
Abstract
Aims/hypothesis
Better understanding of how genetic and epigenetic components control beta cell differentiation and function is key to the discovery of novel therapeutic approaches to prevent beta cell dysfunction and failure in the progression of type 2 diabetes. Our goal was to elucidate the role of histone deacetylase sirtuin 6 (SIRT6) in beta cell development and homeostasis.
Methods
Sirt6 endocrine progenitor cell conditional knockout and beta cell-specific knockout mice were generated using the Cre-loxP system. Mice were assayed for islet morphology, glucose tolerance, glucose-stimulated insulin secretion and susceptibility to streptozotocin. Transcriptional regulatory functions of SIRT6 in primary islets were evaluated by RNA-Seq analysis. Reverse transcription-quantitative (RT-q)PCR and immunoblot were used to verify and investigate the gene expression changes. Chromatin occupancies of SIRT6, H3K9Ac, H3K56Ac and active RNA polymerase II were evaluated by chromatin immunoprecipitation.
Results
Deletion of Sirt6 in pancreatic endocrine progenitor cells did not affect endocrine morphology, beta cell mass or insulin production but did result in glucose intolerance and defective glucose-stimulated insulin secretion in mice. Conditional deletion of Sirt6 in adult beta cells reproduced the insulin secretion defect. Loss of Sirt6 resulted in aberrant upregulation of thioredoxin-interacting protein (TXNIP) in beta cells. SIRT6 deficiency led to increased acetylation of histone H3 lysine residue at 9 (H3K9Ac), acetylation of histone H3 lysine residue at 56 (H3K56Ac) and active RNA polymerase II at the promoter region of Txnip. SIRT6-deficient beta cells exhibited a time-dependent increase in H3K9Ac, H3K56Ac and TXNIP levels. Finally, beta cell-specific SIRT6-deficient mice showed increased sensitivity to streptozotocin.
Conclusions/interpretation
Our results reveal that SIRT6 suppresses Txnip expression in beta cells via deacetylation of histone H3 and plays a critical role in maintaining beta cell function and viability.
Data availability
Sequence data have been deposited in the National Institutes of Health (NIH) Gene Expression Omnibus (GEO) with the accession code GSE104161.
Similar content being viewed by others


Avoid common mistakes on your manuscript.

Introduction
Beta cells, the major cell type in the endocrine pancreas, control glucose homeostasis by secreting insulin in response to blood sugar level [1]. Dysfunction of beta cells and impaired insulin sensitivity of peripheral tissues lead to type 2 diabetes [2]. Histone modifications, including acetylation, methylation, phosphorylation and ubiquitination, affect the accessibility of genetic information by altering the physical interaction of DNA, histones and non-histone proteins [3]. Studies have shown that histone acetylation and deacetylation play pivotal roles in regulating endocrine differentiation and beta cell maturation and function [4]. In addition, histone acetylation and deacetylation are implicated in the pathogenesis of diabetes [5]. Thus, targeting such mechanisms may provide a potential approach to treat diabetes [5]. The biochemical reactions of histone acetylation and deacetylation in cells are executed by catalytic enzymes called histone acetyltransferases (HATs) and histone deacetylases (HDACs) [5]. Despite the identification of more than 20 HATs and 18 HDACs [6, 7], the expression pattern and precise function of these specific enzymes in beta cells are not yet fully understood. Furthermore, despite extensive studies conducted in vitro, the physiological functions of these proteins have not been completely established [8].
Sirtuins, first identified in yeast as ageing regulators, belong to the class III HDACs [9]. There are seven members in the mammalian sirtuin family (sirtuin 1–7) [9], all of which exhibit NAD-dependent deacetylation activity. However, different sirtuins act on different substrates and are localised in specific subcellular compartments [10]. For example, SIRT6 is closely associated with chromatin and works as a transcriptional co-suppressor by catalysing the deacetylation of histone H3 lysine residues acetylated at positions 9, 56 and 18 (H3K9Ac, H3K56Ac and H3K18Ac) [11,12,13]. Prior genetic studies showed that SIRT6 is essential for mouse development and plays versatile roles in regulating glucose homeostasis, lipid metabolism and many other processes [14]. To elucidate the role of SIRT6 in beta cell development and homeostasis, we generated endocrine pancreas-specific (EKO) and beta cell-specific knockout (BKO) mouse models in which Sirt6 was deleted at the embryonic and adult stages, respectively. By taking advantage of transcriptome analysis, we further investigated the underlying mechanisms by which SIRT6 contributes to endocrine pancreas functions.
Methods
Animal studies
All mouse studies were approved by the Institutional Animal Care and Use Committee of the University of Texas Health Science Centre at San Antonio. The Sirt6 flox/flox (f/f) [15], Ngn3-Cre [16] (Ngn3 is also known as Neurog3) and mouse insulin promoter 1-driven, inducible CreERT transgenic line (MIP1-CreERT) [17] mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA) and maintained in a mix genetic background. Mice were housed in a conventional facility with a 12 h night–day cycle and free access to food and water. Mouse genotyping was performed using a dirty-tail method as previously described [18]. Tamoxifen (T5648; Sigma, St Louis, MO, USA) was used to delete the floxed Sirt6 allele via the MIP1-CreERT system [19]. Male mice at 2–11 months old and female mice at 2–9 months old were used in the analysis. For all the experiments, Sirt6 EKO mice or Sirt6 BKO mice and their littermate control mice (see electronic supplementary material [ESM] Fig. 1a and ESM Fig. 4 for the breeding strategy) were selected by genotype and were randomly assigned a unique label in the tail, with the genotype blinding to operators until experiments were completed. No data were excluded in all the analysis. See ESM Table 1 and ESM Methods for genotyping primers and experimental procedures for tamoxifen injection, verapamil (V4629; Sigma) treatment and streptozotocin (STZ) (S0130; Sigma) challenge.
Changes in pancreas and islet architecture were revealed by H&E staining and anti-insulin, anti-glucagon and anti-somatostatin immunostaining. Beta cell apoptosis was determined by anti-cleaved caspase 3 immunostaining. The metabolic studies, including glucose measurement, insulin measurement, IPGTT, ITT and hyperglycaemic clamp were performed as previously reported [20]. See ESM Methods for details.
Pancreatic islet isolation and glucose-stimulated insulin secretion
Islets were isolated by collagenase perfusion and used for determination of glucose-stimulated insulin secretion as previously described [21]. See ESM Methods for details.
Gene expression
Sirt6 mRNA levels in Ngn3-null and Ngn3-positive cells at mouse embryonic day 15.5 (E15.5) were generated using data from Table S6 in Benitez et al [22]. Sirt6 and Txnip mRNA levels in isolated islets were determined by reverse transcription (RT) quantitative (q)-PCR. See ESM Methods for details.
Immunoblotting
SIRT6, thioredoxin-interacting protein (TXNIP), H3K9Ac, H3K27Ac and H3K56Ac levels were determined by immunoblotting. See ESM Methods for details.
RNA-Seq analysis
Islets isolated from 2 weeks post-tamoxifen control (Sirt6f/+; MIP1-CreERT) and Sirt6 BKO (Sirt6f/f; MIP1-CreERT) mice and 2-month-old control (Sirt6+/+; Ngn3-Cre or Sirt6f/f) and Sirt6 EKO (Sirt6f/f; Ngn3-Cre) mice were used for the RNA-Seq analysis. See ESM Methods for details.
ChIP
Chromatin occupancies of SIRT6, IgG, H3K9Ac, H3K56Ac and Active RNA polymerase II in beta cells were determined by chromatin immunoprecipitation (ChIP) [23]. See ESM Methods for details.
Statistical analysis
All results are presented as means ± SEM. Statistical analysis was performed by two-tailed Student’s t test. p values <0.05 were considered statistically significant.
Results
Deletion of Sirt6 in pancreatic endocrine progenitor cell does not affect beta cell development
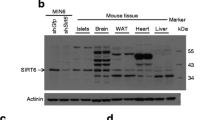
Gene expression profiles of pancreatic cells throughout different stages of mouse pancreas development were analysed previously [22]. Using the data from this study (Table S6) we found that Sirt6 transcripts were expressed at a much higher level in E15.5 neurogenin 3 (NGN3)-expressing endocrine progenitor cells than in Ngn3-null cells (Fig. 1a) [22]. In the adult mouse pancreas, Sirt6 transcripts and proteins were also expressed highly in isolated islets (Fig. 1b, d). To examine the role of SIRT6 in endocrine pancreas development and function, we generated an EKO mouse model (Sirt6f/f; Ngn3-Cre) by crossing Sirt6f/f mice with the Ngn3-Cre strain (ESM Fig. 1a) [24]. As expected, the Sirt6 transcripts and proteins were undetectable in islets isolated from mutant mice (Fig. 1c, d), indicating efficient deletion of the Sirt6 gene in the endocrine pancreas.

Deletion of Sirt6 in pancreatic endocrine progenitor cells does not affect beta cell development. (a) Sirt6 mRNA levels in E15.5 Ngn3-null cells and Ngn3-positive cells, n = 3–4; the graph was generated based on data published previously [22]. (b) RT-qPCR of Sirt6 mRNA levels in the pancreas and isolated islets. The relative expression level of Sirt6 mRNA in islets was normalised to that in the pancreas, n = 3. (c) RT-qPCR of Sirt6 mRNA levels in islets from the control (Sirt6f/f) and Sirt6 EKO mice, n = 3. (d) Immunoblot of SIRT6 protein in the pancreas or islets isolated from the control and Sirt6 EKO mice. P1, P2, pancreas samples from two separate control mice (Sirt6f/f). (e) Immunostaining of pancreatic islets (green: anti-insulin; blue, DAPI; red, anti-glucagon) from control and Sirt6 EKO mice. Scale bar, 50 μm. (f, g) Quantification of the average number of islets per section (f) and beta cell mass (g), n = 3 in control and Sirt6 EKO mice. Data are expressed as means ± SEM. **p < 0.01 for indicated comparisons
The EKO mice were born at the expected Mendelian ratio and displayed no overt phenotype. To test whether loss of Sirt6 affects endocrine morphology, we collected pancreases from 2-month-old control and EKO mice and performed histological analysis. No obvious differences between the pancreases of the two groups were revealed by H&E staining (data not shown). Furthermore, immunostaining for insulin and glucagon showed no apparent changes in islet architecture or islet number (Fig. 1e, f and ESM Fig. 1b). Endocrine cell mass analysis showed no change in beta cell mass in EKO mouse pancreases (Fig. 1g). However, alpha cell mass showed a decrease (not statistically significant) and delta cell mass showed a statistically significant increase (ESM Fig. 1c, d). These data are consistent with the results reported from recent studies in which Sirt6 was knocked out in early pancreas and beta cells [25, 26]. Thus, deletion of Sirt6 in NGN3-expressing progenitor cells does not affect beta cell development or endocrine pancreas gross morphology.
Deletion of Sirt6 in pancreatic endocrine progenitor cell leads to defective glucose-stimulated insulin secretion
Next, we assessed the effect of Sirt6 knockout on glucose homeostasis function of the endocrine pancreas. During fasting or feeding, blood glucose and plasma insulin levels of mutant mice were comparable with those of their littermate controls (Fig. 2a, b). However, an IPGTT revealed a severe glucose intolerance phenotype in 2- to 3-month-old Sirt6 EKO mice (Fig. 2c, d and ESM Fig. 2a, b). We found that insulin levels during the IPGTT were significantly lower in the Sirt6 EKO mice than in control mice at 15 min (Fig. 2e), implying that the defect in the EKO mice may be due to insulin insufficiency. Moreover, we found that the glucose intolerance phenotype became more severe in 5- to 6-month-old Sirt6 EKO mice, as indicated by the AUC of the GTT, which increased from 147% to 205% of the control value (Fig. 2d, g and ESM Fig. 2c, d), suggesting that the phenotype worsened over time. An ITT showed that in mice aged 8–10 months, the response to insulin of peripheral tissues was the same in Sirt6 EKO mice as in control mice (ESM Fig. 3), further pointing to defective insulin production or secretion as being the cause of the impaired glucose tolerance [27]. Sirt6 EKO mouse pancreases had normal total insulin content (Fig. 3a). Thus, our data supported the notion that the phenotype was caused by an insulin secretion defect of the endocrine pancreas. To test this, we isolated islets from the mice and performed a glucose-stimulated insulin secretion assay in vitro. As expected, upon incubation with a high concentration of glucose, the amount of insulin released from Sirt6 EKO cells was only one-third of that release by cells from control mice (Fig. 3b). Furthermore, during a hyperglycaemic clamp procedure to evaluate beta cell function in vivo, we observed that Sirt6 EKO mice required a significantly lower glucose infusion rate to maintain the glucose level at approximately 16 mmol/l (Fig. 3c, d), and that plasma insulin concentrations of EKO mice were much lower than those of their control littermates (Fig. 3e, f). Thus, combined in vitro and in vivo results illustrate that deletion of Sirt6 in the endocrine pancreas leads to defective glucose-stimulated insulin secretion.

Deletion of Sirt6 in pancreatic endocrine progenitor cell leads to glucose intolerance. (a) Blood glucose levels of 2-month-old control (Sirt6f/f) and Sirt6 EKO male mice in the fasting or random-fed state (n = 6). (b) Plasma insulin levels of 2-month-old control and Sirt6 EKO male mice in the fasting or random-fed state (n = 5). (c) Blood glucose after GTTs in 2- to 3-month-old male mice (n = 8 control; n = 13 EKO). (d) Normalised area under the GTT curve. (e) Plasma insulin levels of 2- to 3-month-old control and Sirt6 EKO mice during the GTT (n = 4–6). (f) Glucose tolerance tests of 5- to 6-month-old male mice (n = 4 control; n = 5 EKO). (g) The normalised area under the GTT curve. Black bars and circles, control mice; grey bars and squares, Sirt6 EKO mice. Data are expressed as means ± SEM. *p < 0.05 and **p < 0.01 vs control mice or for indicated comparisons

Deletion of Sirt6 in pancreatic endocrine progenitor cell leads to defective glucose-stimulated insulin secretion. (a) The total insulin content of the pancreas (n = 3 control; n = 4 EKO). (b) Relative insulin levels secreted from four pairs of islets isolated from control (Sirt6f/f) and EKO mice treated with 16.7 mmol/l glucose for 1 h; values were normalised to those of the 2.5 mmol/l glucose treatment group. (c–e) Blood glucose levels (c), glucose infusion rates (d) and plasma insulin concentrations (e) of 4-month-old control and Sirt6 EKO mice during the hyperglycaemic clamp assay (n = 6 control; n = 7 EKO). (f) The normalised AUC for plasma insulin concentration of the 4-month-old control and Sirt6 EKO mice during the hyperglycaemic clamp assay. Black bars and circles, control mice; grey bars and squares, Sirt6 EKO mice. Data are expressed as means ± SEM. *p < 0.05 and **p < 0.01 vs control mice or for indicated comparisons
Beta cell-specific Sirt6 knockout leads to defective insulin secretion
Given the fact that NGN3 progenitor cells generate other cell types besides beta cells during development [22], we sought to determine whether the Sirt6 EKO phenotype was intrinsic to beta cells. To this end, we generated Sirt6 BKO mice, with beta cell-specific Sirt6 deletion, by intercrossing the floxed Sirt6 mice with MIP1-CreERT mice (ESM Fig. 4) [19]. Two weeks following tamoxifen treatment, SIRT6 transcript and protein levels in primary islets isolated from Sirt6 BKO (Sirt6f/f; MIP1-CreERT) mice were reduced to less than 20% of normal (Fig. 4a, b). The remaining signals were likely due to the expression of SIRT6 in alpha cells or other types of endocrine cells [8]. Similar to Sirt6 EKO mice, Sirt6 BKO mice had normal fasting and fed glucose levels (Fig. 4c) but displayed glucose intolerance because of insufficient insulin secretion (Fig. 4d–f). Notably, the insulin levels in Sirt6 BKO mice at 0 min were lower than those in control mice, which was not observed in our Sirt6 EKO mice (Fig. 2e), highlighting the difference between the two models. Moreover, in line with our observations made in Sirt6 EKO mice of different ages, the glucose intolerance phenotype worsened over time in Sirt6 BKO mice as well (Fig. 4g–j), further confirming that the phenotype was largely due to Sirt6 deletion in beta cells. Together, these findings demonstrate that SIRT6 is required for glucose-stimulated insulin secretion in beta cells.

Beta cell-specific deletion of Sirt6 leads to defective insulin secretion. (a, b) RT-qPCR analysis of Sirt6 mRNA (a) and immunoblots of SIRT6 expression (b) in islets isolated from control (Ctrl, a combination of MIP1-CreER and Sirt6f/+; MIP1-CreER mice after tamoxifen treatment) and Sirt6 BKO mice 2 weeks after deletion (n = 3). (c) Fasting and random-fed blood glucose levels of the 2 weeks post-tamoxifen (2-month-old) control and Sirt6 BKO male mice (n = 5–6). (d) Blood glucose during GTTs in 2 weeks post-tamoxifen (2-month-old) control and Sirt6 BKO male mice (n = 5 or 6). (e) AUC for blood glucose levels during GTT shown in (d). (f) Plasma insulin levels in the 2 weeks post-tamoxifen (2-month-old) control and Sirt6 BKO mice during the GTT, related to (d) (n = 4). (g, h) Blood glucose levels during GTTs (g) and normalised AUCs (h) in 2 weeks post-tamoxifen control and Sirt6 female BKO mice (n = 4). (i, j) Blood glucose levels during GTTs (i) and normalised AUCs (j) in 4 months post-tamoxifen control and Sirt6 BKO female mice (n = 4 control; n = 5 BKO). Black bars and circles, control mice; grey bars and squares, Sirt6 BKO mice. Data are expressed as means ± SEM. *p < 0.05 and **p < 0.01 vs control mice or for indicated comparisons
Sirt6 knockout leads to aberrant upregulation of TXNIP
To gain mechanistic insight into how SIRT6 regulates glucose-stimulated insulin secretion, we analysed the transcriptomes of the islets isolated from control and mutant mice (two pairs of Sirt6 BKO mice, 2 weeks post-tamoxifen and two pairs of Sirt6 EKO mice, 2 months old) by RNA-Seq. Out of roughly 20,000 genes evaluated, a total of 99 and 243 genes were upregulated or downregulated more than 1.5-fold in the islets from Sirt6 BKO mice and Sirt6 EKO mice, respectively (GSE104161). In agreement with data shown in Fig. 3a, the levels of insulin-encoding mRNA (Ins1 and Ins2) were comparable in control and mutant islets. Additionally, the expression levels of genes associated with the occurrence of maturity-onset diabetes of the young or type 2 diabetes, such as Pdx1, Hnf4a and Neurod1, were unaltered in mutant islets [1].
To our surprise, only a subset of 23 genes showed a similar trend in both models: 18 were upregulated and 5 were downregulated (Fig. 5a, b). Nevertheless, a hypergeometric probability test revealed that this overlap was highly significant (p < 1.96 × 10−10; Fig. 5a). Among the upregulated genes in mutant islets, Txnip encodes a ubiquitous thioredoxin-binding protein that suppresses the antioxidant activity of thioredoxin, which is essential for eliminating reactive oxygen species (ROS) in cells [28]. It has been reported that TXNIP expression is negatively associated with glucose-stimulated insulin secretion in beta cells and that overexpression of TXNIP inhibits insulin secretion [29,30,31]. To test the hypothesis that TXNIP mediated the insulin secretion defect in the Sirt6 KO beta cells, we first validated the change in TXNIP expression in SIRT6-deficient islets by immunoblot. As expected, TXNIP protein levels were significantly upregulated in islets isolated from 3-month-old Sirt6 EKO mice (Fig. 5c, d). Simultaneously, we also detected a higher level of H3K9Ac in the Sirt6 EKO islets (Fig. 5c), reflecting that loss of the histone deacetylation function of SIRT6 was correlated with elevated expression of Txnip.

Loss of SIRT6 leads to upregulation of TXNIP. (a, b) Venn diagram (a) and heat map (b) of the overlapped genes between the two Sirt6 KO models from the RNA-Seq analysis. p < 1.96 × 10−10. (c, d) Immunoblots and quantification (d) of TXNIP expression levels in islets from 3-month-old control (Sirt6f/f) and Sirt6 EKO mice. Data are expressed as means ± SEM (n = 3–4). **p < 0.01 for indicated comparison
Verapamil has been shown to repress Txnip transcription [32]. We tested whether reducing TXNIP levels by verapamil could partially rescue the impaired glucose tolerance in SIRT6-deficient mice (ESM Fig. 5a). After three weeks oral verapamil treatment (100 mg/kg per day), the immunoblot analysis showed that islets isolated from the two groups expressed similar levels of TXNIP, yet islets from Sirt6 EKO mice still had higher H3K9Ac levels (ESM Fig. 5d). Consistent with the previous findings [32], verapamil lowered the overall glucose level in both control and Sirt6 EKO mice at 8–9 months old (ESM Fig. 5b, f). Analysis of blood glucose during a GTT showed that the Sirt6 EKO mice manifested largely restored tolerance to glucose (ESM Fig. 5e, f). Indeed, five out of six Sirt6 EKO mice showed a similar response to that of control mice. Concurrently, plasma insulin levels of verapamil-treated Sirt6 EKO mice increased 1.5-fold at 15 min after glucose challenge (ESM Fig. 5h, p = 0.05). Note that plasma insulin levels of verapamil-treated control mice increased 1.8-fold and that there was no increase in Sirt6 EKO mice before verapamil treatment (ESM Fig. 5h, d). Together, these data suggest that verapamil treatment partially restored glucose-stimulated insulin secretion in Sirt6 EKO mice, indicating that TXNIP is likely a mediator of the insulin secretion defect in SIRT6-deficient beta cells.
SIRT6 regulates both basal and glucose-induced Txnip transcription
As glucose is a potent inducer of TXNIP [33], we next examined the influence of SIRT6 deficiency on glucose-induced Txnip transcription. To this end, we isolated islets from overnight fasted mice and cultured them in serum-free medium with 2.5 mmol/l glucose. Note that this concentration is lower than the fasting glucose concentration in vivo (~5 mmol/l). Interestingly, we found that in this condition, Txnip mRNA levels in islets isolated from 3-month-old Sirt6 EKO mice were 1.5-fold higher than those in control islets (Fig. 6a), suggesting that there was a basal upregulation of TXNIP in the mutant islets. Upon incubation of control islets with 16.7 mmol/l glucose for 1 h, Txnip mRNA levels increased 1.5-fold and reached the level found in Sirt6 EKO islets in the basal condition (Fig. 6a). In contrast, high glucose treatment of Sirt6 EKO islets induced a 3.3-fold increase in Txnip mRNA beyond its higher basal level (Fig. 6a). TXNIP immunoblot showed consistent results (Fig. 6b, c). These data support the idea that SIRT6 suppresses both basal and glucose-induced TXNIP expression in normal beta cells. In addition, we found that the acute glucose treatment did not affect upregulated H3K9Ac levels in the mutant islets (Fig. 6b). Together, these findings suggest that loss of Sirt6 leads to impaired regulation of TXNIP in beta cells.

SIRT6 regulates both basal and glucose-induced Txnip transcription. (a) RT-qPCR analysis of Txnip mRNA in islets isolated from 3-month-old control (Sirt6f/f) and Sirt6 EKO mice in response to treatment for 1 h with 16.7 mmol/l glucose (n = 3 or 4). (b, c) Immunoblots (b) and quantification (c) of TXNIP and H3K9Ac levels in islets treated as in (a). Black bars, control mouse islets; grey bars, Sirt6 EKO mouse islets. Data are expressed as means ± SEM. *p < 0.05 and **p < 0.01 for indicated comparisons
SIRT6 directly regulates Txnip by controlling H3K9Ac and H3K56Ac levels
To determine whether Txnip is directly regulated by SIRT6, we examined chromatin occupancy of SIRT6 at the Txnip promoter region by ChIP. SIRT6-positive signals were highly enriched at the transcriptional start site (TSS) of Txnip, compared with the region 2 kb upstream of the Txnip TSS (−2 kb) (Fig. 7a, b). Cis-regulatory elements of the Txnip gene are near its TSS (−169 bp to −73 bp) [34]. Thus, this result indicates that SIRT6 is mainly located in the active transcription region of its target, consistent with a previous SIRT6 ChIP-seq study [35].

SIRT6 regulates TXNIP through H3K9Ac and H3K56Ac deacetylation. (a) ChIP assay of SIRT6 at 2 kb upstream of TSS (−2 kb) and at the TSS of the Txnip gene. Islets pooled from four or five mice were used, the result was the average of three experiments; error bars represent SEM. (b) ChIP assay of IgG at −2 kb and the TSS of the Txnip gene. (c–e) Representative immunoblots (c) and quantification (d, e) of H3K9Ac and H3K56Ac in islets from 3-month-old control (Ctrl, Sirt6f/f) and Sirt6 EKO mice (n = 6). (f, g) Representative immunoblots (f) and quantification (g) of H3K27Ac in islets from 3-month-old control and Sirt6 EKO mice (n = 6). (h–j) ChIP assays of H3K9Ac (h), H3K56Ac (i) and Ser5-polymerase (Pol) II (j) at −2 kb and the TSS of the Txnip gene in islets isolated from control and Sirt6 EKO mice. Three biological repeats were used for (d) and (e); for (f), islets pooled from four or five mice were used. The result was the average of three experiments; error bars represent SEM. Black bars, control mouse islets; grey bars, Sirt6 EKO mouse islets. Data are expressed as means ± SEM, unless stated otherwise. *p < 0.05 and **p < 0.01 for indicated comparisons
To further clarify how SIRT6 regulates Txnip expression, we analysed several SIRT6-catalysed histone H3 modifications by immunoblot [14]. Sirt6 EKO mouse islets expressed a significantly higher level of H3K9Ac and H3K56Ac compared with control mouse islets (Fig. 7c–e). In contrast, the level of another histone H3 modification, H3K27Ac, which has been linked to SIRT6 deacetylation activity in vitro [14], was similar in control and EKO islets (Fig. 7f, g). These results show that loss of SIRT6 leads to an aberrant global increase in H3K9Ac and H3K56Ac in beta cells.
Next, we compared H3K9Ac and H3K56Ac ChIP signals at the local promoter regions of Txnip in islets isolated from 3-month-old control and Sirt6 EKO mice under normal feeding. Consistent with the reported role of H3K9Ac and H3K56Ac in transcriptional activation and our earlier SIRT6 ChIP data, we detected significant enrichment of both markers at the TSS of the Txnip gene in the control islets. After loss of SIRT6, the H3K9Ac level at the TSS was further elevated around twofold, whereas it was unaltered at −2 kb (Fig. 7h). In contrast, the H3K56Ac level was upregulated about twofold at −2 kb but not at the TSS of Txnip (Fig. 7i). These results suggest that SIRT6 deficiency resulted in a more open chromatin state around the Txnip promoter region. Since we did not find that SIRT6 was present at the −2 kb region of the Txnip gene (Fig. 7a), it is currently unclear whether this inconsistency was due to low ChIP signal of SIRT6 when chromatins were prepared from a limited number of cells [23]. To further confirm the influence of these changes on Txnip gene transcription, we performed phospho-Ser5-RNA polymerase II ChIP. As expected, active RNA polymerase II intensity in the mutant islets was twofold higher than in control islets at both the TSS and −2 kb (Fig. 7j). Taken together, these findings suggest that SIRT6 directly suppresses Txnip expression in beta cells through deacetylation of H3K9 and H3K56 at the promoter region of Txnip.
Long-term deletion of Sirt6 in beta cells induces susceptibility to STZ
While investigating the impact of Sirt6 knockout on H3 lysine acetylation, we serendipitously found that the increased H3K9Ac and H3K56Ac levels in islets from Sirt6 BKO mice were more evident when they were from mice at a much later time point post-tamoxifen treatment (ESM Fig. 6a). This was correlated with an increase in Txnip mRNA level (ESM Fig. 6b). The time-dependent increase in H3K9Ac, H3K56Ac and TXNIP levels post-tamoxifen treatment or with age were reproduced in both the BKO and EKO model (ESM Fig. 6c and Fig. 8a), excluding the possibility that this was due to prolonged tamoxifen-induced gene recombination, as reported previously [36]. Together, these results suggest that loss of Sirt6 leads to a progressive accumulation of H3K9Ac and H3K56Ac and sustained upregulation of TXNIP in beta cells, which is likely to be the molecular basis of the time-dependent worsening of glucose intolerance in the SIRT6-deficient mice.

Long-term deletion of Sirt6 in beta cells induces susceptibility to STZ. (a) Immunoblots of TXNIP, H3K9Ac and H3K56Ac in islets isolated from 2-month-old and 7-month-old control (Ctrl, Sirt6f/f) and Sirt6 EKO mice. (b) Representative H&E staining of pancreases from 11-month-old control and Sirt6 EKO mice. Scale bar, 1 mm. (c) Quantification of the mass of the islet area in (b). (d) Blood glucose levels in 2-month-old male control and Sirt6 EKO mice after a single STZ treatment (75 mg/kg). (e) Plasma insulin levels in 2-month-old male control and Sirt6 EKO mice 2 days after a single STZ treatment. (f) Blood glucose levels in 7-month-old control and Sirt6 EKO mice after a single STZ treatment (75 mg/kg) (n = 6). (g) Plasma insulin levels of 7-month-old control and Sirt6 EKO mice 2 days after a single STZ treatment. (h) Immunostaining of pancreases from 7-month-old control and Sirt6 EKO mice 2 days after a single STZ treatment. Green, anti-insulin; red, anti-cleaved caspase 3; blue, DAPI. Scale bar, 50 μm. (i) Representative H&E staining and immunostaining of pancreases from 7-month-old control and Sirt6 EKO mice 4 days after a single STZ treatment. Green, anti-insulin; red, anti-glucagon; blue, DAPI. Scale bar, 50 μm. (j) Quantification of the insulin-positive area of 7-month-old control and Sirt6 EKO mice 4 days after a single STZ treatment. Black bars and circles, control mice; grey bars and squares, Sirt6 EKO mice. Data are expressed as means ± SEM (n = 3 or 4, unless stated otherwise). *p < 0.05 and **p < 0.01 vs control condition or for indicated comparisons
As TXNIP expression is associated with beta cell apoptosis [28], we next examined whether TXNIP accumulation affects the viability of SIRT6-deficient beta cells, especially in older Sirt6-knockout mice. Of note, the islet mass of 11-month-old Sir6 EKO mice did not differ appreciably from that of control mice (Fig. 8b, c), indicating that upregulation of TXNIP in SIRT6-deficient beta cells did not cause spontaneous cell death. Next, we tested whether SIRT6-deficient beta cells are sensitive to STZ [37], a genotoxic agent. As STZ selectively acts on beta cells, we directly treated the mice with STZ. Interestingly, the older SIRT6-deficient mice (>6 month old) showed a more dramatic response to STZ treatment than the older control mice or younger mice (2 months old) in both EKO and BKO models (Fig. 8d–g and ESM Fig. 6d, e). Upon a single injection of STZ (75 mg/kg for Sirt6 EKO mice and 140 mg/kg for Sirt6 BKO mice), all older SIRT6-deficient mice displayed hyperglycaemia (blood glucose level >16 mmol/l) after 2 days, due to insulin insufficiency (Fig. 8g). Consistent with this finding, a massive beta cell loss was observed within 2 days of STZ treatment of older SIRT6-deficient mice, as demonstrated by cleaved caspase 3, H&E and insulin staining (Fig. 8h–j). These findings suggest that long-term deficiency of SIRT6 in beta cells affects beta cell survival, which is correlated with sustained upregulation of TXNIP.
Discussion
In this study, we reported that SIRT6 is not required for beta cell development but is essential for insulin secretion function of beta cells. Our unbiased gene expression profiling studies identified Txnip as a SIRT6 target. Concurrently, inhibition of TXNIP by verapamil could partially rescue the insulin secretion defect in Sirt6-knockout mice. In addition, we showed that SIRT6 directly suppresses Txnip expression by catalysing the deacetylation of H3K9 and H3K56 at the Txnip promoter region. Finally, we uncovered an unanticipated time-dependent increase in H3K9Ac, H3K56Ac and TXNIP levels in SIRT6 null beta cells, which correlates with a worsened insulin secretion defect and leads to increased susceptibility to the diabetogenic agent STZ. These findings clearly highlight the important roles of SIRT6 histone deacetylation activity in supporting beta cell function and viability (Fig. 9).

Diagram depicting a proposed role for SIRT6 supporting beta cell function and survival through inhibition of Txnip transcription. Ac, acetylation; Pol II, RNA polymerase II; TF, transcription factor
Glucose-stimulated insulin secretion in beta cells is a complex process that requires coupling of cellular metabolism with ATP-sensitive potassium channels and subsequent activation of voltage-dependent calcium channel-mediated Ca2+ influx [1]. Xiong et al observed a decrease in cellular ATP levels, accumulation of damaged mitochondria, reduction in electron transport chain protein levels and impaired Ca2+ influx in SIRT6-deficient beta cells [25]. However, they did not identify the molecular mechanism linking these phenotypic findings with loss of SIRT6. We found that expression of TXNIP is upregulated in Sirt6 null mouse islets and that reducing TXNIP level by verapamil was able to partially restore the insulin secretion and largely rescue the glucose intolerance in mice. In agreement with our findings, TXNIP expression is negatively correlated with glucose-stimulated insulin secretion capacity in beta cells [29,30,31]. Importantly, Yoshihara et al demonstrated that genetic disruption of Txnip enhanced glucose-stimulated insulin secretion, whereas overexpression of TXNIP in INS1 cells in the first 48 h attenuated this process, likely through an effect on mitochondrial ATP production [29]. Although Ucp2 was identified as a TXNIP transcriptional target in their study and presumably caused the reduction in cellular ATP levels [29], we did not detect significant changes in Ucp2 mRNA levels in our model. In addition, Song et al showed that loss of SIRT6 in beta cells stimulated transcriptional repression activity of forkhead box O1 and led to downregulation of its two targets, Pdx1 and Glut2 (also known as Slc2a2) [26]. However, expression levels of Pdx1 and Glut2 were not changed in either of our models. These discrepancies could be attributed to differences in mouse genetic background and methodologies used in these molecular characterisations. However, it is still unclear how TXNIP affects insulin secretion in our model.
Verapamil is a calcium antagonist and has been shown to inhibit insulin release in islets isolated from rodents [38, 39]. However, verapamil treatment has no significant effect on either glucose tolerance or insulin secretion in vivo [39, 40], consistent with our observations. Thus, our finding that oral verapamil treatment improved glucose tolerance and insulin secretion is likely mediated by Txnip suppression. As verapamil inhibits Txnip through an indirect calcium-dependent calcineurin pathway [32], genetically or pharmacologically attenuating TXNIP activity would be necessary for future studies to more precisely dissect the contribution of TXNIP to the SIRT6-deficient mouse phenotype.
TXNIP mainly functions as an inhibitor of thioredoxin by antagonising its scavenger effect on ROS [28]. Thus, upregulation of TXNIP is implicated in glucose-, endoplasmic reticulum (ER) stress- and STZ-induced cell death [28]. We found that upregulation of TXNIP in SIRT6-deficient beta cells did not cause spontaneous cell death. Consistent with this finding, SIRT6 deficiency did not induce ROS accumulation in islets (data not shown). However, several genes in the oxidation–reduction pathway including Nqo1, Gstm7 and Dusp26 were significantly upregulated in the islets from Sirt6 EKO but not Sirt6 BKO mice (GSE104161). Whether this is a compensatory response to TXNIP-induced oxidative stress or the direct consequence of SIRT6 deficiency requires further investigation. We speculate that the progressive upregulation of TXNIP in SIRT6-deficient beta cells induces a gradual increase in ROS that the cells can cope with by upregulating the genes in the oxidation–reduction pathways.
Genetic background of mice is a contributing factor to phenotypic variation, including metabolic traits and the response to STZ [37, 41]. However, the severity and time-dependent progression of the functional defects in our models, as well as the similarity between our EKO and BKO model, strongly suggest that these phenotypes are independent of mouse genetic background. Multiple studies have reported that the transgene-driven human growth hormone expression in the MIP-CreER mouse strain has a profound effect on gene expression and beta cell integrity [19, 42, 43]. Specifically, islets in MIP1-CreER mice are less sensitive to the cytotoxic effect of STZ [19]. To avoid this potential confounding factor, mice carrying the Cre transgene were used as controls throughout the BKO study (ESM Fig. 4).
In conclusion, our study demonstrates that SIRT6 is required for glucose-stimulated insulin secretion and cell survival in beta cells, partially via suppression of Txnip transcription. As defective insulin secretion and loss of beta cell mass are the main culprits of type 2 diabetes [1], our findings raise the possibility that preserving SIRT6 activity and expression could be beneficial in improving beta cell function and maintaining beta cell mass in individuals with impaired glucose tolerance or type 2 diabetes.
Data availability
Sequence data have been deposited in the NIH Gene Expression Omnibus (GEO) with the accession code GSE104161. All other remaining data are available within the article and ESM files or are available from the authors on request.
Abbreviations
- BKO:
-
Beta cell-specific knockout
- ChIP:
-
Chromatin immunoprecipitation
- EKO:
-
Endocrine pancreas-specific knockout
- HAT:
-
Histone acetyltransferase
- HDAC:
-
Histone deacetylase
- H3K9Ac:
-
Acetylation of histone H3 lysine residue at 9
- H3K18Ac:
-
Acetylation of histone H3 lysine residue at 18
- H3K56Ac:
-
Acetylation of histone H3 lysine residue at 56
- MIP1-CreER:
-
Mouse insulin promoter 1-driven, inducible CreERT transgenic line
- NGN3:
-
Neurogenin 3
- NIH:
-
National Institutes of Health
- ROS:
-
Reactive oxygen species
- SIRT6:
-
Sirtuin 6
- STZ:
-
Streptozotocin
- TSS:
-
Transcriptional start site
- TXNIP:
-
Thioredoxin-interacting protein
References
Ashcroft FM, Rorsman P (2012) Diabetes mellitus and the beta cell: the last ten years. Cell 148:1160–1171
Zaccardi F, Webb DR, Yates T, Davies MJ (2016) Pathophysiology of type 1 and type 2 diabetes mellitus: a 90-year perspective. Postgrad Med J 92:63–69
Xie R, Carrano AC, Sander M (2015) A systems view of epigenetic networks regulating pancreas development and beta-cell function. Wiley Interdiscip Rev Syst Biol Med 7:1–11
Bernstein D, Golson ML, Kaestner KH (2017) Epigenetic control of β-cell function and failure. Diabetes Res Clin Pract 123:24–36
Arguelles AO, Meruvu S, Bowman JD, Choudhury M (2016) Are epigenetic drugs for diabetes and obesity at our door step? Drug Discov Today 21:499–509
Haberland M, Montgomery RL, Olson EN (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10:32–42
Lee KK, Workman JL (2007) Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol 8:284–295
Remsberg JR, Ediger BN, Ho WY et al (2017) Deletion of histone deacetylase 3 in adult beta cells improves glucose tolerance via increased insulin secretion. Mol Metab 6:30–37
Chang HC, Guarente L (2014) SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab 25:138–145
Imai S, Guarente L (2014) NAD+ and sirtuins in aging and disease. Trends Cell Biol 24:464–471
Michishita E, McCord RA, Berber E et al (2008) SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 452:492–496
Yang B, Zwaans BM, Eckersdorff M, Lombard DB (2009) The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle 8:2662–2663
Tasselli L, Xi Y, Zheng W et al (2016) SIRT6 deacetylates H3K18ac at pericentric chromatin to prevent mitotic errors and cellular senescence. Nat Struct Mol Biol 23:434–440
Tasselli L, Zheng W, Chua KF (2017) SIRT6: novel mechanisms and links to aging and disease. Trends Endocrinol Metab 28:168–185
Kim HS, Xiao C, Wang RH et al (2010) Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab 12:224–236
Schonhoff SE, Giel-Moloney M, Leiter AB (2004) Neurogenin 3-expressing progenitor cells in the gastrointestinal tract differentiate into both endocrine and non-endocrine cell types. Dev Biol 270:443–454
Wicksteed B, Brissova M, Yan W et al (2010) Conditional gene targeting in mouse pancreatic ss-cells: analysis of ectopic Cre transgene expression in the brain. Diabetes 59:3090–3098
Pan H, Qin K, Guo Z et al (2014) Negative elongation factor controls energy homeostasis in cardiomyocytes. Cell Rep 7:79–85
Oropeza D, Jouvet N, Budry L et al (2015) Phenotypic characterization of MIP-CreERT1Lphi mice with transgene-driven islet expression of human growth hormone. Diabetes 64:3798–3807
Zhang J, Zhang N, Liu M et al (2012) Disruption of growth factor receptor-binding protein 10 in the pancreas enhances beta-cell proliferation and protects mice from streptozotocin-induced beta-cell apoptosis. Diabetes 61:3189–3198
Neuman JC, Truchan NA, Joseph JW, Kimple ME (2014) A method for mouse pancreatic islet isolation and intracellular cAMP determination. J Vis Exp e50374
Benitez CM, Qu K, Sugiyama T et al (2014) An integrated cell purification and genomics strategy reveals multiple regulators of pancreas development. PLoS Genet 10:e1004645
Dahl JA, Collas P (2008) A rapid micro chromatin immunoprecipitation assay (microChIP). Nat Protoc 3:1032–1045
Goodyer WR, Gu X, Liu Y, Bottino R, Crabtree GR, Kim SK (2012) Neonatal β cell development in mice and humans is regulated by calcineurin/NFAT. Dev Cell 23:21–34
Xiong X, Wang G, Tao R et al (2016) Sirtuin 6 regulates glucose-stimulated insulin secretion in mouse pancreatic beta cells. Diabetologia 59:151–160
Song MY, Wang J, Ka SO, Bae EJ, Park BH (2016) Insulin secretion impairment in Sirt6 knockout pancreatic β cells is mediated by suppression of the FoxO1-Pdx1-Glut2 pathway. Sci Rep 6:30321
Ayala JE, Samuel VT, Morton GJ et al (2010) Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis Model Mech 3:525–534
Shalev A (2014) Minireview: thioredoxin-interacting protein: regulation and function in the pancreatic β-cell. Mol Endocrinol 28:1211–1220
Yoshihara E, Fujimoto S, Inagaki N et al (2010) Disruption of TBP-2 ameliorates insulin sensitivity and secretion without affecting obesity. Nat Commun 1:127
Rani S, Mehta JP, Barron N et al (2010) Decreasing Txnip mRNA and protein levels in pancreatic MIN6 cells reduces reactive oxygen species and restores glucose regulated insulin secretion. Cell Physiol Biochem 25:667–674
Luo Y, He F, Hu L et al (2014) Transcription factor Ets1 regulates expression of thioredoxin-interacting protein and inhibits insulin secretion in pancreatic beta-cells. PLoS One 9:e99049
Xu G, Chen J, Jing G, Shalev A (2012) Preventing β-cell loss and diabetes with calcium channel blockers. Diabetes 61:848–856
De Marinis Y, Cai M, Bompada P et al (2016) Epigenetic regulation of the thioredoxin-interacting protein (TXNIP) gene by hyperglycemia in kidney. Kidney Int 89:342–353
Yu FX, Luo Y (2009) Tandem ChoRE and CCAAT motifs and associated factors regulate Txnip expression in response to glucose or adenosine-containing molecules. PLoS One 4:e8397
Etchegaray JP, Chavez L, Huang Y et al (2015) The histone deacetylase SIRT6 controls embryonic stem cell fate via TET-mediated production of 5-hydroxymethylcytosine. Nat Cell Biol 17:545–557
Reinert RB, Kantz J, Misfeldt AA et al (2012) Tamoxifen-induced Cre-loxP recombination is prolonged in pancreatic islets of adult mice. PLoS One 7:e33529
Deeds MC, Anderson JM, Armstrong AS et al (2011) Single dose streptozotocin-induced diabetes: considerations for study design in islet transplantation models. Lab Anim 45:131–140
Devis G, Somers G, Van Obberghen E, Malaisse WJ (1975) Calcium antagonists and islet function. I. Inhibition of insulin release by verapamil. Diabetes 24:247–251
Semple CG, Smith M, Furman BL (1988) Inhibition of glucose-induced insulin secretion by calcium channel blocking drugs in-vitro but not in-vivo in the rat. J Pharm Pharmacol 40:22–26
Semple CG, Thomson JA, Beastall GH, Lorimer AR (1983) Oral verapamil does not affect glucose tolerance in non-diabetics. Br J Clin Pharmacol 15:570–571
Champy MF, Selloum M, Zeitler V et al (2008) Genetic background determines metabolic phenotypes in the mouse. Mamm Genome 19:318–331
Brouwers B, de Faudeur G, Osipovich AB et al (2014) Impaired islet function in commonly used transgenic mouse lines due to human growth hormone minigene expression. Cell Metab 20:979–990
Carboneau BA, Le TD, Dunn JC, Gannon M (2016) Unexpected effects of the MIP-CreER transgene and tamoxifen on β-cell growth in C57Bl6/J male mice. Physiological reports 4:e12863
Acknowledgements
We thank Y. Liu (Department of Cell Systems & Anatomy, Greehey Children’s Cancer Research Institute, University of Texas Health Science Centre at San Antonio, USA) for assistance in analysing the RNA-Seq data and thank C. Cervantes (Department of Pharmacology, University of Texas Health Science Centre at San Antonio, USA) and C. Dong (Biochemistry Molecular Biology, Indiana University, USA) for critical reading of the manuscript.
Funding
PW and KX are CPRIT Scholars and are supported by the Cancer Prevention and Research Institute of Texas. This work was supported by grants from the NIH (R01-DK80157 and R01-DK089229) and the American Diabetes Association to NM. This research also was supported by the San Antonio Nathan Shock Centre of Excellence in Aging Biology (P30 AG013319) and San Antonio Claude D. Pepper Older Americans Independence Centre (P30 AG044271).
Author information
Authors and Affiliations
Contributions
KQ and PW were responsible for designing the experiments. KQ, NZ, ZZ, MN, ZXZ and JL were responsible for acquisition of data. KQ, NZ, KX, NM and PW analysed and interpreted data. KQ drafted the manuscript. All authors critically revised the manuscript and approved the final version. PW is the guarantor of this work.
Corresponding author
Ethics declarations
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Electronic supplementary material
ESM
(PDF 769 kb)
Rights and permissions
About this article

Cite this article
Qin, K., Zhang, N., Zhang, Z. et al. SIRT6-mediated transcriptional suppression of Txnip is critical for pancreatic beta cell function and survival in mice. Diabetologia 61, 906–918 (2018). https://doi.org/10.1007/s00125-017-4542-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-017-4542-6