
Abstract
The Ming deposit is an early Ordovician, bimodal-mafic Cu–Au volcanogenic massive sulphide (VMS) deposit in the Newfoundland Appalachians that was metamorphosed to upper greenschist/lower amphibolite facies conditions and deformed in the Silurian and Devonian. The Ming deposit consists of several spatially proximal ore bodies of which the 1806 Zone, 1807 Zone, Ming South Up Plunge and Down Plunge and the Lower Footwall Zone are the focus of this paper. The ore bodies have similar stratigraphic sequences. The ore bodies can be divided into (1) a silicified horizon that caps the massive sulphides, (2) semi-massive to massive sulphides and (3) sulphide mineralization in a rhyodacitic footwall. Sulphide mineralization in a rhyodacitic footwall includes (a) sulphide stringers immediately below the semi-massive to massive sulphides and (b) chalcopyrite–pyrrhotite–pyrite stringers distally from semi-massive to massive sulphides in the Lower Footwall Zone. Pyrite, chalcopyrite, pyrrhotite, arsenopyrite and galena were analysed by in situ secondary ion mass spectrometry (SIMS) for sulphur isotope compositions. The isotopic signatures of pyrite, chalcopyrite, pyrrhotite and arsenopyrite fall within a limited range of 2.8 to 12.0 ‰ for semi-massive to massive sulphides and sulphide mineralization in the footwall. The silicified horizon capping the semi-massive to massive sulphides has higher δ 34S (5.8–19.6 ‰), especially for pyrrhotite (mean, 17.2 ± 2.2 ‰, n = 8). The sulphur isotope composition of galena is more heterogeneous, especially within semi-massive to massive sulphides and sulphide stringers, ranging from 0.8 to 17.3 ‰ (mean, 6.1 ± 4.3 ‰, n = 35) and 7.6 to 17.1 ‰ (mean, 13.7 ± 5.3 ‰, n = 3), respectively. Geothermometric calculations give insufficient formation and metamorphism temperatures for neighbouring mineral pairs, because sulphides were not in isotopic equilibrium while deposited in early Ordovician or re-equilibrated during Silurian–Devonian metamorphism, respectively. Therefore, original isotopic compositions of sulphides at the Ming deposit have been preserved. Modelling of the source of sulphur shows that: (1) reduced seawater sulphate and (2) sulphur leached from igneous wall rock and/or derived from magmatic fluids are the main sources of sulphur in the Ming deposit. The influence of igneous sulphur (igneous wall rock/magmatic fluids) increases with temperature and is an important sulphur source for the semi-massive to massive sulphides and footwall mineralization, in addition to a contribution from thermochemical sulphate reduction (TSR) of seawater. It is difficult to distinguish between sulphur leached from igneous rocks and magmatic fluid-related sulphur, and it is possible that both sources contributed to the ores at the Ming deposit. In addition to igneous sulphur, the heavy isotopes of the silicified horizon are consistent with the sulphur in this horizon being derived only from thermochemical sulphate reduction of early Ordovician seawater sulphate.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Volcanogenic massive sulphide (VMS) deposits form on or just below the ocean floor by the complex interaction of hydrothermal fluids with the wall rock resulting in the deposition of massive sulphides containing base and precious metals (Lydon 1984; Franklin 1993, 1996; Galley et al. 2007). The hydrothermal fluids transporting metals and sulphur are predominantly modified seawater (Bischoff and Rosenbauer 1983; Lydon 1984; Franklin 1996; Galley et al. 2007). However, a magmatic fluid contribution may be present in some deposits, resulting in the formation of Au-rich VMS deposits (Sillitoe et al. 1996; Hannington et al. 1999; Huston 2000; Dubé et al. 2007; Mercier-Langevin et al. 2011). During the interaction of circulating hydrothermal ore fluids with wall rock, the physico-chemical conditions (pH, T, ƒO2, ƒS2) of the hydrothermal ore fluids change during the evolution of the hydrothermal system and result in metal zoning and complex mineral assemblages observed in VMS deposits (Large 1992; Lydon 1988; Franklin 1996; Ohmoto 1996).
The source of sulphur in these metal-transporting hydrothermal fluids is variable due to the complex genesis of VMS systems: numerous studies in modern and ancient VMS deposits illustrate that the deposits have one or multiple sulphur sources (Woodruff and Shanks 1988; Goodfellow and Peter 1996; Gemmell et al. 2004; Bradshaw et al. 2008; Alt and Shanks 2011). Possible sources of sulphur reported from modern hydrothermal vents, young seafloor systems and ancient VMS systems include: (1) thermochemical reduction of seawater sulphate (TSR); (2) sulphur from igneous, metamorphic and/or sedimentary wall rock; (3) magmatic fluids that have an isotopic composition towards lower δ 34S due to SO2 disproportionation; and (4) bacterial sulphate reduction of seawater sulphate (BSR; Shanks et al. 1995; Ohmoto and Goldhaber 1997; Huston 1999; Shanks III 2001; Seal II 2006; Hoefs 2009). Despite numerous sulphur isotope studies on ancient VMS deposits, there are relatively few studies on ancient VMS deposits that are significantly metamorphosed to facies equal to or higher than greenschist (Bachinski 1977, 1978; Cook and Hoefs 1997; Bailie et al. 2010) and/or deposits enriched in precious metals (Zaw and Large 1992). Additionally, very few studies have focused on in situ analyses of VMS sulphides that are constrained by paragenesis (Bradshaw et al. 2008).
In this paper, we report in situ secondary ion mass spectrometry (SIMS) sulphur isotope analyses of pyrite, chalcopyrite, pyrrhotite, arsenopyrite and galena from the moderately metamorphosed and precious metal-enriched Cu–Au VMS Ming deposit, Newfoundland Appalachians (Brueckner et al. 2014). This deposit contains five different, actively mined ore bodies with different styles of sulphide mineralization. These different mineralization styles are from top to bottom: (1) silicified horizon that caps massive sulphides; (2) semi-massive to massive sulphides; and (3) discordant ore mineralization in the footwall consisting of (a) the sulphide stringers immediately below massive sulphide; and (b) the Lower Footwall Zone that represents the high temperature, chlorite-rich stockwork zone that occurs distally from the massive sulphides. The mineralization styles differ in mineralogy and mineral assemblages from each other and show variations in mineral textures and compositions. Therefore, the Ming deposit provides an excellent opportunity to study the sulphur isotope variations in an ancient, metamorphosed, precious metal-bearing VMS deposit. Modelling of the sulphur isotope compositions is used to constrain the sources of sulphur in the deposit. The results provide critical information on sulphur sources in Appalachian VMS deposits but have implications for both base metal and precious metal-rich VMS deposits globally.
Geologic background
Baie Verte peninsula
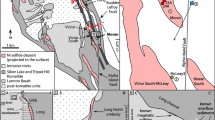
The Baie Verte Peninsula in northwest Newfoundland is part of the Canadian Appalachians and hosts both metamorphosed Precambrian rocks of Laurentia (Humber Zone) and peri-Laurentian Paleozoic rocks of the Notre Dame sub-zone of the Dunnage Zone (Fig. 1). Cambro-Ordovician ophiolitic rocks forming the basement of the Dunnage Zone are crosscut by felsic intrusions and overlain by volcanic sequences. The ophiolitic basement rocks and their cover sequences formed in peri-Laurentian supra-subduction zones due to the closure of the Humber Seaway and were emplaced onto Laurentia during the Taconic Orogeny (Swinden and Thorpe 1984; van Staal 2007; van Staal and Barr 2012). During the Salinic and Acadian orogenies in the Silurian and early Devonian, respectively, the ophiolitic basement rocks and their cover sequences were deformed and metamorphosed to upper greenschist/lower amphibolite facies. Moreover, felsic plutons intruded into the basement (Tuach and Kennedy 1978; Hibbard 1983; Castonguay et al. 2009).

Location of the study area within Newfoundland, Canada (left) and the Baie Verte Peninsula, Newfoundland (right). The geologic zones of Newfoundland are after Williams (1979) with the location of the Baie Verte Peninsula (black). The simplified geology of the Baie Verte Peninsula is after Hibbard (1983). 1, Age data for the Pacquet Harbour Group (PHG) from Castonguay et al. (2009)
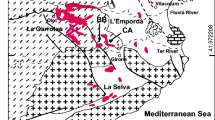
The Pacquet Harbour Group (PHG), Baie Verte Peninsula, is a remnant ophiolitic basement assemblage that hosts the VMS deposits in the Rambler camp (Fig. 2). The PHG is divided into a lower and upper unit. The lower PHG is early Ordovician and consists of dominantly low-Ti boninites and basalts with minor rhyodacite to rhyolite (Rambler rhyolite). The Rambler rhyolite has a U–Pb zircon age of ca. 487 Ma (Castonguay et al. 2009) and is the host to VMS mineralization in the camp. The upper PHG is the cover sequence of the lower PHG, has mafic to felsic, mid-Ordovician volcano-sedimentary rocks and mafic volcanic rocks with an age of 483 to 470 Ma (Hibbard 1983; Castonguay et al. 2009; Skulski et al. 2010). VMS deposition within the lower PHG was coeval with the formation of the PHG rocks in a rifted arc/back-arc setting (Piercey et al. 1997; van Staal 2007; Skulski et al. 2010). The timing of precious metal enrichment in the VMS deposit was argued to be either of syngenetic or of metamorphic origin (Hibbard 1983; Tuach and Kennedy 1978). However, recent research on the Ming deposit has shown that gold and silver are of syngenetic origin and likely of magmatic-hydrothermal origin (Brueckner et al. 2014).

Detailed geologic map of the Pacquet Harbour Group (PHG) hosting the Ming (Main) mine and previously mined volcanogenic massive sulphide deposits (modified after Castonguay et al. 2009). Ore bodies of the Ming deposit are highlighted in black. Coordinates are in UTM 83NAD zone 21 (inside) and WGS 84 (outside)
Consolidated Rambler VMS camp
The consolidated Rambler VMS camp within the lower PHG consists of five VMS deposits (Ming, Ming West, Rambler Main, East Rambler, Big Rambler Pond; Fig. 2). The Ming deposit is the only active producer, although previous production of Cu ± Au occurred at the other deposits (Tuach and Kennedy 1978; Pilgrim 2009). The Ming, Ming West, Rambler Main and East Rambler deposits are stratabound, bimodal-mafic type, Cu(−Au)-bearing VMS deposits hosted in the Rambler rhyolite and dominated by pyrite–chalcopyrite assemblages with lesser pyrrhotite and sphalerite and with variable precious metal enrichment (Tuach and Kennedy 1978; Pilgrim 2009; Brueckner et al. 2014). In contrast, the Big Rambler Pond deposit is a small mafic Cu VMS deposit located south of the Rambler Brook fault within the boninitic Betts Head Formation of the lower PHG and occurs stratigraphically below the Rambler rhyolite. The deposit shows disseminated and stringer sulphide mineralization of pyrrhotite, chalcopyrite and pyrite (Tuach and Kennedy 1978).
Ming mine
The Ming deposit is a bimodal-mafic-type Cu–Au VMS deposit and consists of several, lenticular ore bodies: 1806 Zone, 1807 Zone, Ming South Up Plunge, Ming South Down Plunge, Ming North, Upper Footwall Zone and Lower Footwall Zone (Fig. 3). The 1806 and 1807 zones, Ming South and the Upper and Lower Footwall zones run parallel to an old, previously mined portion of the deposit (Ming Main), trending NE with a plunge of 30–35°. The Ming North ore body is the extension of the previously mined Ming Main ore body (Fig. 3). All ore bodies are hosted within the Rambler rhyolite, a ca. 2.5 km wide sequence of quartz-phyric rhyodacite, felsic tuff, and tuff breccia of the lower PHG (Skulski et al. 2010). Total (measured, indicated and inferred) resources for the Ming deposit are 21.9 Mt with grades of 1.49 wt.% Cu, 0.61 g/t Au, 3.21 g/t Ag and 0.19 wt.% Zn.

Simplified 3-D view of the different ore bodies of the Ming deposit. Of the different ore bodies, only the 1806 Zone, 1807 Zone, Ming South (Up and Down Plunge) and the Lower Footwall Zone are currently mined (in bold) and are reported in this paper
The different ore bodies have similar stratigraphic sequences that are crosscut by mafic dikes. Styles of mineralization (Fig. 4; Table 1) are similar in the 1806 Zone, 1807 Zone and Ming South with concordant to stratabound semi-massive to massive sulphide lenses on the footwall-hanging wall contact and narrow, discordant sulphide stringers immediately below the semi-massive and massive sulphide lenses in the rhyodacitic footwall. Specific for the 1806 Zone is a thin, strongly silicified horizon with discordant sulphide stringers on the footwall-hanging wall contact directly above semi-massive and massive sulphides. The Lower Footwall Zone that occurs beneath Ming South (Fig. 3) lacks both a silicified horizon and semi-massive to massive sulphides and is instead entirely made of thin, discordant sulphide stringers. In contrast to the sulphide stringers occurring in the 1806, 1807, and Ming South ore bodies, alteration of the rhyodacitic footwall as well as sulphide stringer composition is different in the Lower Footwall Zone. Sulphide stringer composition is chalcopyrite–pyrrhotite ± pyrite ± cubanite with trace magnetite, sphalerite and Bi-tellurides. Sulphide stringers make up the entire Lower Footwall Zone, therefore the term Lower Footwall Zone is used in this paper synonymously with ore body (accumulation of low- to high-grade ore in a well defined shape; Fig. 3) and as mineralization style (sulphide mineralization that is similar in ore grade, sulphide abundance, alteration and/or host rock lithology; Fig. 4).

Simplified stratigraphy of the Ming deposit with photographs of the different mineralization styles. a Stratigraphic section with different mineralization styless at the Ming deposit. Horizontal line on top implies grain size of lithologies after Fisher (1961). Maximum thickness of mineralization styles is given in brackets. Sulphide mineralization in semi-massive to massive sulphides and sulphide stringer horizon occur in 1806, 1807 and Ming South ore bodies. The silicified horizon is restricted to the 1806 Zone. Sulphide stringers of the Lower Footwall Zone differ from stringers of the sulphide stringer horizon regarding location, alteration of footwall and sulphide mineralogy. However, sulphide stringers of both the sulphide stringer horizon and the Lower Footwall Zone are part of the footwall mineralization. b Silicified horizon: strongly silicified rock or amorphous quartz with discordant pyrite stringers and disseminated chalcopyrite. c Silicified horizon: strongly silicified rock or amorphous quartz with brecciating pyrite stringer. d Semi-massive sulphides: remnants of altered Rambler rhyolite with homogeneous pyrite-chalcopyrite sulphides containing thin schlieren of sphalerite with galena and tennantite-tetrahedrite. e Massive sulphides: homogeneous pyrite-chalcopyrite with thin pyrrhotite–sphalerite schlieren. f Sulphide stringer horizon: quartz eye-bearing Rambler rhyolite with sericitic alteration (sericite–quartz) and thick, discordant pyrite–sphalerite stringer. g Sulphide stringer horizon: Rambler rhyolite with sericitic alteration (sericite-quartz) with abundant green mica; thin sulphide stringers of pyrite–sphalerite run parallel to foliation. h Lower Footwall Zone: Rambler rhyolite with strong chloritic alteration (chlorite–minor quartz), discordant chalcopyrite–pyrrhotite stringer, and disseminated chalcopyrite. i Lower Footwall Zone: Rambler rhyolite with strong chloritic alteration (chlorite–minor quartz), secondary amphibolite and discordant chalcopyrite–pyrrhotite. Mineral abbreviations: Amp amphibole, Ccp chalcopyrite, Gn galena, Po pyrrhotite, Py pyrite, Sp sphalerite, Tnt tennantite, Ttr tetrahedrite
The silicified horizon, only present in the 1806 Zone, has discordant pyrite–chalcopyrite ± pyrrhotite stringers with minor electrum (Figs. 4b, c and 5a). In all ore bodies with the exception of the Lower Footwall Zone, semi-massive to massive sulphides commonly occur in two spatially proximal horizons, each up to 10 m thick, separated by Rambler rhyolite (Fig. 4a, d, e). Mineral assemblages vary between the ore bodies with pyrite and chalcopyrite as the dominant sulphide phases (Fig. 5b–f; Table 1). Within the rhyodacitic footwall, sulphide mineralization occurs as discordant sulphide stringers. However, footwall mineralization is divided depending on the alteration mineralogy, sulphide content, and location (Table 1; Fig. 4a). Immediately below the massive and semi-massive sulphides, the rhyodacitic footwall contains up to 30 % discordant sulphide stringers and this area is in this paper referred to as sulphide stringer horizon (Fig. 4a). This horizon is up to 80 m thick. Alteration of the footwall in the sulphide stringer horizon is predominantly quartz–sericite ± green mica–sulphide with minor chlorite. The sulphide veinlets show a wide range of sulphide assemblages, but are predominantly of pyrite–chalcopyrite ± sphalerite with minor pyrrhotite–arsenopyrite and traces of tennantite–tetrahedrite–galena–Ag phases (Figs. 4f, g and 5g, h; Table 1). The second type of footwall mineralization is restricted to the Lower Footwall Zone occurring below the Ming South ore bodies (Fig. 3). The Lower Footwall Zone hosts <20 % sulphides and is up to several hundred metres thick (Fig. 4a, h, i). Here, discordant stringers are predominantly chalcopyrite–pyrrhotite–pyrite (Fig. 5i) often with minor cubanite (Fig. 5j), and the alteration assemblage is characterised by intense chlorite–quartz with minor magnetite–sulphide (Fig. 4h, i; Table 1). The Lower Footwall Zone is very similar to high temperature chlorite alteration in footwall stockwork VMS deposits.


Typical sulphide mineral assemblages and textures from the different mineralization styles of the Ming deposit. a Silicified horizon: pyrrhotite with electrum and marginal galena adjacent to sphalerite or chalcopyrite. b Semi-massive sulphides: recrystallized pyrite with interstitial galena in chalcopyrite; pyrrhotite, arsenopyrite and sphalerite with accessory galena in chalcopyrite. c Semi-massive sulphides: pyrrhotite with inclusions of arsenopyrite and galena; accessory galena-sphalerite in chalcopyrite. d Massive sulphides: coarse-grained, recrystallized pyrite in chalcopyrite with interstitial galena and surrounded by sphalerite or pyrrhotite; sphalerite with weak chalcopyrite disease and inclusions of arsenopyrite and galena with Bi-telluride lamellae; pyrrhotite surrounding recrystallized pyrite has inclusions of galena with Bi-telluride lamellae. e Massive sulphides: coarse-grained, recrystallized pyrite in chalcopyrite and surrounded by pyrrhotite has inclusions of galena and fine fractures that are partly filled with chalcopyrite or pyrrhotite; pyrrhotite surrounding recrystallized pyrite has small inclusion of electrum and marginal sphalerite. f Massive sulphides: recrystallized pyrite with marginal galena in contact to gangue; disseminated chalcopyrite with pyrrhotite–galena in gangue. g Sulphide sringer horizon: recrystallized pyrite partly replaced by pyrrhotite; chalcopyrite with recrystallized pyrite–pyrrhotite–sphalerite. h Sulphide stringer horizon: sphalerite–galena schlieren with recrystallized pyrite. i Lower Footwall Zone: recrystallized pyrite at chalcopyrite margins, partly replaced by pyrrhotite; sphalerite at pyrrhotite margins. j Lower Footwall Zone: chalcopyrite with cubanite exsolution lamellae, pyrite and pyrrhotite. Red circles with numbers show SIMS point analysis and δ 34S value, respectively. Mineral abbreviations: Apy arsenopyrite, Cbn cubanite, Ccp chalcopyrite, El electrum, Gn galena, Gn* galena with Bi-telluride exsolution lamellae, Po pyrrhotite, Py pyrite, Sp sphalerite
Sulphide mineralogy
The sulphide mineralogy of the Ming deposit is complex and includes over 17 sulphide and precious metal phases (Brueckner et al. 2014). Pyrite and chalcopyrite are the major sulphide minerals, whereas sphalerite, pyrrhotite and galena are common minor components. Additionally, a variety of trace minerals such as arsenopyrite, various sulfosalts (tennantite–tetrahedrite, boulangerite, stannite, loellingite), tellurides (tsumoite, hessite, altaite) and precious metal phases (electrum, miargyrite, pyrargyrite, stephanite) are present at the Ming deposit (Table 1).
The semi-massive to massive sulphide horizons of the various ore bodies show a complex ore assemblage that varies between the different ore bodies (Table 1). In particular, the abundance of trace sulfosalts, tellurides and precious metals varies. The 1806 Zone is the richest in gold amongst all ore bodies with 2.83 g/t Au. Gold occurs predominantly as electrum and to a much lesser extent in AgHgAu alloys. Silver occurs predominantly in Ag-enriched tetrahedrite, alloys of AgHg ± Au, AgSbS phases and electrum (Brueckner et al. 2014). In the 1806 Zone, electrum occurs typically with chalcopyrite, pyrite, arsenopyrite, sulfosalts and Ag phases. The 1807 and Ming South ore bodies have higher amounts of tellurides that commonly occur texturally proximal to galena; tennantite-tetrahedrite is rare and Ag occurs predominantly as hessite (AgTe). Electrum is also less abundant in these ore bodies compared with the 1806 Zone, where electrum is associated with chalcopyrite, pyrite, arsenopyrite and tellurides.
Sulphide assemblages in the silicified horizon, the sulphide stringer horizon and the Lower Footwall Zone are less complex and dominated by pyrite and chalcopyrite with minor pyrrhotite (Fig. 4g–j). Abundances of sphalerite, galena, arsenopyrite, cubanite, sulfosalts, tellurides and precious metals are highly variable (Brueckner et al., unpublished data). Precious metal assemblages are typically associated with chalcopyrite, arsenopyrite, and/or tellurides, but are rare compared with the semi-massive to massive sulphide lenses.
The sulphides are texturally homogenous and no distinct differences are observed between the different mineralization styles or the ore bodies. Common sulphide mineral textures at the Ming deposit include replacement textures (chalcopyrite disease in sphalerite; pyrrhotite replacing pyrite; Fig. 4h, i) and metamorphic/deformation textures (recrystallization of pyrite, porphyroblasts of pyrite and arsenopyrite; cataclastic texture pyrite; Fig. 4; Table 2).
Sulphur isotope geochemistry
Methodology
Thirty-nine samples from the five ore bodies (1806 Zone, 1807 Zone, Ming South Up Plunge and Down Plunge, Lower Footwall Zone) and representing all mineralization styles (silicified horizon, semi-massive to massive sulphides, footwall mineralization) were selected for sulphur isotope analysis. The most samples come from the 1806 Zone (n = 15), because the sulphide mineralogy is the most complex of all ore bodies. Seven samples each were analysed from the 1807 Zone and Ming South Up Plunge. Four samples from Ming South Down Plunge and six samples from the Lower Footwall Zone were used for sulphur isotope analysis. Amongst the mineralization styles, the semi-massive to massive sulphides had the highest number of samples (n = 23) due to the wide variations in ore mineral assemblages between the ore bodies. Two to three grains of each phase were usually analysed for δ 34S in each sample to observe possible variations in the isotope composition. The grains were chosen depending on mineral assemblage, size and texture. In total, 338 δ 34S spot analyses were obtained from the 39 samples on five different minerals (pyrite, n = 102; chalcopyrite, n = 98; pyrrhotite, n = 52; arsenopyrite, n = 47; and galena, n = 39). Besides samples from the different mineralization styles, one Fe oxide sample on the footwall-hanging wall contact and a sample from a mineralized quartz vein within a mafic dyke were also analysed to investigate possible differences in sulphur isotope composition of pyrite, chalcopyrite and pyrrhotite in these different host lithologies.
Samples were mounted in polished epoxy sections and coated with 300 Å of Au, to mitigate charging under primary ion bombardment. All analyses were performed using the Cameca IMS 4f Secondary Ion Mass Spectrometer (SIMS) at the MAF-IIC Microanalysis Facility of Memorial University, and the described methodology is modified after Toman (2012) and found in Online resource 1. For each sample, determinations of δ 34S were performed on multiple grains of pyrite, chalcopyrite, pyrrhotite, arsenopyrite and galena by bombarding the sample with a primary ion microbeam of 350–1150 pA of Cs+, accelerated by a 10-keV potential and focused into a 5–15 μm diameter spot. The Cs+ current depended on the analysed sulphide phase, the size of the analysed phase and the matrix surrounding the analysed grain (sulphides or non-sulphide gangue). To prevent contamination in the polished surface, each spot was first pre-sputtered for 120 s with a 25 μm square raster. Depending on the minimum diameter of the critically focused analysis, to improve the homogeneity of primary ion delivery, while maintaining lateral resolution at better than 20 μm. Negatively charged sputtered secondary ions were accelerated into the mass spectrometer using a potential of 4.5 keV.
The instrument was operated with a medium Contrast Aperture (150 μm), and Entrance and Exit Slits paired to give flat-topped peaks at a mass-resolving power (MRP) of 2975 (10 % peak height definition)—sufficient to discriminate 33SH− (and 32SH2 −) from 34S−.
Overall reproducibility, based on replicate standard analyses, is typically better than ±0.5 ‰ (1σ). Results for the used sulphide in-house standards, their measured 34S/32S ratio and the calculated instrument mass fractionation (IMF) factors are summarised in Online resources 2 and 3.
Results
Results for all five analysed mineral phases are shown in Figs. 6 and 7 and are given in Online resource 4. For the deposit, most of the data range between +2 and +13 ‰ with a few exceptions (Fig. 6). Galena has the widest range amongst all analysed sulphide phases (δ 34S = +0.8 to +17.3 ‰).

Histogram of δ 34S values of sulphides from the different mineralization styles, a sample of Fe oxide schlieren in Rambler rhyolite on top of massive sulphide and a sample of mineralized quartz vein within a mafic dyke of the Lower Footwall Zone of the Ming deposit; n number of analysed data points

Box–Whisker plot showing the variations in δ 34S values of sulphides from the different mineralization styles of the Ming deposit. The horizontal error bars represent the total range of all spot analyses for each ore zone during the multiple analytical sessions. The horizontal dimension of the box represents the 1σ for all spot analyses during multiple analyses; n number of analysed data points
Silicified horizon of the 1806 Zone
Sulphides in the silicified horizon from the 1806 ore body show the widest range of sulphur isotope values amongst all mineralized zones. In general, δ 34S ranges between 5.8 and 19.6 ‰ (Fig. 6). Pyrrhotite displays the highest values of δ 34S amongst all measured sulphides, ranging between 13.9 and 19.6 ‰ with a mean of 17.2 ± 2.2 ‰ (n = 5). The sulphur isotope signature for pyrite ranges between 5.8 and 17.6 ‰ with a mean of 11.2 ± 4.4 ‰ (n = 8). Chalcopyrite shows similar δ 34S values to pyrite, ranging between 9.6 and 16.2 ‰ with a mean of 13.1 ± 2.9 ‰ (n = 9). Sulphur isotope data for arsenopyrite have a limited variation between 6.9 and 11.5 ‰ and a mean of 9.3 ± 1.7 ‰ (n = 5). The occurrence of galena within the silicified horizon is very limited with only one data point with δ 34S of 9.4 ‰.
Semi-massive to massive sulphides
Most data (n = 232) were obtained from this mineralized horizon. The sulphur isotope composition of semi-massive to massive sulphides is homogenous between the different ore bodies despite variations in the sulphide mineral assemblages (Table 1). The majority of data clusters between 2 and 11 ‰, and the mean δ 34S value for each sulphide phase is relatively lower than those in the silicified horizon and in the footwall (Figs. 6 and 7). The sulphur isotope signature is uniform for pyrite (mean, 7.4 ± 1.5 ‰, n = 69), chalcopyrite (mean, 7.5 ± 1.5 ‰, n = 59), pyrrhotite (mean, 6.3 ± 1.6 ‰, n = 30) and arsenopyrite (mean, 7.2 ± 1.4 ‰, n = 39; Figs. 5b–f, 6 and 7). In contrast, galena has heterogeneous δ 34S values ranging between 0.8 and 17.3 ‰ and a mean of 6.1 ± 4.5 ‰ (n = 35). These variations in the sulphur isotopic composition may be related to paragenetic occurrence, as galena has been deposited in different stages during syngenetic metal deposition (Brueckner et al. 2014). The analysed galena grains are from inclusions in pyrite, in-between groups of annealed pyrite and on margins of pyrite, arsenopyrite or sphalerite. However, there is no relationship between textural occurrence and sulphur isotope composition (Fig. 5b, c, and f).
Footwall mineralization
Forty-two sulphur isotope compositions of pyrite (n = 13), chalcopyrite (n = 16) and pyrrhotite (n = 7) from the sulphide stringer horizon and the Lower Footwall Zone are almost identical to each other and, in general, have slightly higher values than data from the semi-massive to massive sulphides (Figs. 6 and 7). All but two data points from the sulphide stringers range between 6 and 13 ‰. The mean δ 34S value for chalcopyrite is 9.1 ± 1.4 ‰ with a range of 6.4 to 11.2 ‰. Pyrite has a mean δ 34S value of 10.3 ± 1.0 ‰ with a range of 8.4 to 12.0 ‰, and pyrrhotite has a mean δ 34S value of 9.7 ± 0.5 ‰ with a range between 8.8 and 10.4 ‰. Arsenopyrite data were only obtained from one sample collected from the 1806 ore body. The three arsenopyrite values are homogeneous with a mean of 7.5 ± 0.3 ‰, and isotopically these values are lower than those of chalcopyrite, pyrite and pyrrhotite. Galena is a trace phase within the stringer sulphides, and data are from three points in two samples from the 1806 Zone and the Ming South Down Plunge ore body. Similar to the semi-massive and massive sulphides, δ 34S values are heterogeneous (7.6, 16.2 and 17.1 ‰). In contrast to the semi-massive and massive sulphides, there is a relationship between texture and δ 34S. The isotopically heavier signatures are from galena grains associated with coarse-grained sphalerite (Fig. 5h), whereas the low δ 34S value is from a single galena grain within chalcopyrite.
The δ 34S values from the Lower Footwall Zone were obtained only from pyrite, chalcopyrite, and pyrrhotite. These δ 34S values are similar to S isotope signatures in the sulphide stringer horizon. The mean δ 34S values for pyrite, chalcopyrite and pyrrhotite are 9.8 ± 0.6 ‰ (n = 7), 8.4 ± 1.0 ‰ (n = 12) and 8.3 ± 1.1 ‰ (n = 9), respectively (Fig. 5i, j).
In addition to the different sulphide mineralization styles, the sulphur isotope signature of sulphides in a Fe oxide-bearing sample and a mineralized quartz vein were measured (Fig. 6; Online resource 4). The Fe oxide-bearing sample (62133) is from the Ming South Up Plunge ore body within the Rambler rhyolite with magnetite schlieren and traces of pyrite, pyrrhotite, chalcopyrite and sphalerite. Pyrite analyses yielded three values ranging between 5.9 and 8.3 ‰. One pyrrhotite grain yielded a slightly lower value than pyrite with δ 34S of 4.6 ‰. The quartz vein (36113) is within a mafic dyke in the Lower Footwall Zone and contains chalcopyrite-pyrite mineralization with traces of sphalerite. The mean δ 34S values from pyrite and chalcopyrite are similar with 6.3 ± 0.4 ‰ (pyrite, n = 2) and 5.7 ± 0.1 ‰ (chalcopyrite, n = 2). The sulphur isotope data obtained from the Fe oxide-bearing sample and the quartz vein are similar to pyrite, chalcopyrite and pyrrhotite data from the semi-massive to massive sulphides (Fig. 6; Online resource 4).
Discussion
Isotopic equilibrium
Two hundred and three mineral pairs of pyrite–chalcopyrite, pyrite–pyrrhotite, pyrrhotite–chalcopyrite, pyrite–galena, pyrrhotite–galena and chalcopyrite–galena were tested for sulphur isotope equilibrium. Both minerals in each pair were proximal or adjacent to one another in thin section and encased by sulphide phases. The sulphur isotope fractionation factors used are those of Kajiwara and Krouse (1971). Arsenopyrite was not included because of a lack of reliable fractionation values within the temperature range of 250–500 °C that affected the Ming deposit during metal deposition in the early Ordovician and during upper greenschist/lower amphibolite facies metamorphism in the Silurian–Devonian (Tuach and Kennedy 1978; Brueckner et al. 2014).
Neighbouring mineral pairs were plotted in δ–δ plots (Fig. 8; Online resource 5) to test if neighbouring sulphides were in isotopic equilibrium either during early Ordovician deposition or later Silurian–Devonian metamorphism. These plots are described in detail by Gregory and Criss (1986) and Gregory et al. (1989). The purpose of these plots is to identify if neighbouring mineral pairs plot along or parallel to isotherms that would indicate isotopic equilibrium and give temperature of isotopic equilibration or if mineral pairs plot in arrays oblique to isotherms that would indicate isotopic disequilibrium. Isotherms in δ–δ plots are a linear function of the δ 34S values of the two neighbouring mineral pairs with a slope of +1 and an interception with the y-axis of −Δ1–2, which is the difference in isotopic composition between mineral phases 1 and 2.

δ–δ plot for neighbouring pyrite–chalcopyrite mineral pairs. Neighbouring pyrite–chalcopyrite mineral pairs plot in arrays oblique to isotherms of 250–500 °C or above the thick black line indicating zero fractionation between pyrite and chalcopyrite (δ py-ccp = 0). Mineral pairs above this line would have reverse polarity of fractionation, which is not observed in natural systems. Isotherms were calculated for temperatures of 250–500 °C. Isotherms are a linear function (y = mx + n) of the δ 34S values of the two neighbouring mineral pairs; slope m is +1 and intersection n with the y-axis is −Δ1–2, because \( {\delta}_1-{\delta}_2={\varDelta}_{1-2}=\frac{\mathrm{A}\times {10}^6}{T^2} \) and therefore δ 2 = δ 1 − Δ 1 − 2 (Ohmoto and Goldhaber 1997); T is temperature in kelvin; δ 1 and δ 2 is the measured δ 34S value of mineral phases 1 and 2, respectively; Δ1–2 is the difference in isotopic sulphur composition between mineral phases 1 and 2 or the isotopic fractionation between the two minerals; and A is a constant (A = 0.45 for pyrite–chalcopyrite; Kajiwara and Krouse 1971)
In Fig. 8, neighbouring pyrite-chalcopyrite mineral pairs from all mineralization styles are plotted. Isotherms for temperatures of 250–500 °C are given. This temperature range covers both VMS-forming temperatures of the Ming deposit (≈250–350 °C; Brueckner et al. 2014) and upper greenschist/lower amphibolite facies conditions (≈350–500 °C) that affected the deposit in the Silurian–Devonian (Tuach and Kennedy 1978). Pyrite and chalcopyrite were broadly deposited at similar stages during the formation of the Ming deposit (Brueckner et al. 2014), and show arrays that plot oblique to the isotherms (Fig. 8). Some of the mineral pairs plot above the Δpyrite–chalcopyrite = 0 line, and these mineral pairs would have reverse polarity of fractionation, which is not observed in natural sulphur isotope systems. These features argue that neighbouring pyrite-chalcopyrite mineral pairs reflect isotopic disequilibrium. The other five mineral pairs show similar results (Online resource 5). Therefore, isotopic equilibrium for the sulphides at the Ming deposit was not attained during metal deposition in the early Ordovician or by isotopic re-equilibration during Silurian–Devonian metamorphism.
These results are in accordance with other studies on (metamorphosed) hydrothermal systems that showed: (1) that isotopic equilibrium between two, broadly coevally deposited phases is often prevented especially when the hydrothermal, metal-transporting and reduced sulphur-bearing fluids mix with other fluids (cold seawater, magmatic fluid) of different composition (different m H2S/m ∑Metals ratio, ƒO2, ƒS2) and/or temperature (Ohmoto and Goldhaber 1997); and (2) that isotopic re-equilibration during metamorphism is a function of grain size, temperature, amount of fluid and if the neighbouring minerals are in direct contact to each other or not (Crowe 1994; Ohmoto and Goldhaber 1997). For the mineral pairs at the Ming deposit, the metamorphic temperature of upper greenschist/lower amphibolite facies was likely too low to attain isotopic re-equilibration during the Silurian–Devonian, because analysed sulphides are relatively small and completely surrounded by other sulphide phases (Crowe 1994).
Sulphur sources
Sulphur isotope fractionation between mineral phases, aqueous species or organisms is common and widely observed in seafloor hydrothermal systems (Shanks et al. 1995; Ohmoto and Goldhaber 1997; Shanks III 2001; Seal II 2006). In VMS deposits and their modern seafloor equivalents, three main sources of sulphur are common: (1) seawater sulphate with δ 34S≈4–33 ‰ (Claypool et al. 1980; Kampschulte and Strauss 2004; Paytan and Gray 2012) that is present in sulphate minerals or is reduced by thermochemical processes; (2) sulphur leached from igneous footwall rocks and/or direct contributions from magmatic fluids with δ 34S≈−5 to +5 ‰ (averaging 0 ‰); and (3) sulphur from microbial activity in reduced sediments via bacterial sulphate reduction (BSR), with δ 34S≈−50 to +20 ‰ (Shanks et al. 1995; Goodfellow and Peter 1996; Ohmoto and Goldhaber 1997; Gemmell and Sharpe 1998; Canfield 2001; Shanks III 2001; Seal II 2006; Hoefs 2009).
In this section, an attempt to quantify the source(s) of sulphur at the Ming deposit by mass balance modelling is made. There are uncertainties when modelling the sulphur sources in the Ming deposit, including: (1) isotopic fractionation in a partly open vs fully open system; (2) possible evidence for bacterial activity; (3) temperature range at which seawater sulphate reduction took place; (4) range in δ 34S of seawater sulphate at time of formation of the deposit; and (5) range in sulphur isotopic composition of igneous wall rock and magmatic fluid. The uncertainties and the assumptions used in the modelling are outlined below. The modelling results are compared with reported literature data to put these results for the Ming deposit in a broader context to other VMS deposits worldwide.
Evolving hydrothermal systems can be seen as fully open, because there is: (1) a constant seawater (±magmatic fluid/volatile) input (Herzig and Hannington 1995; Seal II 2006); (2) the water/rock ratio changes proximal and distal to the evolving sulphide mound resulting in different alteration assemblages within the wall rock (Seyfried and Bischoff 1981); and (3) physico-chemical parameters (T, pH, ƒO2, ƒS2, aS2) of the hydrothermal fluids change based on the location within the system (proximal/distal to the heat source, the stock work zone or vents; Lydon 1988; Ohmoto 1996). Stratigraphy, alteration and sulphide mineral assemblage show that the criteria for a fully open system existed at the Ming deposit during its formation, because: (1) constant seawater input and the addition of magmatic volatiles/fluids to the hydrothermal fluids resulted in a complex sulphide and precious metal assemblage (Brueckner et al. 2014); (2) changes in the water/rock ratio proximal and distal to the massive sulphides due to changes in the porosity of the Rambler rhyolite resulted in different proximal and distal alteration assemblages (Fig. 4; Table 1; Pilote et al. 2014); and (3) changing physico-chemical parameters of the hydrothermal fluids due to interaction with the footwall and location relative to the hydrothermal heat source resulted in varying distributions of uncommon metal sulphides (tellurides, nisbite, breithauptite), sulfosalts (tennantite–tetrahedrite, stannite, boulangerite) and precious metals (electrum, AgHg ± Au alloy) between the different mineralization styles and ore bodies (Brueckner et al., unpublished data). This complexity of the Ming deposit suggests it is reasonable to assume that sulphur isotope fractionation took place in a fully open system rather than a partly open system.
The generally positive δ 34S values of sulphides in the Ming deposit rule out any significant role for microbial activity in the genesis of the Ming deposit. Moreover, sediments were not directly involved in the formation of the Ming deposit and the sediments of the upper PHG are younger and deposited after the formation of the Ming deposit and therefore could not have provided biogenic sulphur to the deposit.
Sulphates do not occur in the Ming deposit illustrating that seawater sulphate in sulphate mineral form was not important in the deposit; however, the role for thermochemical sulphate reduction (TSR) of seawater sulphate was likely important in the formation of the Ming deposit and has been shown to be important in most Phanerozoic VMS deposits (Shanks et al. 1981, 1995; Shanks and Seyfried 1987; Ohmoto and Goldhaber 1997; Huston 1999; Seal II 2006). To test whether the sulphur in the Ming deposit could have been formed solely from TSR of seawater sulphate, we used the equations of Ohmoto and Rye (1979) and Ohmoto and Goldhaber (1997) and the fractionation between H2S and sulphide of Kajiwara and Krouse (1971) (Online resource 6).
Minimum and maximum temperatures for TSR were assumed to be 250 and 350 °C, based on the results of Shanks et al. (1981) and the interpreted conditions of formation of the sulphide assemblages in the Ming deposit (Brueckner et al. 2014). Although Shanks et al. (1981) proposed that TSR can occur as low as 200 °C, the experimental results at 200 °C did not result in the formation of pyrite. Hence, Shanks et al. (1981) argued that TSR at lower temperatures is too slow and high water/rock ratios are required for successful TSR reactions at temperatures as low as 200 °C. These experiments and the fact that the sulphide and precious metal mineralogy at the Ming deposit indicates formation from fluids at temperatures of ≥250 °C (Brueckner et al. 2014) are the basis for the assumed minimum and maximum temperatures of 250 and 350 °C, respectively.
For TSR modelling, the composition of early Ordovician seawater sulphate was assumed to be ~29 ‰ (Claypool et al. 1980; Kampschulte and Strauss 2004), although evaporates (Claypool et al. 1980) and carbonates associated with sulphates (CAS; Kampschulte and Strauss 2004) show a wide range of sulphur isotopic composition for Ordovician seawater sulphate (15–30 ‰; Kampschulte and Strauss 2004). Nevertheless, Kampschulte and Strauss (2004) and Paytan and Gray (2012) pointed out that the mean sulphur isotopic composition of CAS at the end of the Cambrian and beginning of the Ordovician is 30 ‰, a value that decreased steadily during the Ordovician. The U–Pb zircon age of the Rambler rhyolite is 487 ± 3 Ma (Castonguay et al. 2009), which is close to, but slightly younger, than the age of the Cambro-Ordovician boundary; therefore, the δ 34S composition of seawater sulphate at the time of the formation of the Ming deposit is assumed to be slightly lower (29 ‰) than the mean of δ 34S = 30 ‰.
In Figs. 9 and 10, results of modelled TSR δ 34S values at temperatures of 250 and 350 °C are shown for pyrrhotite in the silicified horizon and chalcopyrite in the semi-massive to massive sulphides, respectively. A comparison of the modelled data with the measured values shows that TSR can account for the isotopic composition of sulphides, especially of the silicified horizon, over VMS-relevant temperatures (225–325 °C; Fig. 9). Modelled pyrrhotite results for the silificied horizon show that at the assumed minimum TSR temperature of 250 °C, up to 45 % seawater sulphate was reduced to sulphide over VMS-relevant temperatures of 225–325 °C (Fig. 9). However, with increasing temperatures, the magnitude of isotopic fractionation between sulphate and sulphide decreases (Fig. 9). Despite this weak decrease of sulphur isotopic fractionation with increasing TSR temperature, it is permissible that TSR was the dominant sulphur source for sulphides within the silicified horizon; other possible sources (sulphur leached from igneous wall rock, magmatic fluid-related sulphur or metamorphic sulphur) have lower δ 34S values and most likely could not have contributed significant amounts of sulphur. TSR modelling for other mineralization styles show that TSR cannot be the sole source of sulphur at the Ming deposit, however. In particular, with increasing TSR temperature, the lower δ 34S values in the semi-massive to massive sulphides cannot be successfully modelled using TSR as a sole source (Fig. 10); therefore, a second sulphur source is required. The lower δ 34S values can be explained by a contribution of sulphur leached from igneous wall rock or magmatic fluid-related sulphur, likely by mixing between TSR and sulphur leached from igneous wall rock/derived from magmatic fluids.

Measured and calculated δ 34S values of pyrrhotite in the silicified horizon of the 1806 ore body. Calculations are thermochemical sulphate reduction of reduced early Ordovician seawater using Eqs. (1)–(4) (Online Resource 6) for minimum and maximum temperatures of TSR of 250 and 350 °C, respectively; δ 34S of early Ordovician seawater sulphate is assumed to be ≈29 ‰ (Kampschulte and Strauss 2004)

Measured and calculated δ 34S values of chalcopyrite from the semi-massive to massive sulphides. Calculations are thermochemical sulphate reduction of reduced early Ordovician seawater by Eqs. (1)–(4) (Online Resource 6) for minimum and maximum temperatures of TSR of 250 and 350 °C, respectively; δ 34S of early Ordovician seawater sulphate is assumed to be ≈29 ‰ (Kampschulte and Strauss 2004)
Mixing between TSR and sulphur leached from igneous wall rock/derived from magmatic fluids (igneous sulphur) was tested using Eqs. (5) and (6) in Online resource 6. Calculations show that mixing between these two end members can account for the measured sulphur isotope compositions of sulphides from semi-massive to massive sulphides, the sulphide stringer horizon, and the Lower Footwall Zone at temperatures of TSR above 250 °C. In Fig. 11, the modelled mixing for chalcopyrite from the semi-massive to massive sulphides (Fig. 11a) and from the footwall mineralization (stringer sulphide horizon and Lower Footwall Zone; Fig. 11b) illustrates that with increasing temperature of TSR, the input of the igneous sulphur source (wall rock/magmatic fluid) must increase as well, and that a high percentage of sulphur in the analysed chalcopyrite can originate from sulphur leached from igneous wall rock/derived from magmatic fluids. Moreover, the average sulphur isotopic composition of chalcopyrite from the semi-massive to massive sulphides suggests that the input of igneous sulphur is higher than for chalcopyrite in footwall rocks (Fig. 11). For example, at a TSR temperature of 350 °C and ƒ = 0.95, the input of igneous sulphur would be higher to produce average δ 34S composition of chalcopyrite in semi-massive to massive sulphides (Fig. 11a) than to produce average δ 34S for chalcopyrite in the footwall (Fig. 11b). Whether the origin of sulphur leached from igneous wall rock/derived from magmatic fluids is either: (1) unaltered Rambler rhyolite; (2) boninitic rocks of the PHG; or (3) magmatic fluids from a subvolcanic intrusion cannot be constrained due to the lack of sulphur isotopic composition of each possible source. However, leached igneous (from Rambler rhyolite, boninitic basement rocks) and magmatic fluid (from subvolcanic intrusion) contributions to the sulphur budget of the deposit would be consistent with the sulphide mineral and hydrothermal alteration assemblages in the Ming deposit (Brueckner et al. 2014).

Measured and calculated δ 34S values for chalcopyrite in a semi-massive to massive sulphides and b in the footwall. Calculations are mixing lines between both end members TSR and S derived from leached igneous wall rock/magmatic fluids using Eqs. (5) and (6) (Online Resource 6). Calculations for fractionation of reduced seawater sulphate to sulphide with ƒ = 0.95, 0.7 and 0.5, for VMS-forming temperature of 300 °C and for assumed minimum and maximum temperatures of TSR of 250 and 350 °C, respectively
The modelling results are also consistent with the observations of Huston (1999), who compared the sulphur isotopic composition of 200 Archean to Tertiary VMS deposits and modern analogues worldwide. Huston (1999) described reduction of seawater sulphate as the dominant source of sulphur for most Paleozoic VMS deposits, especially for sulphides with δ 34S values ≥10 ‰. Most sulphides of the silicified horizon (Fig. 9) and some from the semi-massive to massive sulphides (Fig. 10) have sulphur isotopic compositions greater than 10 ‰. In contrast, Huston (1999) mentioned that sulphur leached from igneous wall rock/derived from magmatic fluids can account for δ 34S values between ≈0 and 5 ‰, but not for δ 34S values above 10 ‰. In this study, the majority of δ 34S values in sulphides, especially from the semi-massive to massive sulphides and the footwall mineralization of the Ming deposit (Figs. 6 and 7) are between +5 and +10 ‰. Hence, a sole igneous sulphur source (igneous wall rock, magmatic fluids) is rather unlikely for the sulphides at the Ming deposit especially in the semi-massive to massive sulphides and footwall mineralization and mixing of the two sulphur sources reduced seawater sulphate and igneous sulphur from igneous wall rock/magmatic fluids is more reasonable for sulphides with δ 34S ≤10 ‰ (Fig. 11).
Changes in sulphur isotope composition with stratigraphic position
Changes in δ 34S as a function of stratigraphic position in VMS deposits are reported from various deposits (Huston 1999). For example, sulphur isotope data from the Iron Mountain Mine, California (South and Taylor 1985), Rio Tinto, Spain (Eastoe et al. 1986) or the Madenköy VMS deposit, Turkey (Çagatay and Eastoe 1995), show higher δ 34Svalues in the footwall compared with the massive sulphide lens. Such an increase in δ 34S from semi-massive and massive sulphides to the footwall is also observed at the Ming deposit (Figs. 6 and 7). Eastoe et al. (1986) and Çagatay and Eastoe (1995) explained the higher δ 34S values in the footwall as being the result of higher inputs of H2S from reduced seawater sulphate over time, correlating with a shift of deposition from seafloor (massive sulphides) to sub-seafloor (stringer in footwall). Eastoe et al. (1986) suggested a decrease in temperature of the hydrothermal fluid or the obstruction of vents by sulphide mounds as a reason for such a shift in the locus of deposition at Rio Tinto, Spain.
At the Ming deposit, alteration and sulphide mineralogy indicate similar temperatures for the semi-massive to massive sulphides and sulphide stringer horizon (≈250–300 °C) but higher temperatures at the Lower Footwall Zone (>300 °C; Pilote et al. 2014). However, the sulphur isotopic composition of sulphides from the sulphide stringer horizon below the massive sulphide and the Lower Footwall Zone are almost identical (Figs. 6 and 7). The occurrence of metal sulphides is similar for all three mineralization styles, although abundances change especially to the higher temperature Lower Footwall Zone. The general metal sulphide assemblage in semi-massive to massive sulphides, sulphide stringer horizon and Lower Footwall Zone is: pyrite–chalcopyrite–pyrrhotite–sphalerite–cubanite ± arsenopyrite ± galena (Brueckner et al., unpublished data). This rather uniform metal sulphide assemblage throughout the Ming deposit suggests that the mineralization styles were formed at the same time. Hence, a change in the locus of deposition over time (Eastoe et al. 1986) is excluded as primary source for the decrease in δ 34S from the footwall to the overlying massive sulphides at the Ming deposit.
The modelling results show that the mixing of TSR with igneous sulphur (igneous wall rock/magmatic fluids) contributed the majority of sulphur to sulphides in both semi-massive to massive sulphides and footwall mineralization (Fig. 11). Therefore, the overall change in the sulphur isotopic composition with stratigraphic position is likely related to changes in the relative contributions from these two sources. Changes in the fractionation of seawater sulphate to sulphide by TSR are directly linked to the temperature of the hydrothermal fluid, where as the proportion of sulphide species in the hydrothermal fluid is a function of several physico-chemical parameters including temperature, pH and redox state. The hotter the hydrothermal fluid is the higher is the fractionation of seawater sulphate to sulphide and the more reduced the hydrothermal fluid becomes the higher is the amount of transported Fe and As species in the fluid. Indications for reduced hydrothermal fluid conditions are: (1) pyrite–pyrrhotite assemblage (Shanks et al. 1981) and (2) the occurrence of arsenopyrite and As-bearing minerals, such as tennantite and loellingite (Heinrich and Eadington 1986). Pyrite–pyrrhotite sulphide assemblage is especially common for the footwall mineralization (pyrrhotite is usually replacing pyrite; Fig. 5g, i). Arsenic-rich assemblages, on the other hand, such as arsenopyrite and As-bearing sulfosalts (tennantite, loellingite), are in greater abundance in the semi-massive to massive sulphides than in the footwall mineralization. Shanks et al. (1981), Lydon (1988) and Hannington et al. (1995) have illustrated that the presence of pyrrhotite in VMS mineral assemblages is consistent with high temperature reduced fluids. Similarly, Heinrich and Eadington (1986) showed that the transport of arsenic as (As)OH3 also requires reduced fluids. Based on the sulphide mineral assemblage indicating reduced conditions, both stratigraphic levels (semi-massive to massive sulphides, footwall mineralization) formed under similarily reducing conditions. Hence, it is assumed that changes in TSR due to variations in redox state and temperature of the hydrothermal fluid during the formation of the Ming deposit are not the primary cause of sulphur isotopic compositional differences between footwall and massive sulphides.
The stratigraphic variations in δ 34S are more likely related to the variable contribution of sulphur leached from igneous wall rock/derived from magmatic fluids relative to TSR with a higher contribution of igneous sulphur resulting in lower δ 34S values in the sulphides (Fig. 11; Huston 1999). Deciphering whether this igneous sulphur is leached from the igneous footwall or a direct contribution from magmatic fluids is difficult; however, it is likely both leaching of igneous rocks and direct magmatic sources were important given the sulphide and precious metal mineral assemblages in the deposit showing both classic VMS and epithermal-like assemblages, and precious metal enrichment in the sulphides (Brueckner et al. 2014). Brueckner et al. (2014) showed there are variations in the abundance of arsenopyrite, sulfosalt and precious metal phases, and they show greater enrichments in the semi-massive to massive sulphides than in the discordant footwall stringer sulphides. Thus, changes in the footwall and semi-massive to massive sulphide horizons are due to varying contributions from TSR and especially from sulphur leached from igneous wall rock/derived from magmatic fluids; similar results were noted in many VMS deposits by Huston (1999). In contrast, the homogeneity and high δ 34S values of the silicified horizon at the Ming deposit are derived solely from TSR and do not require any contribution from igneous sulphur (Fig. 9).
Influence of metamorphism on sulphur isotope composition and comparison with other deposits
Regional metamorphism is common amongst ancient VMS deposits (Galley et al. 2007), and the effects of metamorphism on sulphur isotope composition have been extensively discussed, particularly regarding sulphur isotope fractionation on the syngenetic sulphur isotopic composition of VMS sulphides (Seal II 2006; Hoefs 2009).
The metamorphic influence on the isotopic composition of sulphides is often difficult to distinguish from igneous wall rock/magmatic fluid signatures due to overlapping δ 34S values between igneous and metamorphic rocks (Hoefs 2009). Cook and Hoefs (1997), however, analysed the sulphur isotopic composition of sulphides from Paleozoic metamorphosed Norwegian VMS deposits (Fig. 12). They concluded that sulphur isotopic equilibration caused by metamorphism is locally restricted and most sulphur isotope data represent original δ 34S compositions. Seccombe et al. (1985) showed similar results for metamorphosed VMS mineralization in the Kanmantoo Group, South Australia. The sulphide pairs at the Ming deposit discussed are in isotopic disequilibrium and suggest that metamorphic homogenization of the sulphur isotopic composition of sulphides in the Ming deposit are negligible. Furthermore, despite the Ming deposit containing textures indicative of Silurian–Devonian metamorphism (annealed textures, recrystallization, porphyroblast growth, cataclastic texture; Fig. 5), there is no correlation between metamorphic textures and δ 34S values of the sulphides (Fig. 8; Online resource 6). These arguments alone suggest that metamorphic re-equilibration of δ 34S was not a significant process in the Ming deposit for the sulphides.

Sulphur isotope data for different sulphur sources, metamorphosed VMS deposits including this study and orogenic Au deposits of the Pacquet Harbour Group (PHG); data from:(1) Claypool et al. (1980), (2) Kampschulte and Strauss (2004)and (3) Hoefs (2009), (4) Shanks and Seyfried (1987), (5) Bailie et al. (2010), (6) Wagner et al. (2004), (7) Cook and Hoefs (1997), (8) Zaw and Large (1992) and (9) Bachinski (1977, 1978) with new data for Little Deer* from Toman (2012) and (10) Evans (2004)
In Fig. 12, the sulphur isotopic composition of sulphides from the Ming deposit is compared with the following: (1) sources of reduced sulphur (Claypool et al. 1980, Kampschulte and Strauss 2004; Hoefs 2009); (2) δ 34S values of modern vents (Shanks and Seyfried 1987); (3) metamorphosed VMS deposits worldwide (Zaw and Large 1992; Cook and Hoefs 1997; Wagner et al. 2004; Bailie et al. 2010) and of the Notre Dame Bay, Notre Dame sub-zone, Newfoundland (Bachinski 1977, 1978; Toman, 2012); and (4) orogenic deposits of Silurian age within the PHG (Evans 2004). Sulphur isotope data from the Ming deposit overlap with most δ 34S data from other metamorphosed VMS deposits, especially with sulphides from the Archeap Group, South Africa (Bailie et al. 2010), from Notre Dame Bay (Bachinski 1977, 1978; Toman 2012), Western Tasmania (Zaw and Large 1992) and partly with sulphides from the Norwegian Caledonides (Cook and Hoefs 1997). Sulphur isotopic compositions of sulphides from these deposits are interpreted as being primary with only limited local metamorphic sulphur isotope re-equilibration (Bachinski 1977, 1978; Zaw and Large 1992; Cook and Hoefs 1997; Bailie et al. 2010; Toman 2012). Pyrites from Silurian orogenic Au deposits (Evans 2004) in the vicinity of the Ming deposit have similar δ 34S values to some sulphides from the Ming deposit. However, the data from Evans (2004) are limited and further data for sulphides from orogenic gold deposits in the region would be required to fully evaluate the importance of Silurian–Devonian metamorphic fluid contributions to the sulphur isotope budget of the Ming deposit. At present, the data are not supportive of a significant influence, consistent with previous arguments based on ore mineral assemblages (Brueckner et al. 2014).
Conclusions
The sulphur isotope results on sulphides from the base and precious metal, early Ordovician Ming Cu–Au VMS deposit lead to the following conclusions:
(1) Influence of Siluro-Devonian metamorphism/poly-phase deformation on sulphur isotope composition in the Ming deposit is negligible and did not significantly affect the overall deposit. Instead, measured δ 34S values most likely reflect the original isotopic compositions of VMS mineralization; this is based on (a) isotopic disequilibrium for neighbouring mineral pairs, (b) lack of relation between metamorphic textures and δ 34S composition in pyrites and (c) the lack of evidence for metamorphic isotopic homogenization between pyrite and chalcopyrite and pyrite and pyrrhotite.
(2) Modelling of sulphur isotope compositions illustrate that sulphur was predominantly derived from mixtures of TSR of seawater sulphate combined with sulphur leached from igneous wall rock/derived from magmatic fluids (igneous sulphur). Although quantitative calculations assume many variables, the calculated contribution of sulphur leached from igneous wall rock/derived from magmatic fluids is elevated in semi-massive and massive sulphides and stringer footwall mineralization. Moreover, with increasing temperature, the contribution of igneous sulphur in these mineralization styles is shown to increase. The source of igneous sulphur may be leaching from igneous wall rock or through direct input of magmatic fluids/volatiles, but it is not possible to decipher with sulphur isotope data alone; however, given the variations in mineral assemblages in the deposit, it is permissive that both igneous sulphur sources were important in the Ming deposit. In contrast, the sulphur isotopic compositions in the silicified horizon that caps the 1806 Zone require only TSR of early Ordovician seawater sulphate; and
(3) Changes in δ 34S between the semi-massive to massive sulphides (lower δ 34S values) and the footwall mineralization (higher δ 34S values) are attributed to variations in the ratio of reduced sulphur from TSR to igneous sulphur. Exclusive changes of the fractionation of seawater sulphate to sulphide or a change of the locus of deposition (seafloor to sub-seafloor) during the time of formation are unlikely; this is based on (a) sulphide mineral assemblages in the semi-massive to massive sulphides and footwall that indicate rather reduced conditions for both mineralization styles and (b) broadly coeval deposition of sulphides in the footwall and on the seafloor at the Ming deposit.
References
Alt JC, Shanks WC (2011) Microbial sulfate reduction and the sulfur budget for a complete section of altered oceanic basalts, IODP Hole 1256D (eastern Pacific). EPSL 310:73–83. doi:10.1016/j.epsl.2011.07.027
Bachinski DJ (1977) Sulfur isotopic composition of ophiolitic cupriferous sulfide deposits, Notre Dame Bay, Newfoundland. Econ Geol 72:243–257
Bachinski DJ (1978) Sulfur isotopic composition of thermally metamorphosed cupriferous iron sulfide ores associated with cordierite-antophyllite rocks, Gull Pond, Newfoundland. Econ Geol 73:64–72
Bailie R, Gutzmer J, Strauss H, Stüeken E, McClung C (2010) Sulfur isotope characteristics of metamorphosed Zn–Cu volcanogenic massive sulfides in the Areachap Group, Northern Cape Province, South Africa. Miner Deposita 45:481–496. doi:10.1007/s00126-010-0285-8
Bischoff JL, Rosenbauer RJ (1983) A note on the chemistry of seawater in the range 350°–500°C. Geochim et Cosmochim Acta 47:139–144
Bradshaw GD, Rowins SM, Peter JM, Taylor BE (2008) Genesis of the Wolverine volcanic sediment-hosted massive sulfide deposit, Finlayson Lake District, Yukon, Canada: mineralogical, mineral chemical, fluid inclusion, and sulfur isotope evidence. Econ Geol 103:35–60
Brueckner SM, Piercey SJ, Sylvester PJ, Maloney S, Pilgrim L (2014) Evidence for syngenetic precious metal enrichment in an Appalachian volcanogenic massive sulfide system: The 1806 Zone, Ming Mine, Newfoundland, Canada. Econ Geol 109:1611–1642
Çagatay MN, Eastoe CJ (1995) A sulfur isotope study of volcanogenic massive sulfide deposits of the eastern Black Sea province, Turkey. Min Dep 30:55–66
Canfield DE (2001) Biogeochemistry of sulfur isotopes. In: Valley JW, Cole, D.R. (ed) Stable isotope geochemistry, vol 43. Reviews in Mineralogy and Geochemistry. pp 607–636
Castonguay S, Skulski T, van Staal C, Currie M (2009) New insights on the structural geology of the Pacquet Harbour group and Point Rousse complex, Baie Verte peninsula, Newfoundland. Current Res Newfoundland and Labrador Dept of Natural Resources. Geol Surv Rep 09–1:147–158
Claypool GE, Holser WT, Kaplan IR, Sakai H, Zak I (1980) The age curves of sulfur and oxygen isotopes in marine sulfate and their mutual interpretation. Chem Geol 28:199–260
Cook NJ, Hoefs J (1997) Sulphur isotope characteristics of metamorphosed Cu-(Zn) volcanogenic massive sulphide deposits in the Norwegian Caledonides. Chem Geol 135:307–324
Crowe DE (1994) Preservation of original hydrothermal ∂34S values in greenschist to upper amphibolite volcanogenic massive sulfide deposits. Geol 22:873–876
Dubé B, Gosselin P, Mercier-Langevin P, Hannington M, Galley A (2007) Gold-rich volcanogenic massive sulphide deposits. In: Goodfellow WD (ed) Mineral deposits of Canada: a synthesis of major deposit-types, district metallogeny, the evolution of geological provinces, and exploration methods, Special Publication No. 5. Geological Association of Canada, Mineral Deposits Division, pp 75–94
Eastoe CJ, Solomon M, Garcia Palomero F (1986) A sulfur isotope study of the massive and stockwork pyrite deposits at Rio Tinto, Spain. Trans Inst Min Metall Sect B: Appl Earth Sci 95:201–207
Evans DTW (2004) Epigenetic gold occurrences, Baie Verte Peninsula, (NTS 12H/09, 16 and 12I/01), Newfoundland. Mineral resource report 11. Government of Newfoundland and Labrador, Department of Natural Resources, Geological Survey, St. John's, NL
Fisher RV (1961) Proposed classification of volcaniclastic sediments and rocks. Geol Soc Am Bull 72:1409–1414
Franklin JM (1993) Volcanic-associated massive sulphide deposits. In: Kirkham RV, Sinclair WD, Thorpe RI, Duke JM (eds) Mineral deposit modeling. Geological Association of Canada, Special Paper 40, pp 315–334
Franklin JM (1996) Volcanic-associated massive sulphide base metals. In: Eckstrand OR, Sinclair WD, Thorpe RI (eds) Geology of Canadian Mineral Deposit Types. Geological Survey of Canada, Geology of Canada Series no. 8, Ottawa, ON, pp 158–183
Galley A, Hannington M, Jonasson I (2007) Volcanogenic massive sulphide deposits. In: Goodfellow WD (ed) Mineral Deposits of Canada: a synthesis of major deposit-types, district metallogeny, the evolution of geological provinces, and exploration methods, vol Special publication no. 5. Geological Association of Canada, Mineral Deposits Division, pp 141–161
Gemmell JB, Sharpe R (1998) Detailed sulfur sotope investigation of the TAG hydrothermal mound and stockwork zone, 26° N, Mid-Atlantic Ridge. In: Herzig PM, Humphris SE, Miller DJ, Zierenberg RA (eds) Proceedings of the Ocean Drilling Program, Scientific Results, vol 158. College Station, TX, pp 71–84
Gemmell JB, Sharpe R, Jonasson IR, Herzig PM (2004) Sulfur isotope evidence for magmatic contributions to submarine and subaerial gold mineralization: conical seamount and the Ladolam gold deposit, Papua New Guinea. Econ Geol 99:1711–1725
Goodfellow WD, Peter JM (1996) Sulphur isotope composition of the Brunswick no. 12 massive sulphide deposit, Bathurst Mining Camp, New Brunswick: implications for ambient environment, sulphur source, and ore genesis. Can J Earth Sci 33(2):231–251
Gregory RT, Criss RE (1986) Isotopic exchange in open and closed systems. Rev Min Geochem 16(1):91–127
Gregory RT, Criss RE, Taylor HP Jr (1989) Oxygen isotope exchange kinetics of mineral pairs in closed and open systems: Applications to problems of hydrothemal alteration of igneous rocks and Precambrian iron formations. Chem Geol 75:1–42
Hannington MD, Jonasson IR, Herzig PM, Petersen S (1995) Physical and chemical processes of seafloor mineralization at mid-ocean ridges. In: Humphris SE, Zierenberg RA, Mullineaux LS, Thomson RE (eds) Seafloor hydrothermal systems: physical, chemical, biological, and geological interactions. Geophys Monograph 91, American Geophysical Union, Washington, DC, pp 115–157
Hannington MD, Poulsen KH, Thompson JFH, Sillitoe RH (1999) Volcanogenic gold in the massive sulfide environment. In: Barrie CT, Hannington MD (eds) Volcanic-associated massive sulfide deposits: processes and examples in modern and ancient settings, vol 8. Reviews in Econ Geol. Society of Economic Geologists, Boulder, CO, pp 325–351
Heinrich CA, Eadington PJ (1986) Thermodynamic predictions of the hydrothermal chemistry of arsenic, and their significance for the paragenetic sequence of some cassiterite-arsenopyrite-base metal sulfide deposits. Econ Geol 81:511–529
Herzig PM, Hannington MD (1995) Polymetallic massive sulfides at the modern seafloor: a review. Ore Geol Rev 10:95–115
Hibbard LJ (1983) Geology of the Baie Verte Peninsula, Newfoundland, Department of Mines and Energy, Government of Newfoundland and Labrador, Memoir 2
Hoefs J (2009) Stable isotope geochemistry, 6th edn. Springer, Berlin, Heidelberg
Huston DL (1999) Stable isotopes and their significance for understanding the genesis of volcanic-hosted massive sulfide deposits: a review. In: Barrie CT, Hannington MD (eds) Volcanic-associated massive sulfide deposits: processes and examples in modern and ancient settings, vol 8. Rev in Econ Geol. Society of Economic Geologists, Boulder, CO, pp 157–179
Huston DL (2000) Gold in volcanic-hosted massive sulfide deposits: distribution, genesis, and exploration. In: Hagemann SG, Brown PE (eds) Gold in 2000, vol 13. Rev in Econ Geol. Society of Economic Geologists, Boulder, CO, pp 401–426
Kajiwara Y, Krouse HR (1971) Sulfur isotope partioning in metallic sulfide systems. Can J Earth Sci 8:1397–1408
Kampschulte A, Strauss H (2004) The sulfur isotopic evolution of Phanerozoic seawater based on the analysis of structurally substituted sulfate in carbonates. Chem Geol 204:255–286. doi:10.1016/j.chemgeo.2003.11.013
Large RR (1992) Australian volcanic-hosted massive sulfide deposits: Features, styles, and genetic models. Econ Geol 87:471–510
Lydon JW (1984) Volcanogenic massive sulphide deposits. Part 1: a descriptive model. Geosci Can 11(4):195–202
Lydon JW (1988) Volcanogenic massive sulphide deposits. Part 2: genetic models. Geosci Can 15(1):43–65
Mercier-Langevin P, Hannington MD, Dubé B, BécuV (2011) The gold content of volcanogenic massive sulfide deposits. Miner Deposita 46:509–539. doi:10.1007/s00126-010-0300-0
Ohmoto H (1996) Formation of volcanogenic massive sulfide deposits: the Kuroko perspective. Ore Geol Rev 10:135–177
Ohmoto H, Goldhaber MB (1997) Sulfur and carbon isotopes. In: Barnes HL (ed) Geochemistry of hydrothermal ore deposits, 3rd edn. John Wiley & Sons, New York, pp 517–611
Ohmoto H, Rye RO (1979) Isotopes of sulfur and carbon. In: Barnes HL (ed) Geochemistry of hydrothermal ore deposits, 2nd edn. John Wiley & Sons, New York, pp 509–567
Paytan A, Gray ET (2012) Sulfur Isotope stratigraphy. In: Gradstein FM, Ogg JG, Schmitz M, Ogg G (eds) The geologic timescale 212. Elsevier, Amsterdam, pp 167–180. doi:10.1016/B978-0-444-59425-9.00009-3
Piercey S, Jenner GA, Wilton DHC (1997) The stratigraphy and geochemistry of the southern Pacquet Harbour Group, Baie Verte Peninsula, Newfoundland: implications for mineral exploration. Current Res Newfoundland and Labrador, Dept of Nat Resources. Geol Surv Rep 97–1:119–139
Pilgrim L (2009) Mineral resource estimate for the Ming Mine, Newfoundland, Canada. Rambler Metals and Mining Canada Ltd, Baie Verte, NL
Pilote J-L, Piercey SJ, Mercier-Langevin P (2014) The Ming Cu-Au volcanogenic massive sulphide deposit, Baie Verte Peninsula, Newfoundland: stratigraphy and hydrothermal alteration. Current Res Newfoundland and Labrador Dept of Natural Resources, Geol Surv Report [submitted]
Seal II RR (2006) Sulfur isotope geochemistry of sulfide minerals. In: Vaughan DJ (ed) Sulfide mineralogy and geochemistry, vol 61. Rev in Miner and Geochem. pp 633–677
Seccombe PK, Spry PG, Both RA, Jones MT, Schiller JC (1985) Base metal mineralization in the Kanmantoo Group, South Australia: a regional sulfur isotope study. Econ Geol 80:1824–1841
Seyfried WE Jr, Bischoff JL (1981) Experimental seawater-basalt interaction at 300 °C, 500 bars, chemical exchange, secondary mineral formation and implications for the transport of heavy metals. Geochim et Cosmochim Acta 45:135–147
Shanks III WC (2001) Stable isotopes in seafloor hydrothermal systems: vent fluids, hydrothermal deposits, hydrothermal alteration, and microbial processes. In: Valley JW, Cole, D.R. (ed) Stable isotope geochemistry, vol 43. Rev in Miner and Geochem. pp 469–525
Shanks WC III, Seyfried WE Jr (1987) Stable isotope studies of vent fluids and chimney minerals, southern Juan de Fuca Ridge: sodium metasomatism and seawater sulfate reduction. J Geophys Res 92(B11):11387–11399
Shanks WC III, Bischoff JL, Rosenbauer RJ (1981) Seawater sulfate reduction and sulfur isotope fractionation in basaltic systems: interaction of seawater with fayalite and magnetite at 200–350 °C. Geochim et Cosmochim Acta 45:1977–1995
Shanks WC III, Böhlke JK, Seal RR II (1995) Stable isotopes in mid-ocean ridge hydrothermal systems: interaction between fluids, minerals and organisms. In: Humphris SE, Zierenberg RA, Mullineaux LS, Thomson RE (eds) Seafloor hydrothermal systems: physical, chemical, biological, and geological interactions. Geophys Mon 91, American Geophysical Union, Washington, DC, pp 194–221
Sillitoe RH, Hannington MD, Thompson JFH (1996) High sulfidation deposits in the volcanogenic massive sulfide environment. Econ Geol 91:204–212
Skulski T, Castonguay S, McNicoll V, van Staal C, Kidd W, Rogers N, Morris W, Ugalde H, Slavinski H, Spicer W, Moussallam Y, Kerr I (2010) Tectonostratigraphy of the Baie Verte oceanic tract and its ophiolite cover sequence on the Baie Verte Peninsula. Current Res Newfoundland and Labrador Dept of Natural Resources. Geol Surv Rep 10–1:315–335
South BC, Taylor BE (1985) Stable isotope geochemistry and metal zonation at the Iron Mountain mine, West Shasta district, California. Econ Geol 80:2177–2195
Swinden HS, Thorpe RI (1984) Variations in style of volcanism and massive sulfide deposition in Early to Middle Ordovician island-arc sequences of the Newfoundland Central Mobile Belt. Econ Geol 79(7):1596–1619. doi:10.2113/gsecongeo.79.7.1596
Toman HC (2012) Geology and metallogeny of north-central Newfoundland and the Little Deer VMS deposit: an introduction and overview. M.Sc. thesis, Memorial University of Newfoundland, St. John's, NL [unpublished]
Tuach J, Kennedy MJ (1978) The geologic setting of the Ming and other sulfide deposits, consolidated Rambler mines, Northeast Newfoundland. Econ Geol 73:192–206
van Staal CR (2007) Pre-Carboniferous tectonic evolution and metallogeny of the Canadian Appalachians. In: Goodfellow WD (ed) Mineral deposits of Canada: a synthesis of major deposit-types, district metallogeny, the evolution of geological provinces, and exploration methods, Special publication no. 5. Geological Association of Canada, Mineral Deposits Division, pp 793–818
van Staal CR, Barr SM (2012) Lithospheric architecture and tectonic evolution of the Canadian Appalachians and associated Atlantic margin. In: Percival JA, Cook FA, Clowes RM (eds) Tectonic Styles in Canada: the LITHOPROBE perspective. Geological Association of Canada, Special Paper 49, St. John's, NL, pp 41–96
Wagner T, Boyce AJ, Jonsson E, Fallick AE (2004) Laser microprobe sulphur isotope analysis of arsenopyrite: experimental calibration and application to the Boliden Au–Cu–As massive sulphide deposit. Ore Geol Rev 25:311–325
Williams H (1979) Appalachian orogen in Canada. Can J Earth Sci 16:792–807
Woodruff LG, Shanks WC III (1988) Sulfur isotope study of chimney minerals and vent fluids from 21°N, East Pacific Rise: Hydrothermal sulfur sources and disequilibrium sulfate reduction. J Geophys Res 93(B5):4562–4572
Zaw K, Large RR (1992) The precious metal-rich, South Hercules mineralization, western Tasmania: a possible subsea-floor replacement volcanic-hosted massive sulfide deposit. Econ Geol 87(3):931–952. doi:10.2113/gsecongeo.87.3.931
Acknowledgments
Stefanie Brueckner gives special thanks to staff and miners of the Ming mine, who are explicitly thanked for their help, assistance and support during core logging and underground mapping. Rambler Metals and Mining Canada Ltd. provided logistical support for the project. Stefanie Brueckner also thanks especially Anthony E. Fallick, an anonymous reviewer, co-editor Karen Kelley and editor-in-chief Georges Beaudoin for their constructive, critical and helpful comments, which improved the manuscript greatly. Funding for the project was provided by grants to Stephen Piercey, including an NSERC Discovery Grant and the NSERC Altius Industrial Research Chair in Mineral Deposits supported by NSERC, Altius Minerals Ltd. and the Research and Development Corporation of Newfoundland and Labrador. The installation of the MAF-IIC SIMS Facility at Memorial University was catalysed by a Leaders Opportunity Fund grant to Graham Layne from the Canada Foundation for Innovation. Ongoing support for this facility is also partially derived from an NSERC Discovery Grant to Graham Layne.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editorial handling: K. Kelley and G. Beaudoin
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online resource 1
Methodology (PDF 111 kb)
Online resource 2
Box–Whisker plot of the results for sulphide reference materials used for IMF calibration and quality control of SIMS microanalyses. Data are the recovered δ 34S values for each individual measurement of a reference material once the IMF correction (based on the mean of replicate measurements for each day) was applied. The vertical error bars represent the total range of all spot analyses during the multiple analytical sessions. The vertical dimension of the box represents the 1σ for all spot analyses during multiple analyses. These data demonstrate the overall reproducibility discussed for δ 34S in text, and the relative homogeneity of the reference sulphide materials. All data are compiled in Online resource 3; n = number of analysed data points (PDF 127 kb)
Online resource 3
Results of measured 34S/32S ratio, calculated instrumental mass fractionation (IMF), and δ 34S analysis corrected for IMF of the used in-house standards analysed via SIMS; SEM standard error mean, Stddev standard deviation; asterisk accepted value; n number of analyses (PDF 205 kb)
Online resource 4
Analysed samples from the Ming deposit, their brief description, sulphide mineralogy and results of S isotope analysis (PDF 290 kb)
Online resource 5
Isotopic equilibrium (PDF 928 kb)
Online resource 6
Calculations used for modelling S sources at the Ming deposit (PDF 265 kb)
Rights and permissions
About this article

Cite this article
Brueckner, S.M., Piercey, S.J., Layne, G.D. et al. Variations of sulphur isotope signatures in sulphides from the metamorphosed Ming Cu(−Au) volcanogenic massive sulphide deposit, Newfoundland Appalachians, Canada. Miner Deposita 50, 619–640 (2015). https://doi.org/10.1007/s00126-014-0567-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00126-014-0567-7