acidosis on endogenous nitric oxide (NO) production and hypoxic pulmonary vasoconstriction (HPV) presents conflicting results. The aim of this study is to test the hypothesis that hypercapnic acidosis augments HPV without changing endogenous NO production in both hyperoxic and hypoxic lung regions in pigs.
Methods
Sixteen healthy anesthetized pigs were separately ventilated with hypoxic gas to the left lower lobe (LLL) and hyperoxic gas to the rest of the lung. Eight pigs received 10% carbon dioxide (CO2) inhalation to both lung regions (hypercapnia group), and eight pigs formed the control group. NO concentration in exhaled air (ENO), nitric oxide synthase (NOS) activity, cyclic guanosine monophosphate (cGMP) in lung tissue, and regional pulmonary blood flow were measured.
Results
There were no differences between the groups for ENO, Ca2+-independent or Ca2+-dependent NOS activity, or cGMP in hypoxic or hyperoxic lung regions. Relative perfusion to LLL (Q LLL/Q T) was reduced similarly in both groups when LLL hypoxia was induced. During the first 90 min of hypercapnia, Q LLL/Q T increased from 6% (1%) [mean (standard deviation, SD)] to 9% (2%) (p < 0.01), and then decreased to the same level as the control group, where Q LLL/Q T remained unchanged. Cardiac output increased during hypercapnia (p < 0.01), resulting in increased oxygen delivery (p < 0.01), despite decreased PaO2 (p < 0.01).
Conclusions
Hypercapnic acidosis does not potentiate HPV, but rather transiently weakens HPV, and does not affect endogenous NO production in either hypoxic or hyperoxic lung regions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute respiratory distress syndrome (ARDS) is associated with both hypoxia and hypercapnic acidosis due to pathological gas exchange in the lung and the use of permissive hypercapnia during mechanical ventilation to prevent pulmonary hyperinflation and ventilator-induced lung injury (VILI) [1, 2]. Hypercapnic acidosis affects the pulmonary and systemic circulation and oxygenation. Hypercapnic acidosis causes pulmonary vasoconstriction [3, 4], but reports of the effects on hypoxic pulmonary vasoconstriction (HPV) present conflicting results [5–7]. In systemic circulation, hypercapnic acidosis causes vasodilatation through increased NO production [8, 9], but whether hypercapnic acidosis also causes changes in endogenous NO production in the pulmonary circulation is unclear. NO is an important regulator of pulmonary blood flow, and blockade of enzymatic NO production enhances HPV [10, 11]. In ARDS patients, HPV can contribute up to 20 torr to arterial oxygenation [12]. If hypercapnic acidosis increases NO production in hypoxic lung regions, this would attenuate HPV, and could be detrimental for patients suffering from severe ARDS.
Metabolic acidosis augments HPV, without any change in endogenous pulmonary NO production [13]; therefore, it was hypothesized that hypercapnic acidosis has similar effects. The aim of this study is to test the hypothesis that hypercapnic acidosis augments HPV without any changes in pulmonary NO production in both hyperoxic and hypoxic lung regions in pigs.
Materials and methods
The study was approved by the Animal Research Ethics Committee of Uppsala University, Uppsala, Sweden. Sixteen healthy pigs (Swedish country breed, weight 25–30 kg) were premedicated with intramuscular injection of 6 mg/kg Soletil Forte (Tiletamin and Zolazepam) and 2.2 mg/kg Rompun (Xylazin chloride) before anesthesia induction with 40–100 mg propofol given intravenously. Anesthesia was maintained with propofol infusion at rate of 3 mg/kg/h and infusion of ketamin vet. (5 g), fentanyl (1 mg), and pancuronium (60 mg) in 1,000 ml buffered glucose (25 mg/ml) at rate of 4 ml/kg/h. Oxygen saturation, and inspiratory and end-tidal concentrations (CET) of oxygen (O2) and CO2 were monitored continuously (Datex AS/3™ anesthesia monitor; Datex Ohmeda, Helsinki, Finland) during the whole experiment. Warm buffered Ringer’s solution (10–15 ml/kg/h) was infused, and a suprapubic catheter was inserted for urinary output. Throughout the experiment, the pigs were laid supine on a heating mattress with warm blankets to maintain normal and stable body temperature.
Ventilation
A tracheotomy was performed, and a cuffed endotracheal tube (inner diameter 6.0 mm) was inserted. A second cuffed endotracheal tube (inner diameter 4.5 mm) was inserted through the tracheostoma and positioned in the left lower lobar bronchus. A medial sternotomy allowed the tubes to be guided into a position to separate the left lower lobe (LLL) from the other lung regions, which meant the lungs could be inspected to ensure the left middle and upper lobes and the right lung were ventilated through the main tube. The different and persistent fractions of expired O2 during LLL hypoxia and hyperoxia to the other lung regions were considered additional proof of separation. The lungs were mechanically ventilated by two synchronized Servo 900 C ventilators (Siemens Elema, Lund, Sweden). Both ventilators were set at volume-controlled ventilation of 20 breaths per minute and an inspiration-to-expiration ratio of 1:2. Positive end-expiratory pressure (PEEP) of 5 cmH2O was applied, and a total tidal volume of 8 ml/kg was distributed between the LLL and the other lung regions, aiming at equal end-inspiratory plateau pressures in the LLL and other parts of the lungs. The minute ventilation was then adjusted, if needed, by increasing the respiratory rate to obtain arterial CO2 tension (PaCO2) of 41–49 mmHg (5.5–6.5 kPa), and a corresponding normal pH of 7.35–7.45 in the initial control situation. The ventilation was then kept constant throughout the experiment.
Hemodynamics
An arterial catheter, a central venous catheter, and a pulmonary artery catheter were inserted (Criti Cath™ No 7F; Ohmeda Pte Ltd, Singapore) to record arterial, central venous, and pulmonary blood pressures and temperatures (Datex AS/3™ anesthesia monitor; Datex Ohmeda, Helsinki, Finland). Cardiac output (Q T) and blood flow to the LLL (Q LLL) were measured continuously by enclosing the pulmonary artery and the artery to the LLL in ultrasonic flow probes connected to flow meters (T208 Transonic volume flow meter; Transonic Systems Inc., Ithaca, NY, USA). The relative perfusion of the LLL was calculated as Q LLL/Q T.
Blood gases
Mixed venous and arterial blood samples were collected for analysis of O2 tensions (PvO2 and PaO2), PaCO2, and pH (ABL 625; Radiometer, Copenhagen, Denmark), and arterial O2 saturation (SaO2) and methemoglobin (MetHb) (OSM 3; Radiometer, Copenhagen, Denmark).
Exhaled nitric oxide concentration (ENO)
ENO was measured alternately from the hypoxic LLL and the hyperoxic lung regions by chemiluminescence (analyzer model 42; Thermo Environmental Instruments Inc., Franklin, MA, USA). The measurements were taken in the expiratory limb of the ventilator tubings and more than 100 cm from the endotracheal tubes, which ensured complete mixing and avoided contamination by inspired gas. The average concentration (mean expired values) over ten breaths was used for the statistical analyses.
Nitric oxide synthase activity (NOS)
NOS activity was measured by standard procedure as presented in detail elsewhere [13].
cGMP enzyme immunoassay (EIA)
For quantification of cGMP, lung tissue lysates were analyzed with a commercially available EIA kit (Detect X® Direct Cyclic GMP; Arbor Assays, MI, USA), and according to manufacturer’s instructions. Optical density was read at 450 nm, which was corrected at 570 nm with a Tecan Sunrise instrument (Tecan Nordic AB, Mölndal, Sweden) with Magellan software. For normalization, the protein concentration in the tissue lysates was determined with a commercially available protein assay kit based on the Bradford assay (Coomassie Plus Assay Kit; Thermo Scientific, IL, USA).
Experimental protocol
Ventilatory and hemodynamic parameters were measured and blood was sampled for mixed venous and arterial blood gases 30 min after preparation and ventilation with hyperoxic gas [fraction of inspired O2 (FIO2) 0.8, balance nitrogen] to both lungs (baseline). The inhaled gas was then changed from hyperoxic to hypoxic (FIO2 0.05, balance nitrogen) to the LLL, and data were collected after 30 min of regional LLL hypoxia. The pigs were then randomized into the control group (n = 8) or hypercapnia group (n = 8). In the control group, regional LLL hypoxia was continued throughout the experiment. In the hypercapnia group, the LLL was ventilated with 10% CO2 in 5% O2, balance nitrogen; the rest of the lung was ventilated with 10% CO2 in 80% O2, balance nitrogen throughout the experiment. Data were collected every 30 min during 3.5 h in both groups.
ENO was measured alternately from the hypoxic LLL and the hyperoxic lung regions (HL). Blood flow in the main pulmonary artery (Q T) and Q LLL were measured continuously. At the end of each experiment, with the pig anesthetized, alive, and ventilated, pieces from the hypoxic LLL and HL were excised and immediately frozen in liquid nitrogen for analysis of NOS activity and cGMP. Finally, the pigs were euthanized with an intravenous injection of potassium chloride (KCl).
Statistical analyses
Data in the text and tables are presented as mean (SD). A two-way analysis of variance for repeated measures (ANOVA) on one factor was applied to disclose any interaction effects (pAB) or differences within or between groups (pA). Data collection periods (fixed) and pigs (random) were the two block factors for comparisons within the groups. Groups (fixed) and pigs (random) were the two block factors for comparisons between the groups. Tukey’s test was used as the post hoc test. A probability of <0.05 was accepted as significant. A one-way ANOVA was used for analysis of NOS activity and cGMP. All analyses were performed with Statistica (version 8; Statsoft Inc., Tulsa, OK, USA).
Results
Thirty minutes of CO2 inhalation (hypercapnia group) decreased pH from 7.38 (0.03) to 7.10 (0.02) (p < 0.01); the pH then slowly decreased to 7.01 (0.03). pH was normal in the control group throughout the experiment [7.40 (0.03)–7.37 (0.03)] (Table 1).
ENO and NOS activity
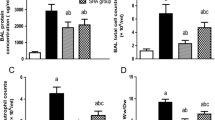
There were no differences between the groups for ENO from the hypoxic LLL or from the hyperoxic lung regions (Table 1), nor were there any differences between the hypercapnia and control groups for Ca2+-independent (iNOS) or Ca2+-dependent (cNOS) activity in hypoxic or hyperoxic lung regions (Fig. 1). In the hyperoxic lung, cGMP was 8.2 (6.5) pmol/mg in the control group and 4.1 (1.6) pmol/mg in the hypercapnia group (p = 0.1), and in hypoxic lung, cGMP was 3.7 (1.5) pmol/mg in the control group and 2.6 (1.4) pmol/mg in the hypercapnia group (p = 0.2).

Nitric oxide synthase activity in lung tissue (pmol g−1 min−1). Mean (standard error of the mean, SEM). There were no significant differences in the Ca2+-dependent or Ca2+-independent NOS activities between the groups
Pulmonary hemodynamics
Q LLL/Q T decreased by 72% (5%) during LLL hypoxia in the hypercapnia group and by 68% (9%) in the control group; there was no further change in Q LLL/Q T throughout the experiment in the control group. Inhalation of CO2 increased Q LLL/Q T (p < 0.01) from 6% (1%) to 9% (2%). Q LLL/Q T remained elevated for 1.5 h in the hypercapnia group and then declined to reach the same level as in the control group after 3.5 h (Fig. 2a). Mean pulmonary arterial pressure (MPaP) increased in the hypercapnia group, compared with the control group (p < 0.01). There were no differences between the groups for PVR, PVRLLL, or PVRHL (Table 2). The pulmonary arterial diastolic-pulmonary capillary wedge pressure (Ppadiast–PcwP) gradient increased after 90 min of CO2 inhalation in the hypercapnia group (p < 0.01) (Table 2).

a, b, c Mean (SEM). p values = significant interaction effect (pAB) of hypercapnia over time between the control group (n = 8) and the hypercapnia group (n = 8). a Q LLL/Q T. Blood flow to the LLL (Q LLL) in relation to cardiac output (Q T). b PaO2 (partial pressure of oxygen in arterial blood). c Cardiac output (Q T)
Blood gases and oxygen delivery
PaO2 decreased in both groups with LLL hypoxia. A further decrease was observed in the hypercapnia group (p < 0.01), but PaO2 remained unchanged in the control group (Fig. 2b). Oxygen delivery and PvO2 increased (p < 0.01) in the hypercapnia group, compared with the control group (Tables 1, 2).
Systemic hemodynamics
In the hypercapnia group, Q T increased after introduction of CO2 inhalation (p < 0.01) and remained elevated throughout the experiment, whereas Q T did not change over time in the control group (Fig. 2c). Central venous pressure (CVP) and PcwP did not differ between the groups, but heart rate (HR) increased (p < 0.01) and systemic vascular resistance (SVR) decreased (p < 0.01) in the hypercapnia group (Table 2). The intrapulmonary shunt (Q S/Q T) increased similarly in both groups with LLL hypoxia. A further increase was observed in the hypercapnia group (p < 0.01), whereas Q S/Q T remained unchanged in the control group (Table 2).
Discussion
The major finding in the present study was that hypercapnic acidosis did not affect endogenous NO production, in either hypoxic or hyperoxic lung regions, as indicated by the lack of changes in ENO, NOS activity, and cGMP. Hypercapnic acidosis did not potentiate HPV during the time span of the study. However, a transient weakening of HPV was observed.
Hypercapnic acidosis and endogenous NO production
ENO measured from a tracheostoma, as in this study, reflects NO production from the lower airways, providing that other factors that influence ENO, such as changes in ventilation, PEEP, FIO2, and total and regional blood flow [14], remain constant. To eliminate any influence on the results, CO2 was added to the inspiratory gas mixture to induce hypercapnic acidosis, and the ventilator settings and PEEP were kept constant.
Q T increased during hypercapnia. An increased pulmonary blood flow increases shear stress, stimulates endothelial NOS (eNOS) activity, and increases ENO [15]. ENO can also be expected to decrease due to increased pulmonary blood flow, as more NO is scavenged by the blood and less escapes into the exhaled air [14]. In the present study, no changes in ENO from either hypoxic or hyperoxic lung regions were observed during hypercapnic acidosis.
Severe hypoxia, not compatible with survival, decreases ENO, which is consistent with NO synthesis from l-arginine requiring molecular oxygen [14, 16, 17]. Moderate hypoxia, compatible with life, does not affect, or increases ENO [16–19]. Increased NO production during hypoxia is proposed [20, 21] as an intrinsic system for protecting the individual from injurious pulmonary hypertension and risk of right heart failure.
iNOS activity was generally lower than cNOS activity in both groups, which concurred with findings from previous studies in healthy and endotoxemic pigs [22, 23]. Enzymes operate at an optimal intracellular pH, which can differ depending on cell type. Endothelial NOS activity increases during alkalosis via influx of extracellular calcium and decreases during acidosis [24]; in cultured cells [24, 25], alkalosis favors cNOS activity and acidosis favors iNOS activity. However, we could not demonstrate that hypercapnic acidosis caused any statistically significant changes in cNOS or iNOS activity in either hypoxic or hyperoxic lung regions in pigs.
Hypercapnic acidosis and HPV
Hypercapnic acidosis increased both Q T and Q LLL/Q T during the first 90 min; HPV was transiently attenuated. There are several mechanisms related to hypercapnia-induced Q T increase: first, hypercapnia can initiate a sympathetically mediated release of catecholamines due to neuroadrenal stimulation [26, 27]; second, hypercapnic acidosis induces adenosine triphosphate (ATP)-sensitive K+ channel-mediated vasodilatation [28]; and, third, preload may be increased via venoconstriction in acidemia [26]. Therefore, Q T may increase because of increased preload, decreased afterload, and increased contractility. Increased heart rate and decreased afterload were the most obvious mechanisms observed in this study.
Intrapulmonary shunt (Q S/Q T) varies directly with Q T. An increased Q T decreases HPV due to a combination of increases in PvO2 [29, 30], pulmonary artery pressure [31], and pulmonary blood flow [32, 33]. All these factors probably contributed to the transient decrease in HPV. The increase in Q T and PvO2 persisted, whereas Q LLL/Q T slowly declined to values similar to the control group. Simultaneous with the transient increase in Q LLL, a decrease in PVRLLL was observed (Table 2), but this decrease did not reach statistical significance (p = 0.07). PVRHL did not increase in the hypercapnic pigs, indicating that the transient redistribution of pulmonary blood flow to the LLL could not be explained by higher impedance in the hyperoxic lung regions.
Acidosis causes dilatation in the systemic vasculature and constriction in the pulmonary vasculature, because pH differentially regulates voltage-gated potassium channels in pulmonary and systemic vascular smooth muscle cells, thus modulating vascular reactivity [34]. Hypercapnic acidosis has two components: the CO2 molecule and the resulting activity of hydrogen ions (pH).
The vasoactive action of CO2 is dependent on the initial PVR. During basal tone condition, CO2 is a mild vasoconstrictor, whereas, at high PVR, such as in hypoxia, it is a potent vasodilator [5, 35–37]. The effect of CO2 dilatation is proposed to have a direct action on smooth muscle, and constriction is caused by decreasing pH [37]. Hence, depending on the balance between the dilating effect of the CO2 molecule and the vasoconstrictive effect of the hydrogen ion, hypercapnia can be expected to both attenuate and augment HPV.
The observed transient decrease in HPV was attributed to both the vasodilating effect of the CO2 molecule and the increase in Q T. This effect was mainly caused by hypercapnia, as acidosis per se augments HPV. However, as the intracellular pH decreased, the vascular tone in the hypoxic lung regions increased and HPV returned to initial values. Extracellular pH decreased rapidly to 7.10, and then slowly to pH 7.01. The rate of change in intracellular pH might differ from the rate of change in extracellular pH, as intracellular pH in the pulmonary capillary bed is regulated by membrane-bound carbonic anhydrase [38]; CO2 freely crosses cell membranes, whereas hydrogen ions do not.
The effects of hypercapnic acidosis on HPV vary depending on species and experimental models [5, 6, 39]. The use of isolated lungs perfused with blood-free solution and constant Q T [6, 7, 40] or the use of intact animals will yield different results. The perfused lung is denervated and isolated from the systemic circulation, with particular concerns being related to the lack of pulmonary–systemic interaction. The intact large animal model has the advantage of more closely resembling the clinical situation and physiology in patients, although data in animals cannot readily be extrapolated to humans.
There was no change in PVR in the hypercapnic pigs in this study; however, the calculation of PVR becomes difficult to interpret as Q T increases. Therefore, the Ppadiast–PcwP gradient may be more accurate for expressing resistance to flow through the pulmonary vascular bed [41]. The Ppadiast–PcwP gradient increased in the hypercapnia group after 90 min of CO2 inhalation, which indicated an increased pulmonary vascular tone. Thus, pulmonary hypertension in the hypercapnic pigs could be explained by increased pulmonary blood flow and increasing pulmonary vascular tone over time.
Hypercapnic acidosis and oxygenation
Hypercapnic acidosis decreased PaO2. The hypercapnia-induced increase in Q T and intravascular pulmonary pressure combined with a vasodilatory effect of CO2 resulted in recruitment of pulmonary vessels in poorly ventilated lung regions and led to an increase in Q S/Q T. A decreased alveolar partial pressure of oxygen (PAO2), due to the increase in inspiratory CO2 partial pressure, was considered an additional explanation for the decrease in PaO2. However, as the calculation of PAO2 revealed no differences between the groups, it was reasonable to conclude that the decrease in PaO2 was explained by increased shunt and ventilation/perfusion (V/Q) mismatch. Although PaO2 decreased, the net effect of hypercapnic acidosis on tissue oxygenation was increased oxygen delivery caused by increased Q T.
Limitations
In this study, the pigs were healthy, which is contrary to the clinical situation, where hypercapnic acidosis typically occurs in hemodynamically and respiratory compromised critically ill patients, where particular concerns are related to oxidative stress, iNOS induction, inflammatory mediators, etc. Hypercapnic acidosis was induced by inhalation of CO2, instead of permissive hypercapnia mimicking the clinical situation. However, any changes in the ventilator settings would have affected ENO, thus rendering it impossible to evaluate the effects of hypercapnic acidosis on endogenous pulmonary NO production, as was the aim of the study. If pH reduction is the same, similar results would be expected irrespective of whether hypercapnia was induced through low minute ventilation or CO2 inhalation. The study design was considered appropriate for eliminating as many confounding factors as possible and standardizing the level of hypercapnic acidosis throughout the lung.
Conclusions
Hypercapnic acidosis did not potentiate HPV; instead, a transient weakening of HPV was observed. This effect was mainly caused by hypercapnia, as acidosis per se augments HPV. Hypercapnic acidosis did not affect endogenous NO production in either hypoxic or hyperoxic lung regions, as indicated by the lack of changes in ENO, NOS activity, or cGMP. Cardiac output increased during hypercapnic acidosis, resulting in increased oxygen delivery. At the same time, PaO2 decreased due to increased shunt and/or V/Q mismatch.
References
Amato MB, Barbas CS, Medeiros DM, Magaldi RB, Schettino GP, Lorenzi-Filho G, Kairalla RA, Deheinzelin D, Munoz C, Oliveira R, Takagaki TY, Carvalho CR (1998) Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med 338:347–354
Feihl F, Perret C (1994) Permissive hypercapnia. How permissive should we be? Am J Respir Crit Care Med 150:1722–1737
Gordon JB, Rehorst-Paea LA, Hoffman GM, Nelin LD (1999) Pulmonary vascular responses during acute and sustained respiratory alkalosis or acidosis in intact newborn piglets. Pediatr Res 46:735–741
Chang AC, Zucker HA, Hickey PR, Wessel DL (1995) Pulmonary vascular resistance in infants after cardiac surgery: role of carbon dioxide and hydrogen ion. Crit Care Med 23:568–574
Brimioulle S, Lejeune P, Vachiery JL, Leeman M, Melot C, Naeije R (1990) Effects of acidosis and alkalosis on hypoxic pulmonary vasoconstriction in dogs. Am J Physiol 258:H347–H353
Balasubramanyan N, Halla TR, Ghanayem NS, Gordon JB (2000) Endothelium-independent and -dependent vasodilation in alkalotic and acidotic piglet lungs. Pediatr Pulmonol 30:241–248
Ketabchi F, Egemnazarov B, Schermuly RT, Ghofrani HA, Seeger W, Grimminger F, Shid-Moosavi M, Dehghani GA, Weissmann N, Sommer N (2009) Effects of hypercapnia with and without acidosis on hypoxic pulmonary vasoconstriction. Am J Physiol Lung Cell Mol Physiol 297:L977–L983
Carr P, Graves JE, Poston L (1993) Carbon dioxide induced vasorelaxation in rat mesenteric small arteries precontracted with noradrenaline is endothelium dependent and mediated by nitric oxide. Pflugers Arch 423:343–345
Najarian T, Marrache AM, Dumont I, Hardy P, Beauchamp MH, Hou X, Peri K, Gobeil F Jr, Varma DR, Chemtob S (2000) Prolonged hypercapnia-evoked cerebral hyperemia via K(+) channel- and prostaglandin E(2)-dependent endothelial nitric oxide synthase induction. Circ Res 87:1149–1156
Archer SL, Tolins JP, Raij L, Weir EK (1989) Hypoxic pulmonary vasoconstriction is enhanced by inhibition of the synthesis of an endothelium derived relaxing factor. Biochem Biophys Res Commun 164:1198–1205
Freden F, Wei SZ, Berglund JE, Frostell C, Hedenstierna G (1995) Nitric oxide modulation of pulmonary blood flow distribution in lobar hypoxia. Anesthesiology 82:1216–1225
Naeije R, Brimioulle S (2001) Physiology in medicine: importance of hypoxic pulmonary vasoconstriction in maintaining arterial oxygenation during acute respiratory failure. Crit Care 5:67–71
Nilsson MC, Freden F, Wiklund P, Hambraeus-Jonzon K (2011) No effect of metabolic acidosis on nitric oxide production in hypoxic and hyperoxic lung regions in pigs. Acta Physiol (Oxf) 202:59–68
Carlin RE, Ferrario L, Boyd JT, Camporesi EM, McGraw DJ, Hakim TS (1997) Determinants of nitric oxide in exhaled gas in the isolated rabbit lung. Am J Respir Crit Care Med 155:922–927
Uematsu M, Ohara Y, Navas JP, Nishida K, Murphy TJ, Alexander RW, Nerem RM, Harrison DG (1995) Regulation of endothelial cell nitric oxide synthase mRNA expression by shear stress. Am J Physiol 269:C1371–C1378
Warren JB, Maltby NH, MacCormack D, Barnes PJ (1989) Pulmonary endothelium-derived relaxing factor is impaired in hypoxia. Clin Sci (Lond) 77:671–676
Kantrow SP, Huang YC, Whorton AR, Grayck EN, Knight JM, Millington DS, Piantadosi CA (1997) Hypoxia inhibits nitric oxide synthesis in isolated rabbit lung. Am J Physiol 272:L1167–L1173
Hambraeus-Jonzon K, Chen L, Freden F, Wiklund P, Hedenstierna G (2001) Pulmonary vasoconstriction during regional nitric oxide inhalation: evidence of a blood-borne regulator of nitric oxide synthase activity. Anesthesiology 95:102–112
Hampl V, Cornfield DN, Cowan NJ, Archer SL (1995) Hypoxia potentiates nitric oxide synthesis and transiently increases cytosolic calcium levels in pulmonary artery endothelial cells. Eur Respir J 8:515–522
Hampl V (1997) The role of endogenous nitric oxide in acute hypoxic pulmonary vasoconstriction. In: Nitric oxide and the lung, Marcel Dekker Inc, pp 113–127
Frasch HF, Marshall C, Marshall BE (1999) Endothelin-1 is elevated in monocrotaline pulmonary hypertension. Am J Physiol 276:L304–L310
Rimeika D, Wiklund NP, Lindahl SG, Wiklund CU (2006) Regional differences in nitric oxide-mediated vasorelaxation in porcine pulmonary arteries. Acta Anaesthesiol Scand 50:947–953
Fujii Y, Goldberg P, Hussain SN (1998) Intrathoracic and extrathoracic sources of exhaled nitric oxide in porcine endotoxemic shock. Chest 114:569–576
Mizuno S, Demura Y, Ameshima S, Okamura S, Miyamori I, Ishizaki T (2002) Alkalosis stimulates endothelial nitric oxide synthase in cultured human pulmonary arterial endothelial cells. Am J Physiol Lung Cell Mol Physiol 283:L113–L119
Huang CJ, Haque IU, Slovin PN, Nielsen RB, Fang X, Skimming JW (2002) Environmental pH regulates LPS-induced nitric oxide formation in murine macrophages. Nitric Oxide 6:73–78
Walley KR, Lewis TH, Wood LD (1990) Acute respiratory acidosis decreases left ventricular contractility but increases cardiac output in dogs. Circ Res 67:628–635
Brofman JD, Leff AR, Munoz NM, Kirchhoff C, White SR (1990) Sympathetic secretory response to hypercapnic acidosis in swine. J Appl Physiol 69:710–717
Nakahata K, Kinoshita H, Hirano Y, Kimoto Y, Iranami H, Hatano Y (2003) Mild hypercapnia induces vasodilation via adenosine triphosphate-sensitive K + channels in parenchymal microvessels of the rat cerebral cortex. Anesthesiology 99:1333–1339
Sandoval J, Long GR, Skoog C, Wood LD, Oppenheimer L (1983) Independent influence of blood flow rate and mixed venous PO2 on shunt fraction. J Appl Physiol 55:1128–1133
Domino KB, Wetstein L, Glasser SA, Lindgren L, Marshall C, Harken A, Marshall BE (1983) Influence of mixed venous oxygen tension (PVO2) on blood flow to atelectatic lung. Anesthesiology 59:428–434
Benumof JL, Wahrenbrock EA (1975) Blunted hypoxic pulmonary vasoconstriction by increased lung vascular pressures. J Appl Physiol 38:846–850
Cheney FW, Colley PS (1980) The effect of cardiac output on arterial blood oxygenation. Anesthesiology 52:496–503
Wang Z, Su F, Bruhn A, Yang X, Vincent JL (2008) Acute hypercapnia improves indices of tissue oxygenation more than dobutamine in septic shock. Am J Respir Crit Care Med 177:178–183
Berger MG, Vandier C, Bonnet P, Jackson WF, Rusch NJ (1998) Intracellular acidosis differentially regulates KV channels in coronary and pulmonary vascular muscle. Am J Physiol 275:H1351–H1359
Viles PH, Shepherd JT (1968) Evidence for a dilator action of carbon dioxide on the pulmonary vessels of the cat. Circ Res 22:325–332
Baudouin SV, Evans TW (1993) Action of carbon dioxide on hypoxic pulmonary vasoconstriction in the rat lung: evidence against specific endothelium-derived relaxing factor-mediated vasodilation. Crit Care Med 21:740–746
Chuang IC, Dong HP, Yang RC, Wang TH, Tsai JH, Yang PH, Huang MS (2010) Effect of carbon dioxide on pulmonary vascular tone at various pulmonary arterial pressure levels induced by endothelin-1. Lung 188:199–207
Geers CGG, Heming TA, Bidani A, Crandall ED (1986) Effects of intra-and extracellular carbonic anhydrase on CO2 excretion and intravascular pH equilibrium in the isolated perfused rat lung. Prog Respir Res 21:26–29
Balanos GM, Talbot NP, Dorrington KL, Robbins PA (2003) Human pulmonary vascular response to 4 h of hypercapnia and hypocapnia measured using Doppler echocardiography. J Appl Physiol 94:1543–1551
Barer GR, Howard P, Shaw JW (1970) Sensitivity of pulmonary vessels to hypoxia and hypercapnia. J Physiol 206:25P–26P
Sibbald WJ, Paterson NA, Holliday RL, Anderson RA, Lobb TR, Duff JH (1978) Pulmonary hypertension in sepsis: measurement by the pulmonary arterial diastolic-pulmonary wedge pressure gradient and the influence of passive and active factors. Chest 73:583–591
Acknowledgments
The authors thank Prof. Göran Hedenstierna M.D. Ph.D., head of the laboratory, Agneta Ronéus, Biomedical Engineer, and Maria Lundqvist, Biomedical Engineer, of the Hedenstierna Laboratory, University Hospital, Uppsala, Sweden, for invaluable support and help with the experiments. The study was supported by grants from the Swedish Research Council (no. 5315); the Swedish Heart–Lung Fund; the Tore Nilsson Research Fund, Stockholm, Sweden; The Selander Research Fund, Uppsala, Sweden; AGA AB Medical Research Fund, Lidingö, Sweden; and, The Laerdal Foundation, Stavanger, Norway.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was carried out at the Department of Clinical Physiology, The Hedenstierna Laboratory, University Hospital, Uppsala, Sweden.
Rights and permissions
About this article
Cite this article
Nilsson, M.C.A., Fredén, F., Larsson, A. et al. Hypercapnic acidosis transiently weakens hypoxic pulmonary vasoconstriction without affecting endogenous pulmonary nitric oxide production. Intensive Care Med 38, 509–517 (2012). https://doi.org/10.1007/s00134-012-2482-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-012-2482-7