
Abstract
Objective
To determine whether pediatric PICU patients with mannose-binding lectin (MBL) gene polymorphisms associated with low levels of the functional protein have an increased risk of developing sepsis and SIRS.
Design and setting
A prospective, observational cohort study in a 22-bed PICU in a tertiary referral centre.
Patients
One hundred consecutive admissions to a PICU with at least one organ system failure longer than 12 h. Patients were classified into those with infectious or non-infectious insults as the primary reason for intensive care admission. Patients were followed to determine which developed sepsis or non-infection related SIRS using standard criteria.
Measurements and results
Of the 100 patients 50 had infectious and 50 had non-infectious insults as the precipitant for admission. 42 patients had variant MBL alleles (determined by MBL-2 gene exon 1 and promoter polymorphisms) and were significantly over-represented amongst the 59 patients that developed SIRS. This effect was not explained by differences in age, sex or ethnicity and was seen in both the infection and non-infection subgroups. In patients with infection, variant MBL alleles were associated with increased systemic response (2/15 with localised infection, 10/19 with sepsis and 12/16 with septic shock). MBL serum levels showed close concordance with the genotype and indicated that MBL levels less than 1000 ng/ml are associated with a greatly increased risk of SIRS.
Conclusions
MBL-2 exon 1 polymorphisms with low serum levels of functional MBL protein are associated with a greatly increased risk of developing SIRS and of progression from infection to sepsis and septic shock in paediatric ICU patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Multiple organ failure resulting from systemic inflammation remains the predominant cause of morbidity and mortality on intensive care, irrespective of the initial illness or insult precipitating admission [1, 2]. Attempts to attenuate the severity of the systemic inflammatory response to infection by neutralising endotoxin or by selective modulation of components of the inflammatory response [3] have proved disappointing [4]. Recent studies have indicated that success may be achieved by intervening in a less specific fashion. Targeted early resuscitation [5], careful glycaemic regulation [6], judicious use of steroids [7] and administration of activated protein C [8] have all proven beneficial. However, the reasons for the success of such strategies are unclear and suggest that further improvements are likely to stem from a greater understanding of the pathogenesis of critical illness.
Mannose-binding lectin (MBL) is a collectin found in serum, which binds to mannose and N-acetyl glucosamine residues found on micro-organisms. On binding, MBL activates the complement system in an antibody-independent manner (the lectin pathway) via an associated serine protease, mannan-binding lectin-associated serine protease 2, which cleaves C4 and then C2 to form the C3 convertase, C4b2a. This enzyme is able to generate significant amounts of opsonic C3b fragments which coat micro-organisms for phagocytosis [9]. There is also evidence of direct interactions of MBL with phagocytic cells to promote phagocytosis [10, 11] and modify cellular activation [12].
Human deficiency of MBL [13] is caused predominantly by point mutations within exon 1 of the MBL-2 gene at codons 52, 54 or 57 resulting in amino acid substitutions which compromise assembly of functional oligomers [9]. Heterozygotes have reduced serum concentrations of MBL, whilst functional multimeric protein is almost absent from the serum of homozygotes and compound heterozygotes [14]. In addition to exon 1 mutations, there are three major polymorphisms in the promoter region of the MBL gene and one of these variants (X/Y) also profoundly influences levels of the protein [15, 16]. More than one-third of the population have haplotypes associated with reduced MBL levels, with very low levels expected in 12% [15]. Numerous studies have now shown that individuals with MBL deficiency are more susceptible to infection [17, 18, 19, 20]. MBL serum levels may also influence cytokine production and therefore the host inflammatory response [12, 21].
We hypothesised that MBL deficiency would increase the risk of the systemic inflammatory response syndrome (SIRS) and severity of sepsis in children admitted to intensive care after a systemic insult. The results from this study indicate that MBL plays a critical role in determining which children develop systemic inflammation and influences the clinical severity of that response irrespective of the underlying illness.
Patients and methods
Consecutive admissions to our tertiary multi-disciplinary paediatric intensive care unit (PICU) were recruited over a 6-month period in 2002. Inclusion criteria were: age up to 17 years and the presence of at least one organ system failure longer than 12 h (or death within the first 12 h). The following exclusions were applied: multiple congenital abnormalities; known congenital immunodeficiency; known central neurological or neuromuscular disease (all considered to represent major risk factors for ICU admission resulting from infection); persistent pulmonary hypertension of the newborn; weight under 2.2 kg; informed consent not available; suspected non-accidental injury; repeat PICU admission during the study period; lack of intravenous or intra-arterial access and anticipated short stay (<24 h) on the PICU.
Of the 211 suitable cases an informed consent discussion was not possible in 101 cases (on-going resuscitation, parents not in attendance, translator not available) and consent was refused in a further 7 cases. Of the 103 remaining cases no blood was available for genotyping in 3 individuals, leaving 100 for full analysis. Of these, serum was also available for MBL determination in 98 patients. Ninety-four children were ventilated for more than 12 h and two died within 12 h of admission. The remaining four were not ventilated but had at least one organ failure. The characteristics of the patients studied in relation to MBL exon 1 polymorphisms are shown in Table 1.
On enrolment, cases were assigned to one of two groups according to the principal reason for ICU admission by ICU physicians not involved in the study. The groups were: (a) infection (presumed or proven), (b) non-infection (trauma, postoperative or ‘other’). Within each group, patients were subdivided into those who did or did not develop SIRS within 48 h of admission by age-adjusted criteria.
Infection was defined as ‘proven’ if a causative organism was isolated and ‘presumed’ in those with a history and examination consistent with infection, for example, fever, cough and coryza combined with chest radiological changes consistent with pneumonia. Diagnoses of SIRS, sepsis and septic shock were made according to the 1992 ACCP/SCCM guidelines modified for age [22, 23]. In brief, SIRS was determined by the presence of at least two of the following: central temperature higher than 38.0°C or lower than 36.0°C, white cell count higher than 12×109/l or lower than 4×109/l and a heart rate outside age-specific ranges. Respiratory rate was not included as a diagnostic criterion because of the high proportion of cases receiving mechanical ventilation. Cases meeting SIRS criteria with ‘proven’ or ‘presumed’ infection were classified as ‘sepsis’. Septic shock was diagnosed in cases of sepsis who were hypotensive, defined against age-specific values for mean blood pressure after fluid resuscitation requiring treatment with inotropic and/or vasopressor therapy (dopamine at 5 µg/kg or more per minute, any dose of epinephrine, norepinephrine or vasopressin).
Patient diagnoses among the 50 patients with infection were: 15 (30%) localised infection only, 19 (38%) sepsis and 16 (32%) septic shock. Among the 50 patients without infections the diagnoses were: trauma (n=25), post-surgical (n=22) and other (n=3; 2 with necrotising enterocolitis and 1 with Stevens-Johnson syndrome). Patients were also divided into those with (n=59, 59%) and those without (n=41, 41%) SIRS, irrespective of aetiology. Infective diagnoses included pneumonia (n=16), bacteraemia (n=10), meningoencephalitis (n=8), central line infection (n=6), tracheitis/epiglottitis (n=4), myocarditis (n=3),), bronchiolitis (n=1), other (n=2). Presumptive causative agents were Gram-negative organisms (n=13), Gram-positive organisms (n=11), viruses (n=7) and mycoplasma (n=2). The remaining 17 patients had ‘presumed’ infection with negative microbiological cultures.
Our electronic patient charting system (Care Vue, Hewlett-Packard) was reviewed daily, and maximum and minimum ventilator and physiological parameters for each 24-h period were recorded prospectively onto a database. Microbiological, biochemical and haematological information was recorded from our PICU and the referring hospital. Paediatric Index of Mortality (PIM) was assessed on first contact with the PICU team [24] and the Paediatric Logistic Organ Dysfunction (PELOD) score [25] was calculated daily. EDTA blood and serum samples were taken within 48 h of admission. Whole blood in EDTA was stored at −20°C until DNA extraction was performed. Serum samples were spun, separated and the serum stored in aliquots at −80°C until analysed. The investigator performing the MBL genotype and serum levels (K.F.) was blinded to the diagnosis of SIRS/non-SIRS. Likewise the clinician acquiring the clinical data (P.W.) was blinded to the MBL data. Local research ethics committee approval was obtained.
DNA extraction
DNA was extracted from 200 µl whole blood using a commercial kit (QIAamp DNA blood mini kit, Qiagen, Crawley, UK) according to the manufacturer’s instructions. The final product was resuspended in 200 µl sterile water (approx. 10 ng/µl) and stored at −20°C until further use.
PCR amplification of genomic DNA and universal hereroduplex generator
Separate polymerase chain reaction (PCR) reactions were performed on the genomic DNA samples, one to amplify the region of the gene containing the exon 1 mutations and one spanning the region of the X/Y promoter polymorphism as previously described [26, 27]. Universal heteroduplex generators (UHGs; synthetic DNA molecules spanning either the exon 1 region, or the X/Y locus of the promoter region) were kindly provided by Dr. N. Wood, University of Bristol, UK, and permitted the detection of all possible known mutations. Separate, similar PCR reactions were performed for each UHG as previously described [26, 27]. The PCR product was confirmed on a 2% agarose gel containing ethidium bromide. Gels were visualised under UV light.
MBL-2 heteroduplexing and genotyping
We added 10 µl UHG PCR product and 5 µl loading dye to 15 µl of each sample. Samples were heteroduplexed by heating at 95°C for 10 min and then cooled to room temperature over 20 min. We loaded 15 µl heteroduplex product onto 20% polyacrylamide gels and run for 16 h at 140 V in Tris-borate-EDTA buffer at 16°C. Gels were stained using 0.5 µg/ml ethidium bromide in Tris-borate-EDTA buffer for 10 min before photography under UV light. Different genotypes produce characteristic patterns [26].
MBL-2 haplotyping
The three MBL-2 structural gene mutations B, C and D are in linkage disequilibrium with the promoter region polymorphism X/Y, and only Y associates with each of the mutations [15]. The data from heteroduplexing procedures were combined to give haplotypes comprising one of the three structural gene mutations (O) or wild-type (A) together with the X/Y promoter polymorphism.
MBL protein levels
MBL levels in serum were determined by a symmetrical sandwich enzyme-linked immunosorbent assay using commercial kits from Antibody Shop (Copenhagen, Denmark) according to the manufacturer’s instructions.
Statistical analysis
Comparisons of the proportions of MBL-deficient genotypes between clinical groups were performed using exact tests (StatXact version 4.0.1). Differences between MBL serum levels were analysed by the Mann-Whitney U test (for two groups) or Kruskal-Wallis test (three or more groups). Multiple logistic regression was used to investigate the association of SIRS with MBL serum level and genotype after adjusting for differences in age, sex and ethnicity and PIM score.
Results
Overall allele frequencies were as follows; wild-type (A) 0.78, codon 54 (variant B) mutation 0.095, codon 57 (variant C) and 52 (variant D) mutations both 0.06. In the non-infection group with a non-white population of 36%, allele frequencies were A 0.82, B 0.05, C 0.06, D 0.07. As expected, these frequencies differ from those published in a recent United Kingdom population of well children, with a non-white component of 2.4% (A 0.775, B 0.144, C 0.015 and D 0.066) [28], reflecting the ethnically diverse population served by our PICU. Figure 1 presents an analysis of the proportion of cases with MBL-2 exon 1 mutations by infection or non-infective reason for admission to PICU (Fig. 1A), the development of SIRS (Fig. 1B), and severity of the systemic response to infection (Fig. 1C). A higher percentage of children who were admitted with infection carried a variant allele (48% vs.36%) but this was not significant (p=0.233, Fisher’s exact test; odds ratio, OR, 1.7; 95% CI 0.68–3.93; Fig. 1A). There was, however, a significant difference in the proportion of children with a variant allele who developed SIRS compared to those who did not (p<0.0001; OR 7.1; 95% CI 2.7–18.6; Fig. 1B). This relationship remained after age, sex and ethnicity were accounted for (adjusted OR 8.2; 95% CI 3.0–22.7; p<0.0005). Addition of the risk of mortality, estimated from admission severity of illness (PIM score), to the prediction model had no effect on this relationship (adjusted OR 8.0; 95% CI 0.28–22; p<0.0005).

MBL exon 1 polymorphisms in children requiring intensive care. A The proportion of cases with a variant allele is not significantly greater in cases with infection as the precipitant to PICU admission than in those with non-infective (trauma, post-operative) reasons for admission. B However, the odds of developing SIRS are greatly increased in the presence of a variant allele. C In patients with infection (n=50) as the reason for ICU admission there was a significant association between clinical severity of the systemic response to infection and the presence of a variant allele (χ2=12.1, d.f. 2, p=0.002). D, E Interestingly, the relationship between MBL variant alleles and the development of SIRS remained significant in both the non-infection (D) and infection (E) groups
A significant association was seen between the severity of the systemic response to infection and the presence of an MBL variant allele (localised infection 2/15 vs. sepsis 10/19 vs. septic shock 12/16; p=0.0007, Kruskal-Wallis test Fig. 1C). Further analyses of groups revealed that in both the ‘non-infection’(Fig. 1D) and ‘infection’ groups (Fig. 1E) the significantly increased odds of developing SIRS in association with an MBL variant allele remained.
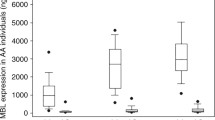
Similar results were seen when MBL levels were analysed. Serum levels according to infection/non-infection, SIRS or non-SIRS, and severity of infection are shown in Fig. 2 panels A, B and C, respectively. The results are consistent with those seen with the genotype above in that there is a non-significant trend for lower MBL levels in patients with infection than in those without (p=0.09). Patients with SIRS had significantly lower median MBL levels than did patients without SIRS (p<0.0001; Fig. 2 B). The median level decreased with severity of infection (infection vs. sepsis vs. septic shock, p<0.0001; Fig. 2 C). Again consistent with the genotype results above, the relationship between lower MBL levels and the development of SIRS was seen in both the non-infection (Fig. 2D) and infection (Fig. 2E) groups.

MBL serum levels in children requiring intensive care. Individual and median values are shown. A MBL serum levels were available in 98 cases and were not significantly reduced in cases in which infection precipitated admission to PICU (p=0.08, Mann-Whitney). B In contrast, MBL levels were significantly lower in cases that developed SIRS early in the ICU admission (p<0.0001, Mann-Whitney). C In patients with infection (n=49) as the reason for ICU admission there was a significant association between clinical severity of the systemic response to infection and lower serum MBL levels (Kruskal-Wallis, p<0.001). D, E The relationship between reduced MBL serum levels and development of SIRS remained significant in both the non-infection (D) and infection (E) groups, confirming the genotype findings illustrated in Fig. 1
As expected, there was a clear relationship between haplotype and MBL level (Fig. 3; Kruskal-Wallis non-parametric analysis of variance, p<0.0001). SIRS occurred more frequently in haplotypes associated with reduced MBL levels. Only two children were homozygous for an MBL-2 exon 1 polymorphism, one D/D (52/52) and one B/C (52/57), both of whom developed SIRS. Interestingly, all 11 patients heterozygous for an MBL exon 1 mutation associated with a low expression promotor (XA/YO) developed SIRS, and the risk of SIRS decreased with increasing prevalence of the A and Y alleles. Previously we have shown that children with MBL levels below 1000 ng/ml are at greater risk of infection than are those with higher levels [18]. Using this cut-off, 37 of 46 (80%) of children with the lower MBL levels developed SIRS whereas only 22 of 52 (42%) with higher MBL levels did so.

Serum MBL levels, MBL short haplotype and development of SIRS. Log10[MBL]serum is displayed against MBL haplotype (exon 1 and promoter polymorphisms) from those associated with the highest (YA/YA), to the lowest, serum levels (YO/YO). As expected there was a clear relationship between Log10[MBL]serum and genotype (one-way analysis of variance, p<0.0001 after correction for multiple comparisons). Filled circles Cases that developed SIRS; open circles cases that did not develop SIRS. The odds ratios (95% confidence intervals) for the development of SIRS for each haplotype are shown as is the risk of mortality derived from the maximum calculated PELOD score. Note that all 13 cases with XA/YO and YO/YO haplotypes developed SIRS
Discussion
Critically ill children with variant alleles for the MBL gene are at greatly (seven-fold) increased risk of developing SIRS within 48 h of presentation to PICU. Our data show this effect to be independent of the patient’s underlying condition, age, sex and ethnicity and not to be confined to children with infection. The importance of MBL genotype in the development of SIRS is underlined by the observation that possession of the wild-type MBL haplotype was associated with a 50% reduction in risk of SIRS, whereas all 13 patients who were either homozygous for a variant structural region allele or heterozygous with a low expression promoter developed SIRS. We have also shown that the presence of an MBL variant allele significantly increases the severity of the systemic response to infection.
Individuals differ considerably in their response to an infectious or traumatic insult. This could be explained by polymorphisms of the genes encoding proteins involved in mediating the innate immune response and inflammatory cascades. Most studies have concentrated on genetic polymorphisms in the genes encoding inflammatory mediators or their receptors in sepsis/septic shock such as tumor necrosis factor α [29, 30], although the influence of these polymorphisms appears small or absent in relation to the enormous spectrum of clinical severity [31]. Whilst this present report was under review, Garred and colleagues [32] in Denmark published a similar study on the role of MBL in critical illness in adults. Considering the differences in the patients studied, there is remarkable concordance between our essential findings and those of the Danish group [32]. Both studies show an increasing over-representation of variant MBL alleles with increased severity of sepsis, although the ratio of WT/variant alleles is more dramatic in children.
We showed a strong relationship between MBL variants and the development of SIRS in the first 48 h on ICU. These findings contrast with the Danish study in which MBL variants occurred with the same frequency amongst SIRS patients admitted to the ICU and healthy controls [32]. Several explanations are possible for these differences. Our study captures the risk of developing early SIRS whilst fully supported on intensive care, which includes more cases than those with established SIRS on admission. The difference might also reflect the stringent criteria that we applied for a diagnosis of SIRS (with exclusion of the respiratory rate as a criterion) or differences between the control populations in the two studies (our ICU cases without SIRS and Danish healthy blood donors and laboratory staff). A remaining possibility is that these findings reflect differences in the pathogenesis of critical illness between children and adults. One example of this is the high proportion of critically ill children who have no major pre-existing morbidity, a situation that will have been refined by our exclusion criteria. In contrast, the adult study reflects a typical pattern, with the majority of cases having very significant pre-morbidity [32]. There may also be differences in the referral patterns to ICU between children and adults, with children being admitted earlier in the course of their illness. Finally, perhaps children and adults also differ in the contribution of the various mechanisms leading to organ dysfunction in SIRS. For example, MBL deficiency is known to be protective against ischaemia-reperfusion injury, which may play a greater role in adults [33]. The Danish study also demonstrated an increased risk of death with MBL variants and SIRS. This could not be directly investigated amongst our cases because of the low numbers of deaths. However, the trend for greater severity of illness with the MBL variants, as assessed by the risk of mortality calculated from the maximum PELOD score (Fig. 3) and the increased risk of septic shock are likely to reflect similar effects.
The high incidence of MBL variant alleles in the general population is compatible with a view that MBL deficiency can be advantageous in some circumstances. MBL deficiency might protect individuals from intracellular pathogens, such as Mycobacteria and Leishmania, as the absence of MBL reduces the uptake of these organisms into their preferred sites of replication [21, 34].
A key question is whether MBL deficiency modulates the initial insult or the subsequent response to this insult. For example, MBL deficiency could alter the host’s response to micro-organisms and/or microbial products, potentially increasing the severity of illness. MBL deficiency could also have an effect on the development of SIRS remote from the primary insult. Two observations in this study point towards the latter as the likely explanation for our observations. Firstly a similar effect exists in trauma and post-operative cases when the role of MBL in opsonophagocytosis would appear to be of little consequence compared to the infection cases. Secondly, the odds ratio for development of SIRS was not altered by correction for initial severity of illness according to PIM.
The reason for the observed effect of MBL on the development of SIRS remains unclear. It is thought that the principal role of MBL is to activate complement, the role of which is complex in critically ill patients [35]. There are numerous pathways by which complement activation can lead to enhanced inflammation, and therefore one might postulate that in fact a low MBL level is actually beneficial as it may reduce inflammation and the development of SIRS. However, a novel mechanism by which MBL could influence the development of SIRS is through a direct effect on pro-inflammatory cytokine production. In a human whole-blood model high levels of MBL have been observed to inhibit tumor necrosis factor α, interleukin 6 and interleukin 1β release from monocytes [12]. It is assumed that in this case MBL operates through a receptor expressed, but as yet unidentified, on monocytes. MBL levels in our patients and those observed in the Danish study were in a range which has been shown to inhibit cytokine production in vitro [12].
Whatever the reason for the influence of MBL on the severity of sepsis and the development of SIRS, this observation could prove to be clinically important. Considerable effort is currently being invested in analyses of risk stratification to identify patients likely to have the most severe patterns of illness. As this study indicates, it is possible to evaluate the host MBL status by partial genotyping (i.e. structural gene variants alone, Fig. 1), extended genotyping (to include the X/Y promoter variant, Fig. 3) or by phenotypic assays of MBL protein levels (Fig. 2). All three approaches showed remarkable concordance, but, as indicated in Fig. 3, 37 of 46 patients (80%) with MBL serum levels below 1000 ng/ml developed SIRS whereas only 22 of 52 (42%) with levels above 1000 ng/ml developed the syndrome. Therefore a simple serum test may provide an indicator of susceptibility to the development of SIRS. MBL is currently being prepared for clinical use and has been used in one patient with cystic fibrosis and end-stage lung disease [36]. We need to understand more clearly the costs and benefits of such therapy during specific episodes of critical illness, but this study raises the possibility that judicious use of MBL in MBL-deficient patients will lead to a reduction in the number that develop SIRS or severe sepsis/septic shock.
References
Marshall JC (2001) Inflammation, coagulopathy, and the pathogenesis of multiple organ dysfunction syndrome. Crit Care Med 29:S99–S106
Vincent JL, Moreno R, Takala J, Willatts S, de Mendonca A, Bruining H, Reinhart CK, Suter PM, Thijs LG (1996) The SOFA (sepsis-related organ failure assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med 22:707–710
Crowther MA, Marshall JC (2001) Continuing challenges of sepsis research. JAMA 286:1894–1896
Opal SM, Cohen J (1999) Clinical gram-positive sepsis: does if fundamentally differ from gram-negative bacterial sepsis? Crit Care Med 27:1608–1616
Rivers E, Nguyen B, Havstad S, Ressler J, Muzzin A, Knoblich B, Peterson E, Tomlanovich M (2001) Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 345:1368–1377
Van den BG, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R (2001) Intensive insulin therapy in the critically ill patients. N Engl J Med 345:1359–1367
Annane D, Sebille V, Charpentier C, Bollaert PE, Francois B, Korach JM, Capellier G, Cohen Y, Azoulay E, Troche G et al (2002) Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA 288:862–871
Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW et al (2001) Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 344:699–709
Turner MW (1996) Mannose binding lectin: the pluripotent molecule of the innate immune system. Immunol Today 17:532–540
Kuhlman M, Joiner K, Ezekowitz RA (1989) The human mannose-binding protein functions as an opsonin. J Exp Med 169:1733–1745
Tenner AJ, Robinson SL, Ezekowitz RAB (1995) Mannose-binding protein (MBP) enhances mononuclear phagocyte function via a receptor that contains the 126:000 m(r) component of the C1q receptor. Immunity 3:485–493
Jack DL, Read RC, Tenner AJ, Frosch M, Turner MW, Klein NJ (2001) Mannose-binding lectin regulates the inflammatory response of human professional phagocytes to Neisseria meningitidis serogroup B. J Infect Dis 184:1152–1162
Super M, Thiel S, Lu J, Levinsky RJ, Turner MW (1989) Association of low levels of mannan-binding protein with a common defect of opsonisation. Lancet II:1236–1239
Lipscombe RJ, Sumiya M, Summerfield JA, Turner MW (1995) Distinct physicochemical characteristics of human mannose binding protein expressed by individuals of differing genotype. Immunology 85:660–667
Madsen HO, Garred P, Thiel S, Kurtzhals JAL, Lamm LU, Ryder LP, Svejgaard A (1995) Interplay between promoter and structural gene variants control basal serum level of mannan-binding protein. J Immunol 155:3013–3020
Madsen HO, Satz ML, Hogh B, Svejgaard A, Garred P (1998) Different molecular events result in low protein levels of mannan-binding lectin in populations from Southeast Africa and South America. J Immunol 161:3169–3175
Koch A, Melbye M, Sørensen P, Homøe P, Madsen HO, Mølbak K, Hansen CH, Andersen LH, Hahn GW, Garred P (2001) Acute respiratory tract infections and mannose-binding lectin insufficiency during early childhood. JAMA 285:1316–1321
Neth O, Hann I, Turner MW, Klein NJ (2001) Deficiency of mannose-binding lectin and burden of infection in children with malignancy: a prospective study. Lancet 358:614–618
Summerfield JA, Sumiya M, Levin M, Turner MW (1997) Association of mutations in mannose binding protein gene with childhood infection in consecutive hospital series. BMJ 314:1229–1232
Peterslund NA, Koch C, Jensenius JC, Thiel S (2001) Association between deficiency of mannose-binding lectin and severe infections after chemotherapy. Lancet 358:637–638
Santos IK, Costa CH, Krieger H, Feitosa MF, Zurakowski D, Fardin B, Gomes RB, Weiner DL, Harn DA, Ezekowitz RA et al (2001) Mannan-binding lectin enhances susceptibility to visceral leishmaniasis. Infect Immun 69:5212–5215
Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ (1992) Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest 101:1644–1655
Hayden WR (1993) Sepsis and organ failure definitions and guidelines. Crit Care Med 21:1612–1613
Shann F, Pearson G, Slater A, Wilkinson K (1997) Paediatric index of mortality (PIM): a mortality prediction model for children in intensive care. Intensive Care Med 23:201–207
Leteurtre S, Martinot A, Duhamel A, Gauvin F, Grandbastien B, Nam TV, Proulx F, Lacroix J, Leclerc F (1999) Development of a pediatric multiple organ dysfunction score: use of two strategies. Med Decis Making 19:399–410
Jack D, Bidwell J, Turner M, Wood N (1997) Simultaneous genotyping for all three known structural mutations in the human mannose-binding lectin gene. Hum Mutat 9:41–46
Turner MW, Dinan L, Heatley S, Jack DL, Boettcher B, Lester S, McCluskey J, Roberton DM (2000) Restricted polymorphism of the mannose-binding lectin gene of indigenous Australians. Hum Mol Genet 9:1481–1486
Mead R, Jack D, Pembrey M, Tyfield L, Turner M and the ALSPAC Study Team (1997) Mannose-binding lectin alleles in a prospectively recruited UK population. Lancet 349:1669–1670
Reid CL, Perrey C, Pravica V, Hutchinson IV, Campbell IT (2002) Genetic variation in proinflammatory and anti-inflammatory cytokine production in multiple organ dysfunction syndrome. Crit Care Med 30:2216–2221
Stuber F, Petersen M, Bokelmann F, Schade U (1996) A genomic polymorphism within the tumor necrosis factor locus influences plasma tumor necrosis factor-alpha concentrations and outcome of patients with severe sepsis. Crit Care Med 24:381–384
Slutsky AS, Russell J (2002) Genetics of critical illness: methodological issues. Crit Care Med 30:2382–2383
Garred P, Strom J, Quist L, Taaning E, Madsen HO (2003) Association of mannose-binding lectin polymorphisms with sepsis and fatal outcome, in patients with systemic inflammatory response syndrome. J Infect Dis 188:1394–1403
Jordan JE, Montalto MC, Stahl GL (2001) Inhibition of mannose-binding lectin reduces postischemic myocardial reperfusion injury. Circulation 104:1413–1418
Garred P, Richter C, Madsen HO, Mtoni I, Svejgaard A, Shao J (1997) Mannan-binding lectin in the sub-Saharan HIV and tuberculosis epidemics. Scand J Immunol 46:204–208
Brandtzaeg P, Hogasen K, Kierulf P, Mollnes TE (1996) The excessive complement activation in fulminant meningococcal septicemia is predominantly caused by alternative pathway activation. J Infect Dis 173:647–655
Garred P, Pressler T, Lanng S, Madsen HO, Moser C, Laursen I, Balstrup F, Koch C, Koch C (2002) Mannose-binding lectin (MBL) therapy in an MBL-deficient patient with severe cystic fibrosis lung disease. Pediatr Pulmonol 33:201–207
Acknowledgements
K.F. is supported by a Training Fellowship from The Wellcome Trust. The work was also supported by: EU grant contract no. QLRT-CT-2001-01039 and by Action Research. Research at the Institute of Child Health and Great Ormond Street Hospital for Children National Health Service (NHS) Trust benefits from research and development funding received from the UK NHS Executive. We thank Clare Booth and Marina Johnson and the staff, parents and children on the PICU.
Author information
Authors and Affiliations
Corresponding author
Additional information
M.T. and N.K. are scientific consultants for NatImmune, a Danish company exploring the therapeutic potential of MBL.
An editorial regarding this article can be found in the same issue (http://dx.doi.org/10.1007/s00134-004-2302-9)
Rights and permissions
About this article
Cite this article
Fidler, K.J., Wilson, P., Davies, J.C. et al. Increased incidence and severity of the systemic inflammatory response syndrome in patients deficient in mannose-binding lectin. Intensive Care Med 30, 1438–1445 (2004). https://doi.org/10.1007/s00134-004-2303-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-004-2303-8