
Abstract
Aims/hypothesis
IL-6 was recently shown to control alpha cell expansion. As beta cells expand following partial pancreatic-duct ligation (PDL) in adult mice, we investigated whether PDL also causes alpha cells to expand and whether IL-6 signalling is involved. As alpha cells can reprogramme to beta cells in a number of beta cell (re)generation models, we examined whether this phenomenon also exists in PDL pancreas.
Methods
Total alpha cell volume, alpha cell size and total glucagon content were evaluated in equivalent portions of PDL- and sham-operated mouse pancreases. Proliferation of glucagon+ cells was assessed by expression of the proliferation marker Ki67. Inter-conversions between alpha and beta cells were monitored in transgenic mice with conditional cell-type-specific labelling. The role of IL-6 in regulating alpha cell proliferation was evaluated by in situ delivery of an IL-6-inactivating antibody.
Results
In response to PDL surgery, alpha cell volume in the ligated tissue was increased threefold, glucagon content fivefold and alpha cell size by 10%. Activation of alpha cell proliferation in PDL pancreas required IL-6 signalling. A minor fraction of alpha cells derived from beta cells, whereas no evidence for alpha to beta cell conversion was obtained.
Conclusions/interpretation
In PDL-injured adult mouse pancreas, new alpha cells are generated mainly by IL-6-dependent self-duplication and seldom by reprogramming of beta cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The generation of beta cells is central to the development of novel therapies for diabetes. In most animal models with increased beta cell formation in response to injury or increased physiological need, proliferation appears a major mechanism of beta cell expansion [1–3]. However, acinar cells can be genetically reprogrammed into beta cells in vivo [4], and partial pancreatic-duct ligation (PDL) injury generates cells positive for Ngn3 (also known as Neurog3) that can differentiate to hormone+ islet cells ex vivo [5] and in vivo, while Ptf1a + acinar cells are a source of Ngn3 + duct cells and beta cells in the injured pancreas of hyperglycaemic mice [6].
Alpha cells can be directly reprogrammed to beta cells following ectopic expression of the developmental transcription factor Pax4 [7, 8] and following extreme ablation of beta cells [9]. Under conditions of beta cell injury or stress, alpha cells play an active role either by glucagon-like peptide-1 (GLP-1) production or by beta to alpha cell conversion [10, 11]. Thus, the fate of alpha cells appears linked to that of beta cells in a number of experimental models for beta cell (re)generation. In mice with PDL we reported expansion of the beta cell mass that could not be entirely explained by increased beta cell proliferation [5, 12].
Lineage tracing studies using Sox9 or Ptf1a as non-endocrine cell markers have so far suggested only limited neogenesis of beta cells from non-endocrine cells in PDL pancreas [6, 13]. Beta cells might in part also derive from endocrine non-beta cells in PDL pancreas. An early study reported simultaneous alpha and beta cell mass expansion in PDL pancreas [14]. Others, however, reported absence of alpha cell growth and beta cell neogenesis [15, 16]. Moreover, while IL-6 is upregulated in PDL pancreas [17], it is unknown whether this cytokine also controls the alpha cell number as was shown for high-fat feeding [18]. Thus, the expansion of alpha cells and their relationship to beta cells in PDL pancreas remains elusive. The present study shows that alpha cells expand, mainly by alpha cell proliferation but also by beta cell reprogramming, following PDL injury.
Methods
Mouse manipulation
All mouse experiments were performed according to the guidelines of our institutional Ethical Committee for Animal Experiments and national guidelines and regulations. BALB/c mice were obtained from Janvier Labs (St Berthevin, France). Transgenic mice were of mixed background and bred in house. Throughout this study, PDL or sham surgery was performed on 8-week-old male mice as described by Xu et al [5]. Collection of mouse serum and measurement of glycaemia were performed as previously described by Grouwels et al [19]. Anti-IL-6 monoclonal antibody (HM100405, Hycult Biotech, Uden, the Netherlands) was injected in the PDL-tail pancreas at day 6 after surgery to inactivate endogenously produced IL-6 [20], with a rat IgG1 antibody isotype control (400402, BioLegend, San Diego, CA, USA). Conversely, recombinant IL-6 (R&D Systems Europe, Abingdon, UK) was injected to examine alpha cell response to this cytokine in the context of PDL.
Cell tracing
Tamoxifen (Tam; T5648, Sigma-Aldrich, Diegem, Belgium) was dissolved at 54 mmol/l in corn oil (C8267, Sigma-Aldrich). For beta cell tracing in PDL studies, RIP CreERT/R26R YFP adult mice were given a total of 54 μmol Tam s. c. in five doses (each 10.8 μmol) over a 2 week period. For alpha cell tracing, triple transgenic Gcg rtTA/TetO Cre/R26R YFP mice [9, 21] were induced for permanent expression of the yellow flourescent protein (YFP) reporter gene by administration of doxycycline (Dox, doxycycline hyclate, 44577 Sigma) via the drinking water (2.2 mmol/l) for 2 weeks.
Alpha cell size, volume and glucagon
The measurement of alpha cell volume (mm3) in PDL or sham tail pancreas was performed as described previously. Analysis of each organ was based on a large number of 5 μm sections, equally spaced and spanning the whole tissue, that together represented 10% of the total volume [12]. The alpha cell size distribution in PDL and sham tail pancreas was documented by area measurement on 100 individual glucagon+ E-cadherin+ cells using NIH ImageJ software (US National Institutes of Health, Bethesda, MD, USA). For determination of alpha/beta cell ratio, 3,150 ± 100 insulin+ cells and 1,000 ± 200 glucagon+ cells (found in 50–100 islets) were counted in each sham-tail or PDL-tail pancreas. Pancreatic glucagon content and serum glucagon levels were measured as previously described [22]. Briefly, the complete sham or PDL pancreatic tail tissue was homogenised in 2 mol/l acetic acid containing 0.25% (wt/vol.) BSA. Glucagon was assayed in the tissue extracts or sera using an RIA kit (Linco Research, St Charles, MO, USA). Islets were isolated from collagenase-treated pancreas tails by handpicking. For secretion experiments, 30 randomly chosen islets were incubated per tube for 1 h in Ham F10 medium (Invitrogen, Merelbeke, Belgium) containing 2 mmol/l glutamine and Ca2+, 1% (wt/vol.) charcoal-treated BSA, and indicated supplements. Total and released glucagon was assayed using the RIA.
RNA analysis
Total RNA was isolated from FACS-sorted beta cells (RNeasy Micro Kit, Qiagen, Venlo, the Netherlands). Quantitative PCR reactions were performed with cDNA corresponding to 6 ng RNA, and using mouse-specific primers and probes for cyclophilin A (Mm02342429), Oct4 (also known as Pou5f1; Mm03053917), L-myc (also known as Mycl; Mm01181427-m1), Pdx1 (Mm00435565_m1), Nkx6.1 (also known as Nkx6-1; Mm.PT.56a.28634562), Neurod1 (Mm01280117-m1), Mafa (Mm00845209-s1), Mafb (Mm00627481-s1), Brn4 (also known as Pou3f4; Mm00447171-s1), Pcsk1 (Mm00479023-m1), Pcsk2 (Mm00500981-m1), Gck (Mm00439129-m1), Foxo1 (Mm00490672), Arx1 (Mm00545903-m1), Ins1 (Mm01950294-s1), Ins2 (Mm00731595-gH) and Gcg (Mm00801712-m1) from Applied Biosystems (Carlsbad, CA, USA) and for Tbp (Mm.PT.39a.22214839), Glut2 (also known as Slc2a2; Mm.PT.45.16276653) and Nanog (Mm.PT.56a.23510265) from Integrated DNA Technologies (Coralville, IA, USA). The mRNA expression data were normalised to cyclophilin A or Tbp.
Immunolocalisation and histochemical procedures
Samples for immunohistochemistry (IHC) were fixed in 4% formaldehyde overnight and embedded in paraffin. Paraffin sections (4–5 μm) were incubated with antisera specific for insulin (1/3,000, guinea pig, generated at the Diabetes Research Center, Brussels, Belgium), glucagon (1/3,000, rabbit), v-maf musculoaponeurotic fibrosarcoma oncogene family, protein B (avian) (MAFB) (1/500, rabbit, Bethyl Laboratories, Montgomery, TX, USA), pro-hormone convertases 1/3 and 2 (PC1/3 and PC2) (1/250, rabbit, Millipore, Temecula, CA, USA), Ki67 (1/1,000, rabbit, Novocastra reagents, Leica Biosystems, Diegem, Belgium), CK19 (1/100, rat, Developmental Studies Hybridoma Bank, University of Iowa, IA, USA), green fluorescent protein (GFP) (1/200, goat, Abcam, Cambridge, UK) or E-cadherin (1/50, mouse, BD Biosciences, Erembodegem, Belgium). Secondary antibodies for the detection of primary guinea pig, rabbit, goat or rat antibodies were labelled by fluorescence (Cy3, Cy2, Cy5) (Jackson ImmunoResearch Labs, Suffolk, UK). Nuclei were labelled by Hoechst 33342 (4 mg/ml, Sigma-Aldrich). Signal transducer and activator of transcription 3 (STAT3) activation was monitored by immunofluorescence staining for p-STAT3 (Y705, 1/250, rabbit, Abcam). This was a read out for IL-6-receptor activation in PDL vs sham, or in response to IL-6 or anti-IL-6 antibody injection in PDL (see mouse manipulation).
Image analysis
Images were acquired with normal or confocal scanning microscopy (normal: Olympus, Aartselaar, Belgium, BX61 with Hamamatsu C10600 ORKA-R2 camera; confocal scanning: Leica, Aartselaar, Belgium, TCS-SP and Zeiss [Carl Zeiss, Zaventem, Belgium] LSM710 NLO).
Ki67- or YFP-expressing cells were counted separately and localisation within insulin+ (beta) cells or glucagon+ (alpha) cells was checked individually. At least 400 alpha cells per sample were counted. Images were analysed using SmartCapture 3 (version 3.0.8, Cambridge, UK), NIH ImageJ (US National Institutes of Health) and Photoshop CS (version 1.3.1, Adobe, San Jose, CA, USA). Confocal images were processed using Volocity LE (PerkinElmer, Zaventem, Belgium) and Zeiss Zen software (Carl Zeiss).
Statistics
Data are expressed as the mean ± SEM of at least three independent experiments. Differences between data were analysed by unpaired two-tailed Student’s t test or two-way Anova with Dunnett’s test, and were considered statistically significant when p < 0.05.
Results
Partial duct ligation generated alpha cells in adult mice
The response of alpha cells to 14 days of PDL was determined by measuring alpha cell volume in the duct-ligated part of the pancreas (‘PDL tail’) and in sham-operated control pancreas (sham tail). This was done by quantification of the glucagon+ cell area in a semi-automated manner on 5 μm thick sections that were 45 μm apart, spanning the whole tissue and thus corresponding to 10% of the total organ volume. The alpha cell volume in the PDL tail increased threefold compared with sham tail (Fig. 1a). The sizes of individual alpha cells were normally distributed (ESM Fig. 1), with an average increase of 10% in PDL pancreas (Fig. 1b). Thus, the increased alpha cell volume in PDL probably results from an increased number rather than larger size. The number of glucagon+ cells relative to insulin+ cells was increased 1.5-fold in PDL tail compared with sham tail (0.37 ± 0.03 vs 0.26 ± 0.04, respectively) (Fig. 1c). In addition, total glucagon content was fivefold higher in complete pancreas tails from PDL-operated compared with sham-operated mice (126 ± 10 vs 23.4 ± 1.3 pmol, respectively) (Fig. 1d). These data suggest that new alpha cells are generated in the 2 weeks that follow PDL surgery in adult mice.

PDL induces alpha cell expansion. Eight-week-old male BALB/c mice underwent PDL or sham surgery (2 weeks). (a) PDL and sham tail total alpha cell volume (mm3) and (b) alpha cell size (μm2) (n = 3; * p < 0.05 and ** p < 0.01, PDL vs sham tail). (c) Alpha/beta cell ratio and (d) total glucagon content (pmol) in sham tail and PDL-tail pancreas (n = 4–6, **p < 0.01 and ***p < 0.001). (e) The plasma glucagon (pmol/l) and (f) blood glucose (mmol/l) levels in day 14 sham and PDL mice with overnight fasting (n = 6). Alpha cell size (see also ESM Fig. 1) and alpha/beta cell ratio were determined as described in Methods
Given the absolute increases in alpha cell volume and glucagon content and the relative increase in the number of glucagon+ to insulin+ cells in PDL pancreas, metabolic variables were examined. No differences in fasting plasma glucagon (Fig. 1e), blood glucose levels (Fig. 1f) or body weight (ESM Table 1) were detected between PDL-operated and sham-operated mice. Freshly isolated islets from PDL tail pancreas were less responsive to secretory stimuli than islets from sham tail pancreas (ESM Fig. 2a), and contained more glucagon (0.318 ± 0.015 pmol/islet [PDL] vs 0.101 ± 0.016 pmol/islet [sham], n = 12, p < 0.0001). There was no difference in expression of PC1 or PC2 between alpha cells in the tail parts of PDL and sham pancreas (ESM Fig. 2b, c).
Proliferation was the major mechanism of alpha cell formation in PDL
To determine the cellular basis of alpha cell expansion in PDL, cell cycle activity was studied by immunofluorescence for glucagon and the proliferation marker Ki67. A threefold increase in the percentage of Ki67+ alpha cells was observed in PDL vs sham tail, both at day 7 (2.45 ± 0.22% vs 0.72 ± 0.17%, respectively) and day 14 (1.29 ± 0.17% vs 0.43 ± 0.05%) after surgery (Fig. 2a, b), indicative of increased alpha cell cycling. To investigate whether, in addition to alpha cell proliferation, new alpha cells were generated by differentiation from non-alpha cells, a pulse–chase experiment was performed in Gcg rtTA/TetO Cre/R26R YFP mice. These mice allow glucagon-driven expression of the reverse tetracycline transactivator (rtTA) resulting in the expression of YFP reporter protein when Dox is administered. Cre recombinase expression and recombination of the R26R YFP locus occurred only in alpha cells and in the presence of Dox [9]. In control mice without Dox treatment, no YFP expression was observed in alpha or non-alpha cells (ESM Fig. 3). At 4 weeks of age, experimental mice received Dox via the drinking water for 2 weeks to label pre-existing alpha cells. After 2 weeks of Dox washout, PDL or sham operation was performed and the pancreas was studied 2 weeks after surgery. Of the glucagon+ cells 89% and 91% expressed YFP in sham and PDL-tail pancreas, respectively (Fig. 2c). Thus, there was no detectable influx of YFP− glucagon+ cells into the pool of pre-existing YFP+ alpha cells in PDL pancreas and therefore proliferation rather than neogenesis is the main mechanism of alpha cell expansion in PDL pancreas.

Alpha cell expansion after PDL is mainly by replication. (a) Immunofluorescence staining for glucagon, Ki67 and DNA in paraffin sections of sham and PDL-tail pancreas. Scale bar, 20 μm. (b) Percentage of alpha cells that expressed Ki67 at day 7 (n = 3; **p < 0.01) and day 14 (n = 4–6; **p < 0.01). White bars, sham tail; black bars, PDL tail. (c) Percentage of glucagon+ alpha cells labelled by YFP in sham and PDL tails of Dox-treated Gcg rtTA /Tet Cre /R26 YFP mice, 2 weeks after surgery (n = 3–6). See also ESM Fig. 3
Alpha cell proliferation is regulated by interleukin-6 in PDL
As IL-6-receptor (IL-6R) is highly expressed in alpha cells, IL-6 can stimulate alpha cell mass expansion [18] and IL-6 production is increased in PDL [17, 23], we investigated whether IL-6 could be responsible for the increase in alpha cell proliferation in PDL pancreas. Phosphorylation of STAT3, functionally coupled to activation of IL-6R, was increased in alpha cells at day 7 following PDL (from 0.6 ± 0.3% in sham to 6.7 ± 1.6% in PDL) (Fig. 3a, b). Importantly, injection of IL-6-inactivating anti-IL-6 antibody in PDL pancreas on day 6 decreased STAT3 activation in alpha cells from 8.4 ± 0.9% to 1.5 ± 0.3% (Fig. 3c, d) and alpha cell proliferation from 3.3 ± 0.3% to 0.95 ± 0.15% (Fig. 3e) compared with PDL pancreas injected with isotype control antibody. These results suggest that endogenously produced IL-6 activates STAT3 and stimulates alpha cell proliferation in PDL. The potential of exogenous IL-6 to further stimulate alpha cell proliferation was examined by intra-PDL-tail injection of recombinant IL-6 on day 6. This exacerbated STAT3 activation in alpha cells 1 h after injection (from 12 ± 4% to 66 ± 5%) compared with vehicle injection (Fig. 3f, g). However, no further increase in alpha cell proliferation was detected on day 7 (Fig. 3h). IL-6 thus activates STAT3 and is required but is not solely sufficient for alpha cell proliferation in PDL.

IL-6 activates STAT3 and proliferation in alpha cells. Eight-week-old male BALB/c mice underwent sham or PDL surgery, and pancreas tail samples were studied at day 7. (a, c) Immunostaining for glucagon (green), p-STAT3 (red) and DNA (blue). The p-STAT3+ glucagon+ cells are indicated (arrows). (b) PDL surgery increased p-STAT3 in alpha cells at day 7 compared with sham surgery (n = 4, **p < 0.01). (c, d, e) Intra-PDL-tail injection of IL-6-inactivating antibody (1.5 μg) or isotype control antibody (1.5 μg) on day 6 post-PDL and analysis of PDL-tail alpha cells on day 7. (c) Immunofluorescence staining for p-STAT3, glucagon and DNA. The p-STAT3+ glucagon+ cells are indicated (arrows) (d) Anti-IL-6 antibody decreased the STAT3 phosphorylation in PDL-tail alpha cells (n = 4; *p < 0.05). (e) Anti-IL-6 antibody decreased expression of proliferation marker Ki67 in PDL-tail alpha cells (n = 4; *p < 0.05). (f, g, h) Intra-PDL-tail injection of recombinant IL-6 (3.0 μg) or an equivalent volume of vehicle on day 6 post-PDL. (f) STAT3 phosphorylation (red) in alpha cells (glucagon, green) 1 h after IL-6 or vehicle injection. p-STAT3+ glucagon+ cells are indicated (arrows). (g) IL-6 increased STAT3 phosphorylation in PDL-tail alpha cells (n = 4, *p < 0.05). (h) The percentage of Ki67+ alpha cells was not changed 24 h after intra-PDL-tail injection of IL-6 (n = 4). At least 800 alpha cells were analysed in each tissue sample. Scale bar, 20 μm. Ab, antibody
Beta cells do not derive from alpha cells but some alpha cells derive from beta cells in PDL pancreas
As mature alpha cells can generate beta cells [7, 9] and we showed that beta cells can be derived from non-beta cells in PDL pancreas [12], alpha cells were genetically labelled and traced. The 4.5 week old Gcg rtTA/Tet Cre/R26R YFP mice underwent Dox labelling, a 2 week washout and PDL surgery. Immunofluorescence staining for glucagon, insulin and YFP was done on three PDL pancreases at day 14 post-surgery (Fig. 4a). In total, 145 islets were examined (1,900 alpha cells in total), and while 92% ± 2% of the alpha cells were YFP+ (Fig. 4a), only two insulin+ cells were YFP+, indicating little evidence for alpha to beta cell conversion in PDL pancreas.

Beta cells derive less frequently from pre-existing alpha cells than alpha cells derive from beta cells. (a) Gcg rtT/TetO Cre /R26R YFP mice pre-labelled with Dox (2 weeks, via drinking water) underwent washout (2 weeks) and PDL when 8 weeks old. Immunostaining for glucagon (magenta), YFP (green), insulin (red) and DNA (blue) on PDL-tail tissue at 14 days post-PDL. Numerous YFP-labelled glucagon+ (alpha) cells were observed, but only very few YFP+ beta cells were encountered in the PDL tails (n = 3). (b, c, d) RIP CreERT /R26R YFP mice were administered Tam (see Methods). After 2 weeks of Tam washout, PDL or sham surgery was performed, and tail pancreas was studied 14 days later. (b) Immunostaining of PDL-tail tissue for glucagon (magenta), insulin (red), YFP (green) and DNA (blue). Some alpha cells were YFP+ and expressed insulin (glucagon+ YFP+ INS+ cells, white arrow), while other YFP+ alpha cells were insulin– (glucagon+ YFP+ INS− cells, yellow arrows). See also ESM Fig. 4. (c) Beta cell YFP labelling efficiency in sham and PDL tail (n = 3; *p < 0.05). (d) PDL increased the percentage of alpha cells that were YFP+ and probably derived from pre-labelled (YFP+) beta cells in these mice (n = 3; *p < 0.05)
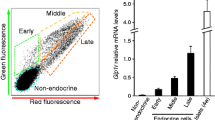
As we occasionally observed glucagon+ insulin+ cells, we genetically labelled and traced beta cells to investigate whether they could be reprogrammed to alpha-like cells in PDL. The 4.5-week-old RIP CreERT ;R26R YFP mice were treated with Tam and underwent Tam washout for 2 weeks and then PDL or sham surgery. Two weeks later, in PDL-tail pancreas 71 ± 4% of beta cells and 5 ± 1% of glucagon+ cells were YFP+ compared with, respectively, 91 ± 1% and 2.0 ± 0.1% in sham tail pancreas (Fig. 4b, c, d), indicating that PDL supported the derivation of glucagon+ cells from YFP-marked beta cells. Simultaneous expression of YFP and glucagon in the same cells was confirmed by confocal microscopy (ESM Fig. 4) and about half of the YFP+ glucagon+ cells in PDL pancreas still contained insulin (Fig. 4b). Thus, in PDL pancreas, beta cells can be converted more frequently into alpha-like or mixed hormone+ cells than alpha into beta cells. Most of the YFP+ glucagon+ cells (96 ± 3%, n = 3) in PDL tail expressed the alpha-cell-specific transcription factor MAFB (ESM Fig. 5a), suggesting that the expression of glucagon in these cells was genetically controlled. To examine whether this reprogramming process was associated with de-differentiation of beta cells, as reported in diabetes [24], mRNA expression of stemness transcription factors Oct4, Nanog and L-myc, of cell-identity regulators Pdx1, Nkx6.1, Neurod1, MafA, Foxo1 and Arx, and of cell-function markers glucose transporter 2 (Glut2), PC1/3 and PC2 (Pcsk1, Pcsk2), insulin 1 and 2 (Ins1, Ins2) and glucokinase (Gck) was studied by quantitative RT-PCR in FACS-sorted beta cells. The expression of stem-like cell markers was not increased in beta cells isolated from PDL-tail pancreas and the expression of markers for functional maturity of beta cells was maintained (ESM Fig. 5b, c). As expected from the protein data, the Gcg and MafB transcript levels were increased in these beta cells (ESM Fig. 5b, c).
Discussion
We investigated whether PDL induces the formation of alpha cells by comparing tail portions of pancreases from PDL- and sham-operated mice, 2 weeks after surgery. Total alpha cell volume increased threefold, while the size of individual alpha cells was increased by 10%. This suggests that many new alpha cells are formed in PDL pancreas. The proportion of alpha cells relative to beta cells increased 1.5-fold in PDL-tail pancreas, suggesting more expansion of alpha than of beta cells. This is consistent with the threefold increase in total alpha cell volume found here and the twofold increase in total beta cell volume reported previously [5, 12]. At present it is uncertain why the glucagon content increased fivefold while alpha cell volume increased threefold.
Differences in glucagonaemia or glycaemia were not observed, suggesting that normal physiological set points were not perturbed in response to PDL surgery. We speculate that the normal glucagonaemia is maintained through a smaller than normal secretion of glucagon per alpha cell (as there are more alpha cells). Alpha cells in islets isolated from PDL-tail pancreas indeed responded less to secretory stimuli than alpha cells in islets isolated from sham tail pancreas, which may in part explain the relatively large glucagon store found in PDL-tail islets and pancreas. Our study suggests proliferation of alpha cells as the main mechanism behind alpha cell expansion. First, the percentage of Ki67+ alpha cells was increased threefold both at 7 and 14 days post-PDL surgery. Second, alpha cells that were conditionally labelled before PDL (pulse) were not diluted by unlabelled alpha cells 2 weeks after surgery (chase period), suggesting little or no contribution by other than pre-existing alpha cells. It must be emphasised that this approach may fail to detect minor contributions of other cell types.
Beta cell ablation in mice showed limited neogenesis of beta cells from alpha cells [9]. Our present data on lineage tracing of alpha cells also suggest no or very little alpha to beta cell conversion in PDL pancreas of normoglycaemic mice. The reduction of YFP label in pre-labelled insulin+ beta cells of PDL tail compared with sham tail (Fig. 4c) confirms previous observations [12], suggesting beta cell neogenesis following PDL. However, the alpha cell is clearly not a major contributor of new beta cells. We do find that beta cells can be reprogrammed to alpha cells in PDL pancreas. The proportion of glucagon+ cells derived from beta cells during a 14 day post-PDL period was limited to 5%. About half of these reporter+ glucagon+ cells no longer expressed insulin, suggesting that these beta-cell-derived cells had progressed towards ‘alpha-like’ cells, giving a net contribution of potentially reprogrammed cells of ∼3%. These cells are different from multiple hormone gene expressing beta cells, which keep the beta cell phenotype [25].
The expression of nuclear MAFB in nearly all reporter+ glucagon+ cells suggests beta cell reprogramming rather than accidental expression of glucagon in beta cells. Beta cell to alpha cell conversion has been reported in mice with diabetes, or following forced expression of the transcription factor Arx [26] or deletion of Foxo1 [24]. In several murine diabetes models, beta cell de-differentiation, increased glucagon content, progressive loss of forkhead box O1 (FOXO1), and beta-cell-derived stem-like cells with high levels of octamer-binding transcription factor 4 (OCT4), NANOG and L-MYC have been described. These stem-like cells give rise to alternative endocrine cell types that do not co-express insulin [24]. In PDL pancreas, however, there is no beta cell or insulin deficit [5] and our present data suggest there is no beta cell de-differentiation or increased expression of stem-cell-like genes Oct4, Nanog or L-myc, and nor is Foxo1 expression decreased or Arx increased in PDL-tail beta cells. In addition, we detect beta-cell-derived glucagon+ cells that still express insulin, suggesting that in PDL pancreas regression of beta cells to a pre-beta cell state is no pre-requisite for conversion into alpha cells. Human beta cells were recently reported to differentiate spontaneously into alpha cells following their re-aggregation in vitro, revealing a previously unsuspected degree of plasticity in this cell type [27]. The finding that beta cells are able to convert to alpha-like cells in PDL pancreas might be of relevance to the (patho)physiology of diabetes. Type 2 diabetes is characterised by an increased proportion of alpha cells relative to beta cells that may, at least in part, result from beta to alpha cell conversion [24] and IL-6 stimulation of alpha cell proliferation [18].
Various cytokines, including IL-6, have been reported to be induced by PDL [17, 28] and increased beta cell proliferation in PDL pancreas was recently shown to be dependent on TGF-β [28]. Increased alpha cell proliferation in PDL pancreas is IL-6-dependent, as shown by the decreased number of Ki67+ alpha cells following IL-6 inactivation in vivo. However, it is not excluded that the altered glucagon release by alpha cells in PDL pancreas partly drives their expansion. In addition, IL-6 may regulate alpha and/or beta cell survival [18, 29], but this remains to be examined in PDL pancreas. In situ delivery of recombinant IL-6 induced extensive STAT3 phosphorylation not only in alpha cells but also in beta cells (Fig. 3f). Our recent data also suggest that PDL beta cells are responsive to IL-6 and that PDL injury triggers STAT3 activation in beta cells (manuscript in preparation). The data suggest that the outcome of PDL, in terms of alpha and beta cell numbers, is controlled by multiple inflammatory cytokines, probably as part of an immune response elicited after the surgery. In type 2 diabetes, IL-6 is required for both proliferation and increased GLP-1 production by alpha cells, and thereby, for beneficial effects on beta cells [18, 29]. In PDL pancreas, IL-6 was required for proliferation of alpha cells, but PC1/3 protein was not increased, suggesting normal pro-glucagon processing. This needs additional investigation, as it has been suggested that other convertases, e.g. furin, might process pro-glucagon to GLP-1 in some hyperplastic alpha cells [11]. However, that the increased formation of alpha cells in PDL-tail pancreas is not associated with PC1/3 protein expression in these cells suggests an interesting difference compared with the PC1/3 expression by alpha cells in obesity and diabetes [29].
In summary, we show that PDL increases alpha cell proliferation through IL-6 signalling, and suggest this to be a major mechanism behind alpha cell expansion.
Abbreviations
- Dox:
-
Doxycycline
- GLP-1:
-
Glucagon-like peptide-1
- GFP:
-
Green fluorescent protein
- IL-6R:
-
IL-6-receptor
- MAFB:
-
V-maf musculoaponeurotic fibrosarcoma oncogene family, protein B (avian)
- PDL:
-
Partial pancreatic-duct ligation
- Pro-hormone convertases 1/3 and 2:
-
PC1/3 and PC2
- rtTA:
-
Reverse tetracycline transactivator
- STAT3:
-
Signal transducer and activator of transcription 3
- Tam:
-
Tamoxifen
- YFP:
-
Yellow fluorescent protein
References
Dor Y, Brown J, Martinez OI, Melton DA (2004) Adult pancreatic β-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429:41–46
Nir T, Melton DA, Dor Y (2007) Recovery from diabetes in mice by β cell regeneration. J Clin Invest 117:2553–2561
Teta M, Rankin MM, Long SY et al (2007) Growth and regeneration of adult β cells does not involve specialized progenitors. Dev Cell 12:817–826
Zhou Q, Brown J, Kanarek A et al (2008) In vivo reprogramming of adult pancreatic exocrine cells to β-cells. Nature 455:627–632
Xu X, D’Hoker J, Stangé G et al (2008) Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 132:197–207
Pan FC, Bankaitis ED, Boyer D et al (2013) Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development 140:751–764
Collombat P, Xu X, Ravassard P et al (2009) The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha- and subsequently beta cells. Cell 138:449–462
Al-Hasani K, Pfeifer A, Courtney M et al (2013) Adult duct-lining cells can reprogram into β-like cells able to counter repeated cycles of toxin-induced diabetes. Dev Cell 26:86–100
Thorel F, Népote V, Avril I et al (2010) Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 464:1149–1154
Habener JF, Stanojevic V (2012) α-Cell role in β-cell generation and regeneration. Islets 4:188–198
Habener JF, Stanojevic V (2013) Alpha cells come of age. Trends Endocrinol Metab 24:153–163
Van de Casteele M, Leuckx G, Baeyens L et al (2013) Neurogenin 3(+) cells contribute to β-cell neogenesis and proliferation in injured adult mouse pancreas. Cell Death Dis 4:e523
Furuyama K, Kawaguchi Y, Akiyama H et al (2010) Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat Genet 43:34–41
Wang RN, Klöppel G, Bouwens L (1995) Duct- to islet-cell differentiation and islet growth in the pancreas of duct-ligated adult rats. Diabetologia 38:1405–1411
Rankin MM, Wilbur CJ, Rak K et al (2013) Beta cells are not generated in pancreatic duct ligation induced injury in adult mice. Diabetes 62:1634–1645
Xiao X, Chen Z, Shiota C et al (2013) No evidence for β cell neogenesis in murine adult pancreas. J Clin Invest 123:2207–2217
Yasuda H, Kataoka K, Ichimura H et al (1999) Cytokine expression and induction of acinar cell apoptosis after pancreatic duct ligation in mice. J Interferon Cytokine Res 19:637–644
Ellingsgaard H, Ehses JA, Hammar EB et al (2008) Interleukin-6 regulates pancreatic alpha-cell mass expansion. Proc Natl Acad Sci U S A 105:13163–13168
Grouwels G, Cai Y, Hoebeke I et al (2010) Ectopic expression of E2F1 stimulates beta-cell proliferation and function. Diabetes 59:1435–1444
Starnes HF, Pearce MK, Tewari A et al (1990) Anti-IL-6 monoclonal antibodies protect against lethal Escherichia coli infection and lethal tumor necrosis factor-alpha challenge in mice. J Immunol 145:4185–4191
Srinivas S, Watanabe T, Lin CS et al (2001) Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol 1:4
Flamez D, Van Breusegem A, Scrocchi LA et al (1998) Mouse pancreatic beta-cells exhibit preserved glucose competence after disruption of the glucagon-like peptide-1 receptor gene. Diabetes 47:646–652
Scoggins CR, Meszoely IM, Wada M et al (2000) p53-dependent acinar cell apoptosis triggers epithelial proliferation in duct-ligated murine pancreas. Am J Physiol Gastrointest Liver Physiol 279:G827–G836
Talchai C, Xuan S, Lin HV et al (2012) Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 150:1223–1234
Katsuta H, Akashi T, Katsuta R et al (2009) Single pancreatic beta cells co-express multiple islet hormone genes in mice. Diabetologia 53:128–138
Collombat P, Hecksher-Sorensen J, Krull J et al (2007) Embryonic endocrine pancreas and mature β cells acquire α and PP cell phenotypes upon Arx misexpression. J Clin Invest 117:961–970
Spijker HS, Ravelli RBG, Mommaas-Kienhuis AM et al (2013) Conversion of mature human β-cells into glucagon-producing α-cells. Diabetes 62:2471–2480
Xiao X, Wiersch J, El-Gohary Y et al (2012) TGFβ receptor signaling is essential for inflammation-induced but not β-cell workload-induced β-cell proliferation. Diabetes 62:1217–1226
Ellingsgaard H, Hauselmann I, Schuler B et al (2011) Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat Med 17:1481–1489
Acknowledgements
We thank A. Demarré, V. Laurysens, J. de Jonge, E. Quartier, R. de Proft and G. Schoonjans (Diabetes Research Center, Vrije Universiteit Brussel, Brussels, Belgium) for technical assistance, and P. Herrera (University of Geneva, Geneva, Suisse) for sharing the Gcg rtTA/TetO Cre mice.
Funding
Financial support was from the VUB Research Council (HH, MVdC), the Institute for the Promotion of Innovation by Science and Technology in Flanders (IWT) (HH, VC), the Chinese Scholarship Council (YY), the Innovative Medicines Initiative Joint Undertaking under grant agreement number 155005 (IMIDIA) composed of financial contributions from the European Union’s Seventh Framework Programme (FP7/2007-2013) and EFPIA companies in kind contribution (HH), Stichting Diabetes Onderzoek Nederland (HH), the Fund for Scientific Research Flanders (FWO) (HH, SDG) and the Interuniversity Attraction Pole networks (HH).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
All authors were involved in the acquisition, analysis or interpretation of data and drafting of the manuscript. YC, YY, MVdC and HH were involved in the study concept and design and critical revision of the manuscript. All authors approved the final version of the manuscript.
HH is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Ying Cai and Yixing Yuchi are joint first authors of this article.
Mark Van de Casteele and Harry Heimberg are joint senior authors of this article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
(PDF 405 kb)
ESM Fig. 2
(PDF 392 kb)
ESM Fig. 3
(PDF 232 kb)
ESM Fig. 4
(PDF 625 kb)
ESM Fig. 5
(PDF 241 kb)
ESM Table 1
(PDF 47 kb)
Rights and permissions
About this article

Cite this article
Cai, Y., Yuchi, Y., De Groef, S. et al. IL-6-dependent proliferation of alpha cells in mice with partial pancreatic-duct ligation. Diabetologia 57, 1420–1427 (2014). https://doi.org/10.1007/s00125-014-3242-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-014-3242-8