
Abstract
Background
Systemic inflammation and sepsis are accompanied by severe metabolic alterations, including insulin resistance together with increased levels of triglycerides (TGs) and decreases in high- and low-density lipoproteins. Clinical studies have clearly established a link between lipid metabolism and systemic inflammation. Lipoproteins were shown to neutralize LPS and to exert direct anti-inflammatory actions. High- and low-density lipoproteins are thus thought to be important regulators of the host immune response during endotoxemia, which may also have the potential of improving the care of patients with Gram-negative sepsis.
Discussion
Nutritional lipids supplied during critical illness have been shown to modulate the host response to inflammation. In particular, inclusion of ω-3 fatty acids seems to have beneficial effects on cellular immunity and helps to maintain the balance between pro- and anti-inflammatory cytokines thereby preventing hyperinflammatory complications. In addition to improvements in the profile of lipid mediators generated, ω-3 fatty acids act as activating ligands of peroxisome proliferator-activated receptors and directly inhibit nuclear factor κB mediated proinflammatory signaling. We present an overview on the alterations in the metabolism of serum lipoproteins during sepsis and present data from clinical studies and discuss the significance of nutritional lipids and their role in immunomodulation with special emphasis on ω-3 fatty acids.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In recent decades intensive collaborative research in the fields of chronic and acute inflammatory disorders has resulted in a better understanding of the pathophysiology and diagnosis of these diseases. Modern therapeutic approaches, however, remain unsatisfactory, and shock, sepsis, and multiple organ failure (MOF) are still great challenges in intensive care medicine. Clinical studies have established a link between lipid metabolism and systemic inflammation. Insulin resistance together with increased levels of triglycerides (TGs) and decreases in high- (HDL) and low-density lipoproteins (LDL) are prominent features of these metabolic disturbances. Lipoproteins were shown to neutralize lipopolysaccharide (LPS) and to exert direct anti-inflammatory actions, as currently reviewed in detail by Murch and coworkers. Thus HDL and LDL lipoproteins are thought to be important regulators of the host immune response during endotoxemia.
Recent research in the field of peroxisome proliferator-activated receptors (PPARs) has opened new perspectives in understanding the interplay between inflammation and lipid homeostasis and may offer additional therapeutic opportunities. Key enzymes of cellular lipid metabolism are regulated by PPARs, and LPS as well as inflammatory cytokines downregulated PPAR-isoforms in liver and heart [1, 2]. In addition, single-nucleotide polymporphisms of the PPAR-δ gene, the predominant PPAR isoform expressed in skeletal muscle, have been demonstrated to be associated with differences in insulin sensitivity by modifying skeletal muscle glucose uptake [3]. However, whether these single-nucleotide polymporphisms are clinically relevant in the context of the systemic inflammation and sepsis has not been determined. In addition to its insulin-sensitizing effect the activation of PPAR-γ was also shown to inhibit nuclear factor (NF) κB induced transcription of proinflammatory genes [4] and a PPAR agonist improved survival in an animal model of endotoxemia [5]. Thus they seem to be attractive therapeutic tools not only in the therapy of type 2 diabetes but also during the systemic inflammation and sepsis.
The first part of this review discusses the changes in lipid metabolism and lipoprotein composition in light of results from clinical studies. As studies in subjects with insulin resistance and type 2 diabetes mellitus also show marked derangements in the regulation of reverse cholesterol transport that can in part be overcome by insulin treatment, we give special emphasis to the recent report by Mesotten and coworkers [6] who demonstrated that the improvements in morbidity and mortality obtained by intensive insulin therapy in critically ill patients are partially attributable to increases in the serum levels of LDL and HDL lipoproteins. The second part of the review discusses the impact of nutritional lipids supplied during critical illness with special emphasis on polyunsaturated ω-3 fatty acids (FA). ω-3 FA have been shown to modulate the host response to inflammation by different mechanisms. In addition to the improvements in the profile of lipid mediators generated, they act as activating ligands of PPARs.
Changes in plasma lipid profile of critically ill patients
During the course of infection significant changes in patients' serum profile of lipids, lipoproteins, and lipoprotein-associated proteins are observed [7, 8, 9, 10]. Whereas moderate (surgical) stress increases TG clearance due to an increased total body fat oxidation, serum TG levels and very low density lipoproteins (VLDL) frequently increase in septic conditions because of reduced TG hydrolysis and fat oxidation. LPS and proinflammatory cytokines such as tumor necrosis factor (TNF) α, interleukin (IL) 1, and IL-6 rapidly induce de novo FA and hepatic TG synthesis [11, 12] which may even result in a fatty liver in severely ill patients. In particular, this was a problem when therapeutic hyperalimentation was common in the care of critically ill patients in the past. At that time nutritional goals were set to limit negative nitrogen balances with up to 4,000 kcal or 1 kg glucose per day. While FA are the primary energy source of hepatocytes, the myocardium, and the skeletal muscle in the critically ill, monitored administration of lipids is crucial in the nutritional care of these patients. Plasma TG levels resulting from lipolysis or from artificial administration further depend on the efficacy by which VLDL TGs are removed from the circulation. Critical determinants of VLDL clearance are the activity of lipoprotein lipase (LPL) and the subsequent tissue uptake of remnant particles. In most surgical patients these mechanisms are not impaired or may even be accelerated and TG levels are normal or lowered [13]. High levels of endotoxin, however, as occurring in severe sepsis depress the activity of LPL leading to elevated TG plasma levels [14]. The increase in free FA induces insulin resistance and thereby contributes to elevated blood glucose levels during systemic inflammation. Diacylglycerol, an intermediate of TG metabolism, leads to sustained activation of protein kinase C (PKC) [15]. This in turn reduces insulin-mediated cellular glucose uptake by interfering with the insulin-receptor signal transduction cascade ultimately leading to reduced translocation of glucose transporter 4 (GLUT 4) to the cell surface [16, 17]. In addition, activated PKC reduced the amount of NF-κB inhibitory protein α, the physiological inhibitor of NF-κB activation [17]). Thus, increased plasma TG levels are critical determinants of inflammation-induced insulin resistance and are capable of amplifying the proinflammatory response.
In contrast to the elevations in plasma TG levels, total cholesterol, HDL and LDL cholesterol, HDL and LDL, are reduced in serum of patients with sepsis [7, 8, 18, 19, 20]. These changes occur early during the course of systemic inflammation with cholesterol content in LDL and HDL decreasing within hours and are attributed to the effects of LPS and cytokines (Fig. 1a). Whether cytokines directly inhibit enzymes involved in cholesterol synthesis is currently unknown. On the side of cholesterol catabolism it is noteworthy that LPS and cytokines inhibit both the classical and the alternative pathways of cholesterol excretion [21, 22].

a HDL levels (mean ± SD) in 83 severely ill patients after surgery; *p < 0.05 vs. day 1 (Bonferroni's adjusted GLM). SAPS II distribution see b. b Scatter plot of the SAPS II score vs. HDL on postoperative day 5; p = 0.005 (Spearman's rank correlation); dotted line individual 95% confidence interval. cOpen circles Survivors; closed circles nonsurvivors. HDL levels in survivors and nonsurvivors in the subgroup with abdominal sepsis. (Data taken from [38], in parts unpublished)
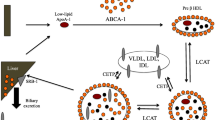
During infection and inflammation there are also important changes in lipoprotein composition (Table 1), especially in the case of HDL that markedly affect HDL metabolism itself [7, 10, 23]. Normal HDL contains the apoproteins apolipoprotein (apo) A-I and apoA-II and associated enzymes and transport proteins that are essential for reverse cholesterol transportlike lecithin cholesteryl acyltransferase (LCAT), cholesteryl ester transfer protein (CETP), and phospholipid transfer protein (PLTP). HDL also carries antioxidative proteins such as paraoxonase, platelet-activating factor acetylhydrolase, protectin, transferrin, and clusterin which serve to protect membranes by scavenging radicals and inhibiting oxidation processes.
Acute-phase HDL is smaller in size and contains less esterified cholesterol, and apoA-I is replaced by serum amyloid A (SAA). SAA also displaces paraoxonase decreasing the antioxidative capacity of HDL [25, 26, 27]. In addition, reductions in HDL-associated LCAT [28] and CETP [29] occur while apoE-containing HDL and PLTP activity were increased in serum from patients with a systemic inflammatory response, despite reduced PLTP mass [8, 9, 27]). The increased PLTP activity together with enhanced HDL TG and reduced LCAT and CETP content were shown to promote HDL conversion to pre-β-HDL with low content of esterified cholesterol and shedding of small apoA-I containing particles [8, 26]. Overexpression of human PLTP in mice has been demonstrated to enhance liver cholesteryl ester content and excretion of bile acid [30]. Finally, the inflammation-induced reduction in LPL activity also contributes to the fall in HDL cholesterol [31] as reduced amounts of free cholesterol are provided to be incorporated into HDL.
In critically ill patients reductions in lipoprotein content were shown to be correlated with the severity of disease [32] and low cholesterol levels were associated with higher mortality [33] and infectious complications [34]. Reduced HDL levels were associated with poor clinical outcome in critically ill [35, 36] and burn patients [37] and negatively correlated with the length of stay in the ICU and serum concentrations of the proinflammatory cytokines TNF-α and IL-6 [36]). Our own observations (Fig. 1a–c) showed significant reduction in HDL levels in severely ill patients over time, an inverse correlation between plasma HDL and the severity of disease and lower levels of HDL in patients with sepsis who died [38]. Reductions in apoA-I serum levels were associated with systemic inflammatory response syndrome exacerbations in surgical ICU patients [39] and increased mortality in patients with severe sepsis [36]. However, these data do not allow determining whether the changes in plasma cholesterol and lipoproteins simply reflect the severity of inflammation, whether they are an epiphenomenon of other regulatory processes, or whether they are directly causative in modifying the host response to inflammation and have a direct influence on mortality. To clarify these questions data from in vitro cell culture experiments and studies in animal models of acute inflammation and sepsis are indispensable.
Impaired insulin signaling is a major feature of the metabolic disturbances during systemic inflammation and sepsis. In addition to the actions of counterregulatory hormones and catecholamines, also increased plasma TGs and free FA impair insulin signaling as outlined above.
As septic patients type 2 diabetics are characterized by increased plasma TG levels, reduced HDL content, and insulin resistance. In both sepsis and type 2 diabetes mellitus PLTP activity is elevated [8, 9, 40]. From studies in patients with type 2 diabetes mellitus it is known that insulin regulates serum lipoprotein levels. Insulin reduced plasma TG levels and lowered PLTP activity [40]. However, the latter effect was blunted under conditions of insulin resistance indicative of impaired insulin action.
Hyperglycemia per se has been demonstrated to affect reverse cholesterol transport. In vitro net cholesterol transport from fibroblasts to severely hyperglycemic plasma was impaired and the effect was reversed when blood glucose was lowered by insulin [41]. However, supraphysiological concentrations of insulin were also shown to decrease reverse cholesterol transport [42]. On the other hand, cell membrane cholesterol depletion reduced the effect of insulin on gene transcription and cell surface expression of GLUT 4 and impaired cellular glucose uptake in adipocytes [43]. Whether insulin regulates the expression of other gate-keeper proteins of cholesterol trafficking such as ATP-binding cassette transporter 1 and the scavenger receptor class B type I is currently unknown, and further research is warranted.
The importance of tight glycemic control by intensive insulin treatment in postoperative ICU patients was recently demonstrated in the randomized controlled trial of intensive insulin therapy by Van den Berghe and coworkers [44]. In a subgroup analysis of the same trial Mesotten and coworkers analyzed patients with prolonged critical illness and an ICU stay of more than 7 days [6]. In this patient group intensive insulin treatment not only normalized blood glucose levels but also modified the serum levels of circulating lipids. In conventionally treated patients TGs increased on day 8 compared to day 1. This increase was not observed in the intensive insulin treatment group. In this group serum TG levels were significantly reduced as early as day 1. Notably, intensive insulin treatment did not affect serum TG in nonsurvivors.
Intensive insulin treatment also affected the serum levels of LDL and HDL lipoproteins. While in both treatment groups HDL levels decreased over time, this decrease was less severe in the intensive insulin treatment group. LDL serum levels were decreased on day 1 and increased on day 8. Again, patients with intensive insulin treatment exhibited higher LDL serum levels on day 8 than conventionally treated patients. While there was a nearly linear correlation between serum TGs and mortality, there seemed to be cutoff levels for both LDL (20 mg/dl) and HDL (15 mg/dl) below which mortality strongly increased. Multivariate regression analysis showed the effect of intensive insulin therapy to be independent of glycemic control and TG levels, but in part this was attributable to the effects of intensive insulin treatment on HDL and LDL serum levels.
Higher insulin dosing, possibly reflecting the severity of insulin resistance, was an independent risk factor for morbidity and mortality. Whether genetic polymorphisms, for example, in the skeletal muscle PPAR-δ determining insulin sensitivity, as recently reported by Vättinen and coworkers [3], contribute to this phenomenon is unknown. Future work in this field will probably enhance our understanding of the underlying mechanisms and help to identify specific at-risk patient subgroups and hopefully allow us to direct therapeutic strategies according to the patient's individual risk profile.
Modulation of LPS-induced inflammation by lipoproteins and apoproteins
The protective roles of lipoproteins in neutralizing LPS have been extensively studied and also direct anti-inflammatory actions such as reductions in cellular adhesion molecule and inducible nitric oxide synthase expression were reported and are reviewed in detail by Murch and coworkers in this issue.
Complexes of LPS with lipoproteins were shown to have little or no stimulatory effect on cytokine production either in vitro or in vivo [36, 45, 46, 47]. Notably, LPS-neutralization efficiency varies for lipopolysaccharides derived from different bacterial species [47]. Kitchens and coworkers [48] showed that lipoproteins are essential for the release of bound LPS from cell membranes, and that HDL is the main lipoprotein involved. Only small amounts of LPS were bound to LDL and even less to VLDL. Soluble CD14 (sCD14) accelerated the release of cell-bound LPS while lipopolysaccharide binding protein (LBP) had only a minor effect and PLTP did not affect LPS binding to HDL [49]. A role for LDL/VLDL and apoB apoprotein in binding LPS in serum has been established by Vreugdenhil and coworkers [50]. They demonstrated that the LBP in serum from both healthy individuals and patients with sepsis is bound mainly on apoB apoprotein present in LDL and VLDL and enhanced LPS binding to these lipoproteins. They also determined a tenfold higher affinity of LBP for apoB than for apoA-I. In addition to these findings, Levels and coworkers [32] reported that in plasma and lymph from patients with systemic inflammatory response syndrome and multiorgan failure, the LPS distribution shifted mainly toward LDL and not VLDL. In summary, these studies suggest that there are at least two different mechanisms of LPS neutralization, i.e., binding of free LPS in serum to apoB-containing LDL and, to a lesser extent, also VLDL lipoproteins, on the one hand, and release of cell-bound LPS from monocytes by HDL and sCD14, on the other. Interestingly, serum from patients with sepsis and severe sepsis induced a higher rate of LPS-release from monocytes than serum from normal individuals or patients with systemic inflammatory response syndrome. This occurred despite reduced HDL lipoprotein levels and was significantly correlated with the serum levels of sCD14 and other acute-phase proteins such as serum amyloid A, LBP, and serum phospholipase A2 [49]. However, while these mechanisms are thought to prevent an exaggerated host response to LPS and are considered to be protective, the role of sCD14 is uncertain as high blood levels of sCD14 were associated with mortality in patients with Gram-negative sepsis [51].
Substitution of lipoproteins by infusion of reconstituted HDL (rHDL) has been proposed as therapeutic option. In animal models of endotoxemia the administration of rHDL decreased mortality [52, 53]. In humans the infusion of rHDL blunted endotoxin-induced procoagulant activation without adverse side effects [54]. However, the safety of rHDL administration is a concern as infusion of higher doses of rHDL has been demonstrated to promote growth of Candida albicans ex vivo [55] although the clinical significance of this finding is currently unknown.
One ongoing worldwide multicenter trial recruiting more than 1,800 patients with Gram-negative sepsis is attempting to establish the therapeutic efficacy of intravenous lipid administration on LPS neutralization. The proposed mechanism of action is to expand the phospholipid surface on plasma lipoproteins, for example, HDL and thereby facilitating LPS neutralization. Similar to rHDL, soy bean oil emulsions (ω-6 FA) have been claimed to be associated with infectious complications in a meta-analysis by Heyland et al. [56]. Thus the concept of administration of soybean oil as well as of rHDL seems to be double edged and must be considered cautiously, in particular in patients with suspected infection. In this regard, lipid emulsions with a reduced content of ω-6 FA in favor of medium chain TG, olive oil, or fish oil rich in ω-3 FA may be the better choice. Recently Lekka and coworkers [57] reported that total parenteral nutrition with medium-chain and long-chain TGs aggravated lung inflammation and deteriorated gas exchange in patients with acute respiratory distress syndrome. However, the adverse effects observed in their study may at least in part be due to to the rapid infusion rate used. From animal experimental data it seems that the inclusion of ω-3 FA may be useful in attenuating lipid induced pulmonary inflammation in acute lung injury [58, 59]. In recent years apolipoprotein mimetic peptides have been developed. Based on the observation that infusion of apoA-I is protective against LPS in mice [52], Gupta and coworkers [60] studied the effects of an apoA-I mimetic peptide (L-4F) on LPS-induced inflammatory responses in vitro and in vivo. They observed reductions in monocyte adhesion to endothelial cells and reduced expression of inflammatory cytokines and adhesion molecules. They further elucidated the mechanisms involved and demonstrated that L-4F reduces the association of LPS with LBP and consecutive adhesion of the LPS-LBP complex with endothelial cell surface receptors. In addition to their effectiveness in reducing atherosclerosis as shown previously [61], apolipoprotein mimetic peptides may therefore offer a therapeutic potential in modulating the host response to LPS.
Although the LPS-neutralizing effects of lipoproteins and apolipoprotein mimetic peptides are well characterized, the significance of these mechanisms in the context of bacterial infections is not as clear. Flegel and coworkers [47] demonstrated that the LPS-neutralizing capacity of lipoproteins is not correlated with the inhibition of cytokine release induced by whole Gram-negative bacteria in vitro. Thus the efficacy of therapeutic approaches aiming at neutralizing LPS by the use of lipoproteins and apolipoprotein mimetic peptides must be evaluated carefully in the clinical setting.
Modulation of LPS-induced inflammation by oxidized phospholipids
While quantities of phospholipids present in lipoproteins do not significantly change during inflammation, they are qualitatively altered by oxidation. Increased oxidative stress during systemic inflammation and a decrease in the antioxidative capacity due to decreases in paraoxonase activity in HDL particles contribute to oxidation of phospholipids. In addition, oxidized phospholipids are also released by necrotic and apoptotic cell death [62]. Oxidated phospholipids have been demonstrated to exert proinflammatory effects and to induce IL-8 and monocyte chemotactic peptide 1 expression in endothelial cells [63]. They are considered to mediate chronic inflammatory processes and thereby to contribute to the pathogenesis of atherosclerosis. In the context of acute bacterial inflammation, however, it is important to note that oxidized phospholipids inhibit LPS-mediated induction of NF-κB and consecutive gene expression [64, 65, 66]. This inhibition seems to involve at least two different mechanisms dependent on the cell type under investigation. Bochkov and coworkers [67] reported that oxidation products of 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (ox-PAPC) inhibit LPS induction of E-selectin in human umbilical vein endothelial cells, and that ox-PAPC prevents binding of LPS to immobilized LBP and CD14. From these data they concluded that ox-PAPC extracellularly sequester LPS and prevent activation of the LPS receptor complex. However, in a recent report Walton and coworkers [68] reported that ox-PAPC do not prevent LPS binding to Toll-like receptors (TLRs) in human aortic endothelial cells. In their study the inhibitory effect of ox-PAPC on LPS-mediated signal transduction was due to inhibition of ligand activation of TLR 2 and 4. They provided evidence that ox-PAPC prevent effective recruitment of some of the components of the LPS receptor complex to the lipid rafts/caveolae thereby preventing LPS-mediated signal transduction. In the context of systemic inflammation and sepsis these findings have two implications. First, the inhibition of the primary host response to LPS can result in protection from septic shock. However, it can also lead to impaired bacterial elimination resulting in enhanced susceptibility to bacterial infections. In addition, due to their chronic proinflammatory effects oxidized phospholipids may also contribute to the clinical course of protracted sepsis.
Modulation of LPS-induced inflammation by polyunsaturated fatty acids: therapeutic potential
ω-3 FA were shown to modify the serum lipid profile and to exert beneficial cardiovascular effects. They seem to be capable of lowering the plasma-TG levels, while HDL slightly increases, and LDL and cholesterol remain constant or even decrease slightly [69, 70, 71]. In addition to their impact on the development of atherosclerosis, ω-3 FA, as with eicosapentaeonic acid (EPA) and docosahexaeonic acid (DHA) are also recognized to modulate the systemic inflammatory response. As a result, improved outcome was observed in a broad range of diseases ranging from chronic disorders to critical care conditions. Reduction in tumor risk, however, could not be substantiated [72]. Experimental data demonstrate that ω-3 FA attenuate an overwhelming inflammatory reaction [73], ameliorate host defense [59, 73], and improve splanchnic blood flow and gut barrier function [74] in septic states. Diets enriched with ω-3 FA result in a change in the ω-3/ω-6 ratio in the membranous fatty acid composition of multiple cells [75, 76, 77] in favor of pentaenoic acids. In states of inflammation EPA is released from cellular membrane sources to compete with arachidonic acid for enzymatic metabolism producing less inflammatory and chemotactic derivatives. The EPA-derived metabolites have a lower biological activity [78, 79] than the analogous arachidonic acid derivatives. In addition, the generation of proinflammatory PAF is reduced by EPA via interference with the PAF precursor pool [80, 81].
Recent data suggest that in the context of LPS-induced inflammation ω-3 FA exhibit beneficial effects beyond their modulatory effect on the profile of inflammatory lipid mediators. Lee and coworkers [82, 83, 84] demonstrated that saturated fatty acids promote the activation of general proinflammatory pathways such as NF-κB and cyclooxygenase 2 expression (Fig. 2). They function as ligands for TLRs and activate TLR4 as well as dimers of TLR2 with TLR6 or TLR1 while polyunsaturated FA were inhibitory. ω-3 FA also affect proinflammatory signal transduction cascades downstream of TLR receptor signaling. ω-3 FA, as with EPA and DHA and their derivatives, are kown natural ligands of PPARs. In human kidney cells Li and coworkers [85] demonstrated that the EPA and DHA inhibit LPS-induced NF-κB activation via PPAR-γ.

Effects of ω-3 FA on the Toll-like receptor (TLR) nuclear factor κB (NFκB)-axis. While TLR-4 communicates the presence of a Gram-negative infection to the inner cell, TLR-2 reports the presence of Gram-positive bacteria. Saturated fatty acids (SFA) may likewise activate the TLR-4 cascade. Via inhibitory factor κB (IκB) kinase the blockade the function of IκB is postponed and NFκB may translocate into the nucleus to promote transcription of inflammatory genes. Consequently inflammatory receptors, enzymes, and cytokines are expressed; ω-3 eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) may competitively interfere with this sequence on various levels of the cascade. LBP Lipopolysaccharide-binding protein; LPS lipopolysaccharide; PPARs peroxisome proliferator-activated receptors
On the other hand, improved production of cytokines, increases in cell-mediated immunity, and opsonization index, and an increase in antigen-induced lymphocyte generation were induced by diets enriched in ω-3 FA [86, 87, 88, 89]. In clinical studies enteral nutrition with diets containing fish oil had restorative effects on the depressed cellular immunity of patients with critical illness and after major abdominal surgery [90, 91]. While these studies did not demonstrate a significant decrease in infectious complications or mortality, a prospective study in burn patients demonstrated that a diet containing fish oil significantly reduced wound infection, shortened hospital stay, and reduced mortality than standard enteral nutrition [92].
Most recently Pontes–Arruda reported improved survival in septic patients after ω-3 FA treatment [93].
Following major abdominal surgery we found that even short-term intravenous administration of ω-3 FA improves liver function without untoward effects on platelet function and coagulation [94]. Moreover, ω-3 FA helped to maintain the balance between pro- and anti-inflammatorycytokines, thereby preventing hyperinflammatory complications. Improved liver function in the group receiving a daily fish oil supplement of 0.2 g/kg resulted in significantly higher levels of VLDL than to sole soy bean oil infusion (Fig. 3). Because VLDL may likewise serve as an LPS scavenger, fish oil supplementation should be considered for this purpose rather than plain soybean oil which prolongs bacterial elimination compared to fish oil [59]. Beneficial effects of fish oil on outcome were confirmed in 661 patients who received fish oil infusion. Fish oil dose-dependently lowered the occurrence of comorbid infection and length of hospital stay and improved survival (Fig. 4) [38]. In a multifactor regression model the two main factors contributing to the length of stay were the amount of soybean oil (+1.6 d/100 g) and the delay in nutrition onset (+1.42 d/day of delay). These findings clearly point out the immunological impact of lipids in addition to being simple components of nutrition. Moreover, in line with the most recent guidelines of the German Society of Nutrition Medicine, these data show that the administration of soybean oil (ω-6 FA) should be limited to its essential dose. A combination of medium-chain TG, olive oil, and fish oil should then complete the lipid component of nutrition to cover 30–40% of daily nonprotein calorie load.

Levels of VLDL (means ± SEM) after major abdominal cancer surgery followed by total parenteral nutrition (TPN) supplemented with soybean oil (SO; 1 g/kg per day; gray columns ω-6) or with SO 0.8 g/kg per day and a fish oil (0.2 g/kg per day black columns ω-3 supplement); hatched columns baseline before TPN

Fish oil dose-related survival in the percentage of 661 ICU patients receiving total parenteral nutrition for at least 3 days (sepsis any cause n = 292; postoperative n = 255; others n = 114) significant differences from Tukey post hoc multiple comparison CANOVA with covariates age, Simplified Acute Physiology Score (SAPS) II score, and daily calorie intake, as indicated; *p < 0.05
Conclusion
Recent clinical studies have established a link between lipid metabolism and systemic inflammation. Lipoproteins were shown to neutralize LPS and consequently blunt its proinflammatory effects. Thus HDL and LDL lipoproteins are important regulators of the host immune response during endotoxemia. In line with this exogenous administration of lipoproteins may have beneficial therapeutic effects in the course of Gram-negative sepsis. To what extent this concept in fact improves clinical outcome needs to be confirmed in further studies.
References
Beigneux AP, Moser AH, Shigenaga JK, Grunfeld C, Feingold KR (2000) The acute phase response is associated with retinoid X receptor repression in rodent liver. J Biol Chem 275:16390–16399
Vänttinen M, Nuutila P, Kuulasmaa T, Pihlajamäki J, Hällsten K, Virtanen KA, Lautamäki R, Peltoniemi P, Takala T, Viljanen APM, Knuuti J, Laakso M (2005) Single nucleotide polymorphisms in the peroxisome proliferator-activated receptor δ gene are associated with skeletal muscle glucose uptake. Diabetes 54:3587–3591
Feingold K, Kim MS, Shigenaga J, Moser A, Grunfeld C (2004) Altered expression of nuclear hormone receptors and coactivators in mouse heart during the acute-phase response. Am J Physiol Endocrinol Metab 286(2):E201–207
Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK (2005) A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 437:759–763
Liu D, Zeng BX, Zhang SH, Yao SL (2005) Rosiglitazone, an agonist of peroxisome proliferator-activated receptor γ, reduces pulmonary inflammatory response in a rat model of endotoxemia. Inflamm Res 54:464–470
Mesotten D, Swinnen JV, Vanderhoydonc F, Woulters PJ, Van den Berghe G (2005) Contribution of circulating lipids to the improved outcome of critical illness by glycemic control with intensive insulin therapy. J Clin Endocrinol Metab 89:219–226
Khovidhunkit W, Kim MS, Memon RA, Shigenaga JK, Moser AH, Feingold KR, Grunfeld C (2004) Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences for the host. J Lipid Res 45:1169–1196
Pussinen PJ, Metso J, Malle E, Barlage S, Palosuo T, Sattler W, Schmitz G, Jauhiainen M (2001) The role of plasma phospholipid transfer protein (PLTP) in HDL remodeling in acute-phase patients. Biochim Biophys Acta 1533:153–163
Barlage S, Fröhlich D, Böttcher A, Jauhiainen M, Müller HP, Noetzel F, Rothe G, Schütt C, Linke R, Lackner KJ, Ehnholm C, Schmitz G (2001) ApoE-containing high density lipoproteins and phospholipids transfer protein activity increase in patients with a systemic inflammatory response. J Lipid Res 42:281–290
Carpentier YA, Scruel O (2002) Changes in the concentration and composition of plasma lipoproteins during the acute phase response. Curr Opin Clin Nutr Metab Care 5:153–158
Feingold KR, Grunfeld C (1987) Tumor necrosis factor-alpha stimulates hepatic lipogenesis in the rat in vivo. J Clin Invest 80:184–190
Nonogaki K, Fuller GM, Fuentes NL, Moser AH, Staprans C, Grunfeld C, Feingold KR (1995) Interleukin-6 stimulates hepatic triglyceride secretion in rats. Endocrinology 136:2143–2149
Akgün S, Ertel NH, Mosenthal A, Oser W (1998) Postsurgical reduction of serum lipoproteins: interleukin-6 and the acute-phase response. J Lab Clin Med 131:103–108
Feingold KR, Staprans I, Memon RA, Moser AH, Shigenaga JK, Doerrler W, Dinarello CA, Grunfeld C (1992) Endotoxin reapidly induces changes in lipid metabolism that produce hypertriglyceridemia: low doses stimulate hepatic triglyceride production while high doses inhibit clearance. J Lipid Res 33:1765–1776
Bronfman M, Morales MN, Orellana A (1988) Diacylglycerol activation of protein kinase C is modulated by long-chain acyl-CoA. Biochem Biophys Res Commun 152:987–992
Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, Cline GW, Slezak LA, Andersen DK, Hundal RS, Rothman DL, Petersen KF, Shulman GI (1999) Effects of free fatty acids on glucose transport and IRS-associated phosphatidylinositol 3-kinase activity. J Clin Invest 103:253–259
Itani SI, Ruderman NB, Schmieder F, Boden G (2002) Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and Iκ-Bα. Diabetes 51:2005–2011
Kitchens RL, Thompson PA, Munford RS, O'Keefe GE (2003) Acute inflammation and infection maintain circulating phospholipid levels and enhance lipopolysaccharide binding to plasma lipids. J Lipid Res 44:2339–2348
Fraunberger P, Schaefer S, Werdan K, Walli AK, Seidel D (1999) Reduction of circulating cholesterol and apolipoprotein levels during sepsis. Clin Chem Lab Med 37:357–362
Leeuwen HJ van, Heezius EC, Dallinga GM, van Strijp JA, Verhoef J, can Kessel JP (2003) Lipoprotein metabolism in patients with severe sepsis. Crit Care Med 31:1359–1366
Feingold KR, Spady DK, Pollock AS, Moser AH, Grunfeld C (1996) Endotoxin, TNF, and IL-1 decrease cholesterol 7 alpha-hydroxylase mRNA levels and activity. J Lipid Res 37:223–228
Memon RA, Moser AH, Shigenaga JK, Grunfeld C, Feingold KR (2001) In vivo and in vitro regulation of sterol 27-hydroxylase in the liver during the acute phase response. Potential role of hepatocyte nuclear factor-1. J Biol Chem 276:30118–30126
Wu A, Hinds CJ, Thiemermann C (2004) High-density lipoproteins in sepsis and septic shock: metabolism, actions, and therapeutic applications. Shock 21:210–221
Pruzanski W, Stefanski E, de Beer FC, de Beer MC, Vadas P, Ravandi A, Kuksis A (2000) Comparative analysis of lipid composition of normal and acute-phase high density lipoproteins. J Lipid Res 41:1035–1047
Cotzee GA, Strachan AF, van der Westhuyzen DR, Hoppe HC, Jeenah MS, de Beer FC (1986) Serum amyloid A-containing human high density lipoprotein 3. Density, size, and apolipoprotein composition. J Biol Chem 261:9644–9651
Cabana VG, Lukens JR, Rice KS, Hawkins TJ, Getz GS (1996) HDL content and composition in acute phase response in three species: triglyceride enrichment of HDL a factor in its decrease. J Lipid Res 37:2662–2674
Cabana VG, Reardon CA, Feng N, Neath S, Lukens J, Getz GS (2003) Serum paraoxonase: effect of the apolipoprotein composition of HDL and the acute phase response. J Lipid Res 44:780–792
Ettinger WH, Miller LD, Albers JJ, Smith TK, Parks JS (1990) Lipopolysaccharide and tumor necrosis factor cause a fall in plasma concentration of lecithin: cholesteryl acyltransferase and lipase deficiency in cynomolgus monkeys. J Lipid Res 31:1099–1107
Masucci-Magoulas L, Moulin P, Jiang XC, Richardson H, Walsh A, Breslow JL, Tall A (1995) Decreased cholesteryl ester transfer protein (CETP) mRNA and protein and increased high density lipoprotein following lipopolysaccharide administration in human CETP transgenic mice. J Clin Invest 95:1587–1594
Post SM, de Crom R, van Haperen R, van Tol A, Princen MG (2003) Increased fecal bile acid excretion in transgenic mice with elevated expression of human phospholipid transfer protein. Arterioscler Thromb Vasc Biol 23:892–897
Feingold KR, Marchall M, Gulli R, Moser AH, Grunfeld C (1994) Effect of endotoxin and cytokines on lipoprotein lipase activity in mice. Arterioscler Thromb Vasc Biol 14:1866–1872
Levels JHM, Lemaire LCJM, van den Ende AE, van Deventer SJH, Lanschot JJB (2003) Lipid composition and lypopolysaccharide binding capacity of lipoproteins in plasma and lymph of patients with systemic inflammatory response syndrome and multiple organ failure. Crit Care Med 31:1647–1653
Schatz IJ, Masaki K, Yano K, Chen R, Rodriguez BL, Curb JD (2001) Cholesterol and all-cause mortality in elderly people from the Honolulu Heart Program: a cohort study. Lancet 358:351–353
Iribarren C, Jacobs, DR Jr, Sidney S, Claxton AJ, Feingold KR (1998) Cohort study of serum total cholesterol and in-hospital incidence of infectious diseases. Epidemiol Infect 121:335–347
Gordon BR, Parker TS, Levine DM, Saal SD, Wang JC, Sloan BJ, Barie PS, Rubin AL (2001) Relationship of hypolipidemia to cytokine concentrations and outcomes in critically ill surgical patients. Crit Care Med 29:1563–1568
Chien JY, Jerng JS, Yu CJ, Yang PC (2005) Low serum level of high-density lipoprotein is a poor prognostic factor for severe sepsis. Crit Care Med 33:1688–1693
Vanni HE, Gordon BR, Levine DM, Sloan BJ, Stein DR, Yurt RW, Saal SD, Parker TS (2003) Cholesterol and interleukin-6 concentrations relate to outcomes in burn-injured patients. J Burn Care Rehabil 24:133–141
Heller AR, Rössler S, Litz RJ, Stehr SN, Heller SC, Koch R, Koch T (2006) Omega-3 fatty acids improve the diagnosis-related clinical outcome. Crit Care Med 34:972–979
Chenaud C, Merlani PG, Roux-Lombard P, Burger D, Harbarth S, Luyasu S, Graf JD, Dayer JM, Ricou B (2004) Low apolipoprotein A-I level at intensive care unit admission and systemic inflammatory response syndrome exacerbation. Crit Care Med 32:632–637
Riemens SC, van Tol A, Sluiter WJ, Dullaart RPF (1998) Plasma phospholipid transfer protein activity is related to insulin resistance: impaired acute lowering by insulin in obese NIDDM patients. Diabetologia 41:929–934
Fielding CJ, Reaven GM, Fielding PE (1982) Human noninsulin-dependent diabetes. Identification of a defect in plasma cholesterol transport normalized in vivo by insulin and in vitro by selective immunoadsorption of apolipoprotein E. Proc Natl Acad Sci USA 79:6365–6369
Oppenheimer MJ, Sundquist K, Bierman EL (1989) Downregulation of high-density lipoprotein receptor in human fibroblasts by insulin and IGF-1. Diabetes 38:117–122
Lay SL, Krief S, Farnier C, Lefrère I, Le Liepvre X, Bazin R, Ferré P, Dugail I (2001) Cholesterol, a cell size-dependent signal that regulates glucose metabolism and gene expression in adipocytes. J Biol Chem 276:16904–16910
Van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R (2001) Intensive insulin therapy in critically ill patients. N Engl J Med 345:1359–1367
Flegel WA, Wolpl A, Mannel DN, Northoff H (1989) Inhibition of endotoxin-induced activation of human monocytes by human lipoprotein. Infect Immun 57:2237–2254
Harris HW, Johnson JA, Wigmore SJ (2002) Endogenous lipoproteins impact the response to endotoxin in humans. Crit Care Med 30:23–31
Sprong T, Netea MG, van der Ley P, Verver-Jansen TJG, Jacobs LEH, Stalenhoef A, van der Meer JWM, van Deuren M (2004) Human lipoproteins have divergent neutralizing effects on E. coli LPS, N. meningitidis LPS, and complete Gram-negative bacteria. J Lipid Res 45:742–749
Kitchens RL, Wolfbauer G, Albers JJ, Munford RS (1999) Plasma lipoproteins promote the release of bacterial lipopolysaccharide from the monocyte cell surface. J Biol Chem 274:34116–34122
Kitchens RL, Thompson PA, Viriyakosol S, O'Keefe GE, Munford RS (2001) Plasma CD14 decreases monocyte responses to LPS by transferring cell-bound LPA to plasma lipoproteins. J Clin Invest 108:485–493
Vreugdenhil ACE, Snoek AMP, Van't Veer C, Greve JWM, Buurman WA (2001) LPS-binding protein circulates in association with apoB-containing lipoproteins and enhances endotoxin-LDL/VLDL interaction. J Clin Invest 107:224–234
Landmann R, Zimmerli W, Sansano S, Link S, Hahn A, Glauser MP, Calandra T (1995) Increased soluble CD14 is associated with high mortality in gram-negative septic shock. J Infect Dis 171:639–644
Levine DM, Parker TS, Donnelly TM, Walsh A, Rubin AL (1993) In vivo protection against endotoxin by plasma high density lipoprotein. Proc Natl Acad Sci USA 90:12040–12044
McDonald M, Dhadly P, Cockerill GW, Cuzzocrea S, Mota-Filipe H, Hinds CJ, Miller NE, Thiemermann C (2003) Reconstituted high-density lipoprotein attenuates organ injury and adhesion molecule expression in a rodent model of endotoxic shock. Shock 20:551–557
Pajkrt D, Lerch PG, van der Poll T, Levi M, Illi M, Doran JE, Arnet B, van den Ende A, tenCate JW, Deventer SJ (1997) Differential effects of reconstituted high-density lipoprotein on coagulation, fibrinolysis and platelet activation during human endotoxemia. Thromb Haemost 77:303–307
Netea MG, Curfs JH, Demacker PN, Van der Meer JW, Kullberg BJ (1999) Infusion of lipoproteins into volunteers enhances the growth of Candica albicans. Clin Infect Dis 28:1148–1151
Heyland DK, MacDonald S, Keefe L, Drover JW (1998) Total parenteral nutrition in the critically ill patient: a meta-analysis. JAMA 16:280:2013–2019
Lekka ME, Liokatis S, Nathanail C, Galani V, Nakos G (2003) The impact of intravenous fat emulsion administration in acute lung injury. Am J Respir Crit Care Med 169:638–644
Koch T, Heller A (2002) Effects of intravenous fish oil on pulmonary integrity and function. Clin Nutr 21:41–45
Kelbel I, Koch T, Prechtl A, Heller A, Schlotzer E, Schiefer G, Neuhof H (1999) Effects of parenteral application of fish oil versus soy oil emulsions on bacterial clearance functions. Beitr Infusionsther Transfusionsmed 26:226–232
Gupta H, Dai L, Datta G, Garber DW, Grenett H, Li Y, Mishra V, Palgunachari MN, Handattu S, Gianturco SH, Bradley WA, Anantharamaiah GM, White CR (2005) Inhibition of lipopolysaccharide-induced inflammatory responses by an apolipoprotein AI mimetic peptide. Circ Res 97:236–243
Navab M, Anantharamaiah GM, Reddy ST, van Lenten BJ, Data G, Garber D, Fogelman AM (2004) Human apolipoprotein A-I and A-I mimetic peptide: potential for atherosclerosis reversal. Curr Opin Lipidol 15:654–649
Huber J, Vales A, Mitulovic G, Blumer M, Schmid R, Witztum JL, Binder BR, Leitinger N (2002) Oxidized membrane vesicles and blebs from apoptotic cells contain biologically active oxidized phospholipids differentially regulate endothelial binding of monocytes and neutrophils. Arterioscler Thromb Vasc Biol 22:101–107
Berliner JA, Subbanagounder G, Leitinger N, Watson AD, Vora D (2001) Evidence for a role of phospholipids oxidation products in atherogenesis. Trends Cardiovasc Med 11:142–147
Eligini S, Brambilla M, Banfi C, Camera M, Sironi L, Barbieri SS, Auwerx J, Tremoli E, Colli S (2002) Oxidized phospholipids inhibit cyclooxygenase-2 in human macrophages via nuclear factor-κB/IκB-and ERK2-dependent mechanisms. Cardiovasc Res 55:406–415
Chung SW, Kang BY, Kim SH, Pak YK, Cho D, Trinchieri G, Kim TS (2000) Oxidized low density lipoprotein inhibits interleukin-12 production in lipopolysaccharide-activated mouse macrophages via direct interactions between peroxisome proliferators-activated receptor-gamma and nuclear factor-κB. J Biol Chem 275:32681–32687
Hamilton TA, Major JA, Armstrong D, Tebo JM (1998) Oxidized LDL modulates activation of NF-κB in mononuclear phagocytes by altering degradation of IκBs. J Leukoc Biol 64:667–674
Bochkov VN, Kadl A, Huber J, Gruber F, Binder BR, Leitinger N (2002) Protective role of phospholipid oxidation products in endotoxin-induced tissue damage. Nature 419:77–81
Walton, KA Cole AL, Yeh M, Subbanagounder G, Krutzik SR, Modlin RL, Lucas RM, Nakai J, Smart EJ, Vora DK, Berliner JA (2003) Specific phospholipid oxidation products inhibit ligand activation of Toll-like receptors 4 and 2. Arterioscler Thromb Vasc Biol 23:1197–1203
Grundt H, Nilsen DW, Hetland O, Aarsland T, Baksaas I, Grande T, Woie L (1995) Improvement of serum lipids and blood pressure during intervention with n-3 fatty acids was not associated with changes in insulin levels in subjects with combined hyperlipidaemia. J Intern Med 237:249–259
Leaf A (1990) Cardiovascular effects of fish oils. Beyond the platelet. Circulation 82:624–628
Mensink RP, Katan MB (1990) Effect of dietary trans fatty acids on high-density and low-density lipoprotein cholesterol levels in healthy subjects. N Engl J Med 323:439–445
MacLean CH, Newberry SJ, Mojica WA, Khanna P, Issa AM, Suttorp MJ, Lim YW, Traina SB, Hilton L, Garland R, Morton SC (2006) Effects of omega-3 fatty acids on cancer risk: a systematic review. JAMA 295:403–415
Pscheidl E, Schywalsky M, Tschaikowsky K, Boke-Prols T (2000) Fish oil-supplemented parenteral diets normalize splanchnic blood flow and improve killing of translocated bacteria in a low-dose endotoxin rat model. Crit Care Med 28:1489–1496
Weiss G, Meyer F, Matthies B, Pross M, Koenig W, Lippert H (2002) Immunomodulation by perioperative administration of n-3 fatty acids. Br J Nutr 87(Suppl 1):S89–S94
Brown AJ, Pang E, Roberts DC (1991) Persistent changes in the fatty acid composition of erythrocyte membranes after moderate intake of n-3 polyunsaturated fatty acids: study design implications. Am J Clin Nutr 54:668–673
Lehr HA, Hubner C, Finckh B, Nolte D, Beisiegel U, Kohlschutter A, Messmer K (1991) Dietary fish oil reduces leukocyte/endothelium interaction following systemic administration of oxidatively modified low density lipoprotein. Circulation 84:1725–1731
Urakaze M, Hamazaki T, Makuta M, Ibuki F, Kobayashi S, Yano S, Kumagai A (1987) Infusion of fish oil emulsion: effects on platelet aggregation and fatty acid composition in phospholipids of plasma, platelets, and red blood cell membranes in rabbits. Am J Clin Nutr 46:936–940
Lee TH, Sethi T, Crea AE, Peters W, Arm JP, Horton CE, Walport MJ, Spur BW (1988) Characterization of leukotriene B3: comparison of its biological activities with leukotriene B4 and leukotriene B5 in complement receptor enhancement, lysozyme release and chemotaxis of human neutrophils. Clin Sci (Lond) 74:467–475
Needleman P, Raz A, Minkes MS, Ferrendelli JA, Sprecher H (1979) Triene prostaglandins: prostacyclin and thromboxane biosynthesis and unique biological properties. Proc Natl Acad Sci USA 76:944–948
Sperling RI, Robin JL, Kylander KA, Lee TH, Lewis RA, Austen KF (1987) The effects of N-3 polyunsaturated fatty acids on the generation of platelet-activating factor-acether by human monocytes. J Immunol 139:4186–4191
Weber C, Aepfelbacher M, Lux I, Zimmer B, Weber P. C (1991) Docosahexaenoic acid inhibits PAF and LTD4 stimulated [Ca2+]i-increase in differentiated monocytic U937 cells. Biochim Biophys Acta 1133:38–45
Lee JY, Sohn KH, Rhee SH, Hwang D (2001) Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cylooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem 276:16683–16689
Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, Sizemore N, Hwang DH (2003) Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/Akt by saturated and polyunsaturated fatty acids. J Biol Chem 278:37041–37051
Lee JY, Zhao L, Youn HS, Wheatherill AR, Tapping R, Feng L, Lee WH, Fitzgerald KA, Hwang DH (2004) Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerization with Toll-like receptor 6 or 1. J Biol Chem 279:16971–16979
Li H, Ruan XZ, Powis SH, Fernando R, Mon WY, Wheeler DC, Moorhead JF, Varghese Z (2005) EPA and DHA reduce LPS-induced inflammation responses in HK-2 cells: evidence for a PPAR-γ-dependent mechanism. Kidney Int 67:867–874
Ertel W, Morrison MH, Ayala A, Chaudry IH (1993) Modulation of macrophage membrane phospholipids by n-3 polyunsaturated fatty acids increases interleukin 1 release and prevents suppression of cellular immunity following hemorrhagic shock. Arch Surg 128:15–20
Bertolini G, Iapichino G, Radrizzani D, Facchini, Simini B, Bruzzone P, Zanforlin G, Tognoni G (2003) Early enteral immunonutrition in patients with severe sepsis: results of an interim analysis of a randomized multicentre clinical trial. Intensive Care Med 29:834–840
Faist E, Hartl WH, Baue AE (1994) Immune mechanisms of post-traumatic hyperinflammation and sepsis. Immun Infekt 22:203–213
Falconer JS, Fearon KC, Ross JA, Elton R, Wigmore SJ, Garden OJ, Carter DC (1995) Acute-phase protein response and survival duration of patients with pancreatic cancer. Cancer 75:2077–2082
Friedman G, Silva E, Vincent JL (1998) Has the mortality of septic shock changed with time? Crit Care Med 26:2078–2086
Gadek JE, DeMichele SJ, Karlstad MD, Pacht ER, Donahoe M, Albertson TE, Van Hoozen C, Wennberg AK, Nelson JL, Noursalehi M (1999) Effect of enteral feeding with eicosapentaenoic acid, gamma-linolenic acid, and antioxidants in patients with acute respiratory distress syndrome. Enteral Nutrition in ARDS Study Group. Crit Care Med 27:1409–1420
Galban C, Montejo JC, Mesejo A, Marco P, Celaya S, Sanchez-Segura JM, Farre M, Bryg DJ (2000) An immune-enhancing enteral diet reduces mortality rate and episodes of bacteremia in septic intensive care unit patients. Crit Care Med 28:643–648
Pontes–Arruda A, Aragao AM, Albuquerque JD (2006) Effects of enteral feeding with eicosapentaenoic acid, gamma–linolenic acid, and antioxidants in mechanically ventilated patients with severe sepsis and septic shock. Crit Care Med 34(9):2325–2333
Heller AR, Rossel T, Gottschlich B, Tiebel O, Menschikowski M, Litz RJ, Zimmermann T, Koch T (2004) Omega-3 fatty acids improve liver and pancreas function in postoperative cancer patients. Int J Cancer 111:611–616
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is discussed in the editorial available at: http://dx.doi.org/10.1007/s00134-006-0434-9.
Rights and permissions
About this article
Cite this article
Wendel, M., Paul, R. & Heller, A.R. Lipoproteins in inflammation and sepsis. II. Clinical aspects. Intensive Care Med 33, 25–35 (2007). https://doi.org/10.1007/s00134-006-0433-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-006-0433-x