
Abstract
Objective: To investigate the effect of propofol and its solvent Intralipid on the adhesion of activated platelets to leukocytes in vitro. Design and setting: Prospective study in an experimental laboratory. Participants: Sixteen healthy volunteers. Interventions: Whole blood was incubated for 60 min with propofol (4, 40 µg/ml), an equal volume of Intralipid 10% or phosphate-buffered saline (PBS). After stimulation with adenosine-5-diphosphate (ADP) platelet–leukocyte adhesion and platelet surface expression of P-selectin, GPIb and fibrinogen-binding to platelets were evaluated by flow cytometry. Measurements and results: The 4 µg/ml concentration of propofol did not alter binding of platelets to leukocytes, expression of P-selectin, GPIb and fibrinogen binding to platelets. The 40 µg/ml concentration of propofol reduced spontaneous and ADP-induced formation of platelet–neutrophil conjugates compared with PBS and the equal volume of Intralipid. In addition, binding of ADP-activated platelets to monocytes were also inhibited by 40 µg/ml propofol. Following incubation with propofol, platelets showed reduced binding of fibrinogen in the unstimulated and ADP-stimulated blood samples as well as a lower percentage of platelets with bound fibrinogen. Effects dependent on the solvent Intralipid were enhanced adhesion of platelets to monocytes in comparison with propofol (40 µg/ml) and PBS. Conclusion: In clinically used concentrations, propofol does not alter the adhesion of platelets to leukocytes in vitro. At ten-fold anesthetic concentration propofol reduced the formation of platelet–neutrophil and platelet–monocyte conjugates. We suggest that this effect is due to an inhibition of fibrinogen-binding to platelets by propofol. These effects were all independent of the propofol carrier Intralipid.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Evidence suggests that activated platelets are closely involved in the regulation of inflammation, hemostasis, and thrombosis. Furthermore, interaction of platelets with leukocytes and endothelium may contribute to the pathogenesis of local and systemic inflammation processes. It is well established that platelets modulate leukocyte function upon direct adhesion primarily via an interaction between P-selectin on the platelet surface and P-selectin ligand (PSGL-1) on the surface of leukocytes [1, 2] as well as by soluble mediators [3]. In addition, adhesion of platelets to leukocytes seems not only to be an in vitro phenomenon, since increased platelet–leukocyte conjugates have been reported in sepsis [4], myocardial infarction [5], postischemic reperfusion damage [6], and thrombosis [7].
Anesthetic agents such as propofol are frequently used for sedation of critically ill patients. The effect of propofol on the cellular function of various types of leukocytes has been intensively investigated over the last years [8]. Most studies have been performed with isolated neutrophils or monocytes. Recent studies favored a whole blood assay to investigate the effect of anesthetics on neutrophil function [9, 10]. However, the effect of propofol or other sedative agents on the interaction between different blood cells of the human immune system has not been investigated. Since binding of activated platelets to leukocytes plays an important role in the regulation of neutrophil and monocyte function as well as in the pathogenesis of inflammatory diseases such as sepsis, we investigated whether propofol alters adhesion of activated platelets to leukocytes to gain further insight into the mechanism of anesthetic-induced immunomodulation. Using activation-dependent monoclonal antibodies and flow cytometry, we studied the effect of propofol as well as its solvent Intralipid on platelet–leukocyte adhesion, expression of platelet adhesion membrane receptors, and binding of fibrinogen to platelets in vitro.
Materials and methods
After approval of the local institutional ethics committees and the obtaining of informed consent, venous blood samples were taken from 16 healthy volunteers who had not ingested nonsteroidal antirheumatics for at least 2 weeks prior to donation. Blood was drawn via a 21-gauge butterfly cannula without tourniquet from an antecubital vein into blood collection tubes (Sarstedt, Nümbrecht, Germany) to a final concentration of 0.32% sodium citrate. The first 2 ml of blood were discarded. Blood samples of each donor were immediately diluted 1:1 with 37 °C prewarmed Dulbecco's phosphate-buffered saline (PBS without Ca2+ and Mg2+; Sigma Chemicals, St. Louis, Mo.), which contained the desired concentration of either propofol or Intralipid. The concentration of propofol tested was that providing clinical anesthesia (4 µg/ml) and ten times this concentration (40 µg/ml) [11]. The appropriate volume of Intralipid 10% equal to the concentration of propofol being investigated was also tested, because parenteral lipid solutions are reported to influence leukocyte function. One blood sample was diluted only with PBS to serve as control. Subsequently, the blood samples were incubated for 60 min at 37 °C in a water bath. After incubation, blood samples were immediately processed for stimulation procedures and flow cytometric analysis.
Whole blood stimulation and flow cytometric analysis were performed as previously described [12, 13]. In brief, adenosine-5-diphosphate (ADP; Sigma Chemicals, St. Louis, Mo.) was used at a final concentration of 2.5 µM. After 5 min stimulation at room temperature, 100 µl of the blood samples were added to polypropylene tubes containing saturating concentrations of fluorochrome-conjugated antibodies. The staining procedure was stopped after 15 min with 2 ml red cell lysing solution (Lysing Solution; Becton-Dickinson). After centrifugation (5 min, 350×g, 4°C), the samples were washed with 2 ml PBS containing 1% bovine serum albumin (BSA), centrifugated, and the remaining pellet resuspended in 500 µl PBS containing 1% BSA and 1% paraformaldehyde. The cells were stored for up to 30 min at 4 °C in the dark until flow cytometric measurements were performed.
The following antibodies (Mab) were used to measure platelet–leukocyte adhesion and expression of specific platelet surface glycoproteins: anti-CD45-FITC (clone HI30) Mab for leukocyte common antigen; anti-CD14-PerCP (clone MϕP9, Becton-Dickinson) Mab recognizing a human monocyte antigen; anti-CD42b-PE (clone HIP1) Mab recognizing the α-subunit of the GPIb complex; anti-CD62P-FITC (clone AK-4) Mab directed against P-selectin expressed on platelet surface; and negative IgG1-FITC and IgG1-PE antibodies (clone MOPC-21) for nonspecific binding (all from Pharmingen, San Jose, Calif.). A FITC-conjugated IgG fraction of rabbit anti-human-fibrinogen (DAKO) was used as a nonspecific isotype control in the fibrinogen-binding assay.
Flow cytometric analysis
Flow cytometric analysis was performed on a FACSCalibur flow cytometer and analyzed using CellQuest 3.1 software (Becton-Dickinson, San Jose, Calif.). The cytometer was calibrated daily with fluorescence microbeads (Calibrite Beads, Becton-Dickinson).
Leukocytes were identified and differentiated into subgroups by their cell size and granularity in the forward and side scatter (SSC), as well as by their anti-CD45-FITC fluorescence (FL 1). As previously described [5, 12, 13], the histogram generated by the anti-CD45-FITC fluorescence versus SSCserved to identify neutrophils (Fig. 1), because this panleukocyte antigen is not present on erythroid cells, platelets, or platelet aggregates. Furthermore, monocytes were also identified with an anti-CD14-PerCP Mab (Fig. 2). Platelet adhesion to leukocytes was defined as neutrophils or monocytes positive for anti-CD42b-PE (FL2). The percentage of neutrophils and monocytes with bound platelets was measured (Figs. 1, 2). For each sample, 40 000 leukocytes were measured.

Adjustment of the aquisition dot plots for analysis of the percentage of neutrophils with bound platelets. Neutrophils were identified in the sideward scatter (SSC) as well as their specific binding characteristics of anti-CD45-FITC (FL 1) as acquired on the flow cytometer (A). The events in the neutrophil gate above the horizontal line in the right upper quadrant (B) were considered to represent neutrophil–platelet conjugates and those below the line were considered to be platelet-free neutrophils, as determined by isotype control. C and D show a representative result of the inhibiting effect of 40 µg/ml propofol on the binding of ADP-activated platelets to neutrophils in comparison with PBS control

Adjustment of the aquisition dot plots for analysis of the percentage of monocytes with bound platelets. Monocytes were identified in the sideward scatter (SSC) versus CD14-PerCP signal as acquired on the flow cytometer (A). The events above the horizontal line in the right upper quadrant (B) were considered to represent monocyte–platelet conjugates and those below the line were considered to be platelet-free monocytes, as determined by isotype control. C and D show a representative result of the inhibiting effect of 40 µg/ml propofol on the binding of ADP-activated platelets to monocytes in comparison with PBS control
To determine GPIb and P-selectin expression as well as fibrinogen binding on the surface of platelets, individual platelets were identified by size (forward scatter) and anti-CD42b-PE immunofluorescence in a logarithmic scaled dot plot. Results are expressed as percentage of platelets positive for P-selectin and fibrinogen, and the mean fluorescence intensity (MFI). The anti-CD42b MFI and anti-CD62P MFI reflect the number of GPIb and P-selectin epitopes expressed on the platelet surface membrane, and the anti-fibrinogen MFI the amount of fibrinogen bound to the GPIIb/IIIa receptor per individual platelet. For each sample, 10 000 platelets were collected.
Statistical analysis
The Kolmogorov-Smirnov test showed that the flow cytometric data were not normally distributed. Thus, results are expressed as median (25th–75th percentile) unless otherwise indicated. Differences between control, Intralipid, and propofol samples were tested using the Friedman test followed by post hoc analysis (Wilcoxon test) with Bonferroni correction. A P value below 0.05 from the post hoc analysis using the Wilcoxon test was regarded as significant.
Results
At the sedating concentration of 4 µg/ml, propofol did not influence the basal or ADP-induced formation of leukocyte-platelet conjugates (Figs. 3, 4), GPIb and P-selectin expression on the platelet surface membrane, and fibrinogen binding to platelets (Tables 1, 2).

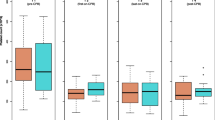
Spontaneous and ADP-induced neutrophil–leukocyte adhesion following incubation with either 4 or 40 µg/ml propofol in comparison with PBS and the corresponding volume of Intralipid. Box plots show 25th and 75th percentiles, median, and range of eight independent experiments for each concentration of propofol and Intralipid. Values are presented as percentage of monocytes with bound platelets (% CD42b positive neutrophils). * P<0.05 versus PBS and 400 µg/ml Intralipid

Spontaneous and ADP-induced monocyte–leukocyte adhesion following incubation with either 4 or 40 µg/ml propofol in comparison with PBS and the corresponding volume of Intralipid. Box plots show 25th and 75th percentiles, median, and range of eight independent experiments for each concentration of propofol and Intralipid. Values are presented as percentage of monocytes with bound platelets (% CD42b positive monocytes). * P<0.05 versus PBS control and 400 µg/ml Intralipid; # P<0.05 versus PBS and 40 µg/ml propofol
After increasing the propofol concentration to 40 µg/ml we observed a significant reduction in the percentage of neutrophils with bound platelets in the unstimulated blood samples, and of neutrophils and monocytes in ADP-stimulated blood samples in comparison with PBS control and the equivalent volume of Intralipid 10% (Figs. 3, 4).
The effect of 40 µg/ml propofol on the expression of GPIb and P-selectin on the platelet surface membrane, and fibrinogen binding to platelets are shown in Table 2. After incubation with 40 µg/ml propofol, binding of fibrinogen to GPIIb/IIIa was significantly reduced compared with PBS control and the equivalent volume of Intralipid 10%. After stimulation with ADP, we observed a reduction in the percentage of platelets with bound fibrinogen as well as the amount of fibrinogen per individual platelet incubated with 40 µg/ml propofol.
Effects attributable to the propofol carrier Intralipid 10% were seen only at the higher concentration, which is equivalent to the amount of lipid present at the concentration of 40 µg/ml propofol (Fig. 4). Effects dependent on Intralipid were an enhanced adhesion of unstimulated platelets to monocytes, whereas platelet glycoprotein expression and fibrinogen-binding to GPIIb/IIIa was not affected (Tables 1, 2).
Discussion
Adhesion of platelets to neutrophils and monocytes represents an important event in physiological and pathological inflammation, as demonstrated in vitro [14, 15, 16] and observed in vivo [4, 5, 6, 7]. Propofol is known to affect several aspects of leukocyte [8] and platelet function [17]. However, most studies investigating the effects of propofol on neutrophil or monocyte function used isolated cells, eliminating the impact of platelets on the immunological function of these cells. Therefore, we were interested to evaluate the effect of propofol on platelet–leukocyte adhesion.
The major findings of the present study are: (1) at the clinically important concentration of 4 µg/ml, propofol does not alter adhesion of activated platelets to leukocytes. (2) The ten-fold propofol concentration inhibits the percentage of neutrophils and monocytes with bound platelets after stimulation with ADP. (3) Fibrinogen-binding to platelets is suppressed at 40 µg/ml propofol. (4) In the 40 µg/ml propofol group, expression of GPIb, which might also be a counterreceptor for neutrophil CD11b, is increased.
Propofol is known to inhibit neutrophil chemotaxis [18, 19], phagocytosis [20], respiratory burst activity [21], and IL-8 release [22]. In monocytes, propofol augmented the endotoxin-induced release of TNF-α and inhibited the release of IL-1ra [10]. However, these studies used isolated neutrophils, despite the study of Larsen et al. [10], eliminating the influence of other blood cells and intercellular mechanisms present in whole blood. Recent investigations revealed that activated platelets binding to neutrophil CD11b modulate respiratory burst activity [23] and recruitment of neutrophils to sites of inflammation [15]. Furthermore, interaction of platelets with neutrophils and monocytes enhances the ability of other mediators to induce the release of IL-1β, IL-8, and monocyte chemotactic protein (MCP-1) [14, 24]. The results of the present study demonstrated that propofol inhibits binding of activated platelets to neutrophils and monocytes in human whole blood only at concentrations that exceed the clinical relevant concentration ten times. Accordingly, we conclude that in clinically used concentrations propofol does not influence neutrophil and monocyte immune function by modifying the mutual cellular interaction between platelets and leukocytes.
Binding of platelets to leukocytes is supposed to occur in several sequential steps. First, binding of activated platelets to leukocytes is initiated by an adhesion cascade in which P-selectin binds to PSGL-1[2]. Engagement of P-selectin with PSGL-1 triggers tyrosine kinase-dependent mechanisms that lead to CD11b activation, enabling subsequent tight adhesion to platelets [25]. The CD11b counterreceptor on the platelet surface has not been definitely characterized. Two proposed receptors are fibrinogen bound to platelet GPIIb/IIIa [26, 27] and the platelet glycoprotein GPIb [28]. However, data on the role of GPIIb/IIIa as a CD11b counterreceptor are controversial. Kirchhofer et al. demonstrated complete inhibition of binding of activated platelets to leukocytes using a P-selectin blocking antibody in combination with a GPIIb/IIIa antagonist, but only partial inhibition when using the P-selectin antibody alone [15]. Weber and Springer showed that binding of activated platelets was partially blocked by an antibody against GPIIb/IIIa [26]. In contrast, results of Ostrovsky et al. did not support the role of GPIIb/IIIa-fibrinogen as the counterreceptor of CD11b [29]. Therefore, the existence of another CD11b receptor on the platelet surface has been proposed, which might be the constitutively expressed platelet glycoprotein GPIb. Since propofol inhibited the formation of platelet–leukocyte conjugates, we tried to clarify the underlying effect by measuring platelet P-selectin and GPIb expression as well as fibrinogen-binding. Propofol had no effect on P-selectin and GPIb expression. However, 40 µg/ml propofol inhibited fibrinogen binding to platelets and the percentage of platelets with bound fibrinogen following activation with ADP. Since adhesion of activated platelets to leukocytes might depend in part on the interaction of leukocyte CD11b with fibrinogen bound to platelet GPIIb/IIIa, this finding may provide an explanation for the inhibiting effect of propofol on the formation of platelet–leukocyte conjugates. However, our data do not rule out the possibility of additional effects of propofol on P-selectin-triggered mechanisms via PSGL-1 leading to activation of CD11b-dependent adhesion events.
Propofol inhibits platelet aggregation, probably by inhibiting the transient rise in intracellular calcium [17]. Furthermore, propofol is assumed to interact with G-protein, phospholipase C, protein kinase C, and mitogen-activated protein kinase p42 [30, 31]. It is possible that the inhibitory effect of propofol on platelet–leukocyte adhesion may also be mediated by interacting with these proteins and enzymes, which are involved in platelet and leukocyte signal transduction and function.
Heine et al. reported an inhibitory effect of the solvent of propofol (Intralipid 10%) on the neutrophil respiratory burst activity [21, 32]. To differentiate between the specific effect of propofol and its solvent, we compared propofol-diluted blood samples with PBS-diluted and Intralipid-diluted blood samples. Therefore, our results show that the described propofol effects are independent of its solvent Intralipid. A specific lipid effect, enhanced adhesion of unstimulated platelets to monocytes, was only seen in the 400 µg/ml Intralipid group.
In conclusion, at the concentrations in clinical use propofol does not alter the adhesion of platelets to leukocytes in vitro. Accordingly, we suggest that the propofol concentrations achieved during sedation of critically ill patients may not inhibit leukocyte immune function by modifying the mutual cellular interaction between platelets and leukocytes. Only at supraphysiological concentrations did propofol reduce the formation of platelet–neutrophil and platelet–monocyte conjugates, which is due to an inhibition of fibrinogen binding to platelets by propofol. However, since in vitro conditions do not reproduce in vivo conditions identically, additional clinical studies in critically ill patients should be continued.
References
Larsen E, Celi A, Gilbert GE, Furie CE, Erban JK, Bonfati R, Wagner DD, Furie B (1989) PADGEM protein: a receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell 59:305–312
Yang J, Furie BC, Furie B (1999) The biology of P-selectin glycoprotein ligand-1: its role as a selectin counterreceptor in leukocyte-endothelial and leukocyte-platelet interaction. Thromb Haemost 81:1–7
Brunetti M, Martelli N, Manarini S, Mascetra N, Musiani P, Cerletti C, Aiello FB, Evangelista V (2000) Polymorphonuclear leukocyte apoptosis is inhibited by platelet-released mediators, role of TGFß-1. Thromb Haemost 84:478–483
Gawaz M, Dickfeld T, Bogner, C, Fateh-Moghadam S, Neumann FJ (1997) Platelet function in septic multiple organ dysfunction syndrome. Intensive Care Med 23:379–385
Ott I, Neumann FJ, Gawaz M, Schmitt M, Schömig A (1996) Increased neutrophil-platelet adhesion in patients with unstable angina. Circulation 94:1239–1246
Carden DL, Granger DN (2000) Pathophysiology of ischaemia-reperfusion injury. J Pathol 190:255–66
Kirchhofer D, Riederer MA, Baumgartner HR (1997) Specific accumulation of circulating monocytes and polymorphonuclear leukocytes on platelet thrombi in a vascular injury model. Blood 89:1270–1289
Galley HF, DiMatteo MA, Webster NR (2000). Immunomodulation by anaesthetic, sedative and analgesic agents: does it matter? Intensive Care Med 26:267–274
Van der Poll T, Jansen J, Endert E, Sauerwein HP, van Deventer SJH (1994). Noradrenalin inhibits lipopolysaccaride-induced tumor necrosis factor and interleukin 6 production in human whole blood. Infect Immun 62:2046–2050
Larsen B, Hoff G, Wilhelm W, Buchinger H, Wanner GA, Bauer M (1998) Effect of intravenous anesthetics on spontaneous and endotoxin-stimulated cytokine response in cultured human whole blood. Anesthesiology 89:1218–1227
Cockshott ID (1985) Propofol (Diprivan) pharmacokinetics and metabolism: an overview. Postgrad Med J 61(Suppl 3):45–50
Rinder HM, Bonan JL, Rinder CS, Ault KA, Smith BR (1991) Dynamics of leukocyte-platelet adhesion in whole blood. Blood 78:1730–1737
de Rossi LW, Horn NA, Hecker KE, Robitzsch T, Hutschenreuter G, Rossaint R (2002) Effect of halothane and isoflurane on binding of ADP- and TRAP-6- activated platelets to leukocytes in whole blood. Anesthesiology 96:117–24
Weyrich AS, Elstad MR, McEver RP, McIntyre TM, Moore KL, Morrissey JH, Prescott SM, Zimmerman GA (1996) Activated platelets signal chemokine synthesis by human monocytes. J Clin Invest 97:1525–1534
Kirchhofer D, Riederer MA, Baumgartner HR (1997) Specific accumulation of circulating monocytes and polymorphonuclear leukcocytes on platelet thrombi in a vascular injury model. Blood 89:1270–1278
Peters MJ, Dixon G, Kotowicz KT, Hatch DJ, Heyderman RS, Klein NJ (1999) Circulating platelet-neutrophil complexes represent a subpopulation of activated neutrophils primed for adhesion, phagocytosis and intracellular killing. Br J Haematol 106:391–399
Aoki H, Mizobe T, Nozuchi S, Hiramatsu N (1998) In vivo and in vitro studies of the inhibitory effect of propofol on human platelet aggregation. Anesthesiology 88:362–370
Jensen AG, Dahlgren C, Eintrei C (1993) Propofol decreases random and chemotactic stimulated locomotion of human neutrophils in vitro. Br J Anaesth 70:99–100
Nagata T, Kansha M, Irita K, Takahashi S (2001) Propofol inhibits FMLP-stimulated phosphorylation of p42 mitogen-activated protein kinase and chemotaxis in human neutrophils. Br J Anaesth 86:853–858
Krumholz W, Endrass J, Hempelmann G (1994) Propofol inhibits phagocytosis and killing of Staphylococcus aureus and Escherichia coli by polymorphonuclear leukocytes in vitro. Can J Anesth 41:446–449
Heine J, Leuwer M, Scheinichen D, Arseniev L, Jaeger K, Piepenbrock S (1996) Flow cytometric evaluation of the in vitro influence of four i.v. anaesthetics on respiratory burst of neutrophils. Br J Anaesth 77:387–392
Galley HF, Dubbels AM, Webster NR (1998) The effect of midazolam and propofol on interleukin-8 from human polymorphonuclear leukocytes. Anesth Analg 86:1289–1293
Ruf A, Patscheke H (1995) Platelet-induced neutrophil activation: platelet-expressed fibrinogen induces the oxidative burst in neutrophils by an interaction with CD11c/CD18. Br J Haematol 90:791–796
Neumann FJ, Marx N, Gawaz M, Brand K, Ott I, Rokitta C, Sticherling C, Meinl C, May A, Schömig A (1997) Induction of cytokine expression in leukocytes by binding of thrombin-stimulated platelets. Circulation 95:2387–2394
Evangelista V, Manarini S, Rontondo S, Martelli N, Polischuk R, McGregor JL, de Gaetano G, Cerletti C (1996) Platelet/polymorphonuclear leukocyte interaction in dynamic conditions: evidence of adhesion cascade and cross talk between P-selectin and the β2-integrin CD11b/CD18. Blood 88:4183–4194
Weber C, Springer TA (1997) Neutrophil accumulation on activated, surface adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to αMβ2 and stimulated by platelet-activating factor. J Clin Invest 100:2085–2093
Spangenberg P, Redlich H, Bergmann I, Lösche W, Gotzrath M, Kehrel B (1993) The platelet glycoprotein IIb/IIIa complex is involved in the adhesion of activated platelets to leukocytes. Thromb Haemost 70:514–521
Simon DI, Chen Z, Xu H, Li CQ, Dong J, McIntire LV, Ballantyne CM, Zhang L, Furman MI, Berndt MC, Lopez JA (2001) Platelet glycoprotein Ibα is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J Exp Med 192:193–204
Ostrovsky L, King AJ, Bond S, Mitchell D, Lorant DE, Zimmermann GA, Larsen R, Niu XF, Kubes P (1998) A juxtacrine mechanism for neutrophil adhesion on platelets involves platelet-activating factor and a selectin-dependent activation process. Blood 91:3028–3036
Weiss M, Birkhahn A, Krone M, Schneider EM (1996) Do etomidate and propofol influence oxygen radical production of neutrophils? Immunopharmacol Immunotoxicol 18:291–307
Nagata T, Kansha M, Irita K, Takahashi S (2001) Propofol inhibits FMLP-stimulated phosphorylation of p42 mitogen-activated protein kinase and chemotaxis in human neutrophils. Br J Anaesth 86:853–858
Heine J, Scheinichen D, Jaeger K, Andre M, Leuwer M (1999). In vitro influence of parenteral lipid emulsions on the respiratory burst of neutrophils. Nutrition 15:540–545
Acknowledgment
This study was supported in part by START, a research grant from the Rheinisch-Westfälische Technische Hochschule Aachen, and the Department of Anaesthesiology, University Hospital Aachen, Germany.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
de Rossi, L., Wessiepe, M., Buhre, W. et al. Effect of propofol on adhesion of activated platelets to leukocytes in human whole blood. Intensive Care Med 29, 1157–1163 (2003). https://doi.org/10.1007/s00134-003-1814-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-003-1814-z