
Abstract
The aim of this study was to determine the toxic effect of atrazine at the ovarian cellular level. Chinese Hamster Ovary (CHO-K1) cell line was used to evaluate the degree of in vitro atrazine cytotoxicity and the morphological changes were followed during the cell death. Application of four bioassays confirmed that atrazine decreases ovarian cell proliferation and IC50 were determined with each assay after 72 h of exposure. The level of apoptosis in atrazine treated cells was low.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
During the past 50 years, tens of thousands of various chemicals have been synthesized and released into the environment. Some of these chemicals inadvertently interfere with hormone function in animals and in humans, and are so-called endocrine disruptors with implications to the public health (National Research Council 1999). Low-level exposures to endocrine-disrupting chemicals are ubiquitous in today’s environment. As hormones act at extremely low levels (in parts per trillion), even exposure to low levels of hormonally active agents may be of concern, because endocrine-mediated effects may be subtle and be manifested primarily in populations rather than in individuals. Reported abnormalities in laboratory animals and wildlife exposed to endocrine-disrupting chemicals include feminization of males, abnormal sexual behaviour, birth defects, altered sex ratio, decreased sperm density, decreased size of testes, breast cancer, testicular cancer, reproductive failure and thyroid dysfunction (Crisp et al. 1998).
Atrazine (2-chloro-4-ethylamino-6-isopropylamino-s-triazine) selectively controls broadleaf weeds and certain grass weeds in corn and sorghum fields. Estimated 3.4 × 107 kg of atrazine (Gianessi and Marcelli 2000) are applied to cropland each year in the United States, but it is banned in the European Union. Effects on other physiological processes point out atrazine as an endocrine disruptor. Atrazine inhibits the activities of androgen metabolizing enzymes and the formation of androgen-receptor complex in male anterior pituitary level (Kniewald et al. 1979), but also in females induces repetitive pseudopregnancies by disrupting ovarian function (Cooper et al. 2000). Also, atrazine alters the functions of the endocrine system by interfering with the synthesis, metabolism, binding or cellular responses of natural hormones (Fujimoto and Honda 2003), inhibits formation of a specific oestradiol receptor complexes in the rat uterus cytosol (Težak et al. 1992). In addition atrazine elicits a broad range of effects associated with neuroendocrine disruption. These include disruption of normal ovarian cycling patterns in adult rats (Šimić et al. 1994; Peruzović et al. 1995), delayed puberty in male rats (Stoker et al. 2000) and prenatal developmental effects such as suppression of suckling induced prolactin release (Stoker et al. 1999).
Application of in vitro assays in evaluation of the toxicity should be an important contribution in elucidating cellular and molecular mechanisms. There has been a great deal of research and debate during the past 20 years concerning the use of alternative on laboratory animals for testing the potential reproductive toxicity of chemicals and physical agents and mixtures (Kniewald et al. 2005). Some aspects of female reproductive function can be modelled in vitro, and several cellular components of the female reproductive organs can be maintained in culture as ovarian somatic cells. The adverse effects can be assessed by examining cell morphology, and by determining cell viability and hormonal responsiveness (OECD 1993). Of course at nowadays, it is impossible to reduce, refine and replace all animal experiments by applying biomarkers in environmental bioassays for toxicological evaluation. But it is increasingly apparent that the development and incorporation of stepwise testing strategies, combining experimental data for a range of alternative methods, provide the most progressive way to predict toxicity, reducing at the same time the number of laboratory animals used for testing purposes.
The aim of this study was to determine the toxic effect of atrazine at the ovarian cellular level. Chinese Hamster Ovary (CHO-K1) cell line was used as the test system for evaluation of atrazine cytotoxicity followed by four bioassays. The morphological changes of atrazine exposed cells were detected by fluorescent microscopy.
Materials and Methods
Chinese Hamster Ovary (CHO-K1; CCL-61) cells originated from the American Type Cultures Collection (ATCC) were a hypoploidal subclone of the parental CHO cell line, which derived from the ovary of an adult Chinese hamster. Cells were grown in Dulbecco’s Modified Eagle’s Medium (D-MEM) and Ham’s F12 (1:1) supplemented with l-glutamine and 15 mM HEPES (GIBCO, Paisley, Scotland, UK) with addition of 5% of newborn calf serum (NCS) (GIBCO, Paisley, Scotland, UK). The cells were cultured in 75 cm2 cell culture flasks in a humidified atmosphere of 95% air and 5% CO2 at 37°C. For experimental purposes cells in log phase of growth were seeded in 6-well plates (2 mL of cell solution /well) at the initial concentration of 2 × 104 cells/mL and allowed 24 h to attach before treatment with atrazine.
Atrazine [2-chloro-4-ethylamino-6-isopropylamino-s-triazine; CAS# 1912-24-9] (HERBOS d. d., Sisak, Croatia) was prepared in absolute ethanol insomuch that the addition of 20 μL resulted in the final concentrations of 10–160 μg atrazine/mL of cell suspension (46–742 μM). Cells were treated with atrazine for 24, 48 or 72 h. The cell viability was determined by four different bioassays: Trypan blue exclusion method, Kenacid blue, Neutral red and MTT assays.
Trypan blue exclusion method was used in estimating the proportion of viable cells. Cells were separated from the well surface by adding 0.5 mL Trypsine-EDTA solution (Sigma-Aldrich, Co Ltd., UK) and 0.5 mL of medium was added. Twenty microlitre of cell suspension was mixed with 20 μL of 0.4% trypan blue dye (Sigma-Aldrich Chemie, GmbH, Germany; cell culture tested) solution. Cells were resuspended and dye excluding viable cells were microscopically counted on Fuchs-Rosenthal haemocytometer (Freshney 2005).
The total cell protein content was determined by the Kenacid blue assay according to O’Hare and Atterwill (1995) with minor modifications. After exposure to atrazine, the medium was discarded from cells and culture was rinsed twice with phosphate buffered saline (PBS) to remove any remaining medium. When PBS solution was removed, 1 mL of the fixative (1% glacial acetic acid; 50% ethanol; 49% distilled water) was added to each well. Plates were shaken gently for 20 min, fixative was removed and 1 mL of Kenacid blue stain was added. After 25 min the stain was discarded, each well was filled with 1 mL of washing solution (10% ethanol; 5% glacial acetic acid; 85% distilled water), replaced after 1 min with new aliquote and shaken again for 20 min. Washing solution was replaced with 2 mL of desorbing solution (9.8 g potassium acetate, 70 mL absolute ethanol and 30 mL demineralized water) and shaken for 20 min. The absorbance was measured spectrophotometrically at 577 nm (Helios, Thermo Electro Corporation).
The Neutral red uptake assay (O’Hare and Atterwill 1995) was applied to determine the accumulation of the neutral red dye in the lysosomes of viable uninjured cells. Neutral red dye (Merck, Darmstadt, Germany) was added and the mixture was incubated at 37°C for 3 h. Neutral red solution was discarded and cells were washed with PBS. The cells were destained with 2 mL of destaining solution (1% glacial acetic acid; 50% ethanol; 49% distilled water) and shake for 20 min. The absorbance was measured at 540 nm using a spectrophotometer.
The viability of cells was further followed by MTT assay (Freshney 2005). The MTT (SIGMA-Aldrich Chemie, Steinheim, Germany) was prepared by dissolving in PBS. After treatment with atrazine, 200 μL of MTT solution was added to each well. The plates were incubated for 4 h at 37°C, and at the end of incubation period the media were discarded and 2 mL of DMSO was added to each well. The plates were mechanically shaken until purple formazan crystals were dissolved and the absorbance was determined at 570 nm.
The fluorescent microscopy was applied to determine the viability of attached and detached cells and possible morphological changes during cell death using the staining of cells with fluorescein diacetate-propidium iodide (FDA-PI). After treatment with atrazine for 72 h, the cell solution and detached cells were pooled and centrifuged at 1,000 rpm for 3 min. The supernatant was discarded and the pellets were resuspended in residual medium. Stock solutions of FDA (1 mg in 100 μL acetone and 900 μL demineralized water) (Sigma, USA) and PI (50 μg/mL in PBS) (Sigma, USA) were prepared and stored at 4°C in the dark. Staining was achieved by the addition of 6 μL FDA and 3 μL PI to the cell suspension. The solution was gently mixed and stained suspension was then mounted on a microscope slide. The cell morphology was determined on the fluorescent microscope (Olympus BX51; Olympus American Inc., Central Valley, USA) using 488 nm filters (Freshney 2005).
The results are reported as means ± SEM. A two-tailed Student’s t-test was applied to evaluate the significance of differences between means: p < 0.05 was considered significant.
Results and Discussion
CHO-K1 cells were exposed to atrazine (10–160 μg/mL of cell suspension or 46–742 μM) for 24, 48 and 72 h and cytotoxicity was determined by using four different bioassays: Trypan blue exclusion method, Kenacid blue, Neutral red and MTT methods. All applied methods indicated a clear dose-dependent cytotoxic effects. Figure 1 represents the results of atrazine toxicity determination in the concentrations of atrazine 20–80 μg/mL followed by Trypan blue method. After 72 h of incubation period, even the lowest tested concentration of atrazine (20 μg/mL) significantly (p < 0.05) decreased proliferation of cells for 19% in CHO-K1 cell line and the toxicity progressively increased with higher atrazine concentrations. The highest applied dose of 80 μg atrazine/mL decreased the cell viability for 55% (p < 0.001) as well as the dose of 60 μg atrazine/mL for 49% (p < 0.001).

The proliferation of CHO-K1 cells (Control – intact), cells in the presence of 20 μL of absolute ethanol in the cultivating medium (Control – EtOH), and the cytotoxic effect of atrazine in concentrations 20–80 μg/mL (A20–A80) during 72 h followed by Trypan blue exclusion method. *p < 0.05, **p < 0.01, ***p < 0.001 – Significant difference versus control EtOH (Student’s t-test)
As Fig. 1 represents atrazine cytotoxicity followed by Trypan blue method, on the same way other three colorimetric methods were applied and the dose–response curves were observed. For each method at least three experiments were performed and within single experiment each applied atrazine concentration was tested in triplicate. Figure 2 presents the summarized results of atrazine toxicity evaluation with four bioassays in CHO-K1 cells for 24, 48 and 72 h of exposure. Data are expressed as the percentage of control value (control with ethanol) with standard error for cell viability in the presence of atrazine concentrations of 10–160 μg/mL (46–742 μM). IC values were derived from the equations of related exponential trend lines for each applied method. In Table 1 are summarized the IC20 and IC50 values obtained with four applied bioassays (Fig. 2). After 24 h IC20 values were possible to calculate, while after 48 h beside IC20 values, the IC50 was possible to determine only with Trypan blue method. With all applied methods both IC20 and IC50 values were calculated after 72 h.

Comparison of Trypan blue exclusion method (TB), Neutral red (NR), Kenacid blue (KB) and MTT assays in CHO-K1 cells after exposure to 46–742 μM atrazine for 24 (A–B), 48 (C–D) and 72 h (E–F). Data are presented as percentage of control: mean ± standard error of at least three experiments with at least three measurements within each experiment for each atrazine concentration. *Equation of pertaining exponential trendline and its R2 value
When the four cytotoxicity assays, employed to assess atrazine toxicity in vitro are compared, it is obvious that the results obtained are not in agreement. Those differences can be explained by the nature of each assay. Reactivity of trypan blue dye is based on the cell membrane damage, and in other three assays it is possible to detect the injury within the cell. So, the MTT assay is mainly based on the enzymatic conversion of MTT in the mitochondria under the influence of succinate dehydrogenase, the Neutral red assay is a colorimetric assay measuring the uptake of the dye by functional lysosomes and the Kenacid blue assay determines the total cellular protein content by measuring the dye uptake. Comparing the IC20 values either after 48 h or IC20 and IC50 after 72 h of incubation, it is obvious that Neutral red, Kenacid blue and MTT assays show the similar rate of toxicity (Table 1). Results from this table show the different degree of atrazine toxicity, if Trypan blue exclusion assay is applied IC50 value after 72 h is two times lower. It means that atrazine toxicity detected by this method is two times higher in comparison with other applied colorimetric assays. The results for IC50 values after 72 h from 735–760 μM (Table 1) are comparable with previously published results concerning the cytotoxicity of technical grade atrazine in the same mammalian cells (Rayburn et al. 2005). Also, the comparison of different assays as well as IC50 values after 72 h (Fig. 2 and Table 1) shows that the sensitivity of trypan blue dye by measuring the cell membrane damage in the presence of atrazine is the highest in CHO-K1 cells.
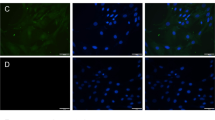
In addition, in order to determine possible morphological changes in cells after atrazine exposure the fluorescent microscopy was applied. As FDA is able to cross the plasma membrane, the cell will present a green fluorescence if the plasma membrane remains intact. In opposite, PI is hydrophilic and only cells with a compromised plasma membrane are able to stain red. PI binds to DNA by intercalating between the bases with little or no sequence preference, so PI stain can also be used to detect chromatin condensation and fragmentation typical for apoptotic cell morphology. Figure 3A–D presents the obtained morphological changes in CHO-K1 cells after 72 h exposure to atrazine. Exposure either to 20, 40 or 60 μg of atrazine/mL evoked the damage and the necrotic cell were detected, and as the concentration is increased the number of necrotic cells is higher. The level of apoptosis in atrazine treated cells was low, and the presence of apoptotic cell is visible at atrazine concentration of 40 μg/mL (Fig. 3C). Also, experiments (Manske et al. 2004) to measure apoptosis in human fibroblasts exposed to low levels of atrazine did not show DNA fragmentation or increased caspase activity.

Morphological analysis of apoptosis/necrosis induced by atrazine in CHO-K1 cells. Cells were seeded in D-MEM/MIX F12 supplemented with 5% NCS at the initial concentration of 2 × 104 cells/mL. Seventy-two hours later cells were stained by fluorescent dyes FDA-PI, then observed using a fluorescence microscope at 488 nm filters. Control (A: mag. 400×), and after treatment with atrazine 20 μg/mL (B: mag. 400×), 40 μg/mL (C: mag. 1,000×) and 60 μg/mL (D: mag. 1,000×). vc, Viable cell; nc, necrotic cell; ac, apoptotic cell
In conclusion, four bioassays applied were found to be the reproducible techniques for atrazine toxicity assessment in ovarian cells. The general cytotoxicity by trypan blue dye, and the effects on specific organelles by Neutral red, Kenacid blue and MTT assays were determined. More than one assay should be always used for determination of cell proliferation in in vitro studies, in order to increase the reliability of the obtained results. The presence of apoptotic cells in atrazine treated cells was found at the low level, so the further studies should be conducted to examine different components of the cell to provide evidence at the cellular level for the changes in cell proliferation.
References
Cooper RL, Stoker TE, Tyrey L, Goldman JM, McElroy WK (2000) Atrazine disrupts the hypothalamic control of pituitary-ovarian function. Toxicol Sci 53:297–307
Crisp TM, Clegg ED, Cooper RL, Wood WP, Anderson DG, Baetcke KP (1998) Environmental endocrine disruption: an effects assessment and analysis. Environ Health Perspect 106:11–56
Freshney RI (2005) Culture of animal cells: a manual of basic techniques, 5th edn. John Wiley & Sons Inc., Hoboken, NJ
Fujimoto N, Honda H (2003) Effects of environmental estrogenic compounds on growth of a transplanted estrogen responsive pituitary tumor cell line in rats. Food Chem Toxicol 41:1711–1717
Gianessi LP, Marcelli MB (2000) Pesticide use in U.S. crop production: 1997, National Summary report, USDA, Washington DC
Kniewald J, Mildner P, Kniewald Z (1979) Effects of s-trazine herbicides on hormone-receptor complex formation, 5alpha-reductase and 3alpha-hydroxysteroid dehydrogenase activity at the anterior pituitary level. J Steroid Biochem 11:833–838
Kniewald J, Kmetič I, Gaurina Srček V, Kniewald Z (2005) Alternative models for toxicity testing of xenobiotics. Arh Hig Rada Toksikol 56:195–204
Manske MK, Beltz LA, Dhanwada KR (2004) Low-level atrazine exposure decreases cell proliferation in human fibroblasts. Arch Environ Contam Toxicol 46:438–444
National Research Council (1999) Committee on hormonally active agents in the environment. Hormonally active agents in the environment. National Academy Press, Washington
OECD (1993) Guidelines for testing of chemicals, guideline 422: combined repeated dose toxicity study with the reproduction/developmental toxicity screening test. OECD, Paris
O’Hare S, Atterwill CK (eds) (1995) In vitro toxicity testing protocols, methods in molecular biology, vol 43. Humana Press, Totowa, NJ
Peruzović M, Kniewald J, Čapkun V, Milković K (1995) Effect of atrazine ingested prior to mating on rat females and their offsprings. Acta Physiol Hung 83:79–89
Rayburn AL, Moody DD, Freeman JL (2005) Cytotoxicity of technical grade versus formulations of atrazine and acetochlor using mammalian cells. Bull Environ Contam Toxicol 75:691–698
Šimić B, Kniewald J, Kniewald Z (1994) Effects of atrazine on reproductive performance in the rat. J Appl Toxicol 14:401–404
Stoker TE, Robinette CL, Cooper RL (1999) Maternal exposure to atrazine during lactation suppresses suckling-induced prolactin release and results in prostatitis in the adult offspring. Toxicol Sci 52:68–79
Stoker TE, Laws SC, Guidici DL, Cooper RL (2000) The effect of atrazine on puberty in male Wistar rats: an evaluation in the protocol for the assessment of pubertal development and thyroid function. Toxicol Sci 58:50–59
Težak Ž, Šimić B, Kniewald J (1992) Effect of pesticides on oestradiol-receptor complex formation in rat uterus cytosol. Food Chem Toxic 30:879–855
Acknowledgments
This study was supported by Ministry of Science, Education and Sports of the Republic of Croatia, grants no. 0582184-2232 and no. 0582184-2414.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kmetič, I., Gaurina Srček, V., Slivac, I. et al. Atrazine Exposure Decreases Cell Proliferation in Chinese Hamster Ovary (CHO-K1) Cell Line. Bull Environ Contam Toxicol 81, 205–209 (2008). https://doi.org/10.1007/s00128-008-9425-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00128-008-9425-6