
Abstract
Aims/hypothesis
The growing obesity epidemic necessitates a better understanding of adipocyte biology and its role in metabolism. The Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway mediates signalling by numerous cytokines and hormones that regulate adipocyte function, illustrating the physiological importance of adipose JAK–STAT. The aim of this study was to investigate potential roles of adipocyte JAK2, an essential player in the JAK–STAT pathway, in adipocyte biology and metabolism.
Methods
We generated adipocyte-specific Jak2 knockout (A-Jak2 KO) mice using the Cre-loxP system with Cre expression driven by the Ap2 (also known as Fabp4) promoter.
Results
Starting at 2–3 months of age, male and female A-Jak2 KO mice gradually gained more body weight than control littermates primarily due to increased adiposity. This was associated with reduced energy expenditure in A-Jak2 KO mice. In perigonadal adipose tissue, the expression of numerous genes involved in lipid metabolism was differentially regulated. In addition, adipose tissue from A-Jak2 KO mice displayed impaired lipolysis in response to isoprenaline, growth hormone and leptin stimulation, suggesting that adipose JAK2 directly modulates the lipolytic program. Impaired lipid homeostasis was also associated with disrupted adipokine secretion. Accordingly, while glucose metabolism was normal at 2 months of age, by 5–6 months of age, A-Jak2 KO mice had whole-body insulin resistance.
Conclusions/interpretation
Our results suggest that adipocyte JAK2 plays a critical role in the regulation of adipocyte biology and whole-body metabolism. Targeting of the JAK–STAT pathway could be a novel therapeutic option for the treatment of obesity and type 2 diabetes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Adipose tissue mass is maintained by the balance between lipid synthesis and catabolism. Disruption in this equilibrium has been implicated in the pathophysiology of various metabolic disorders, particularly obesity and type 2 diabetes [1–3]. A better understanding of the regulatory mechanisms governing adipocyte biology and homeostasis is therefore crucial, given the growing worldwide epidemic of obesity [4].
The Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway mediates the signal transduction of numerous cytokines, growth factors and hormones that regulate adipocyte development and function [5]. Several cytokines secreted by adipocytes also use this signalling pathway, illustrating the physiological importance of JAK–STATs in adipocyte biology. We and others have recently shown that hepatocyte-specific deletion of Jak2, a ubiquitously expressed member of the JAK kinase family, results in profound lipid accumulation in liver tissue, suggesting a role for JAK2 in lipid metabolism [6, 7]. JAK2 is present in pre-adipocytes and mature adipocytes [8], as well as in adipose tissue [9]. Its mRNA abundance has been shown to be downregulated in human omental adipose tissue under obese conditions [10], suggesting a potential role in the regulation of adipose tissue physiology.
During adipogenesis, JAK2 is activated within 2 h of adipogenic induction and has been shown to act upstream of STAT3 activation [11]. Inhibition of JAK2 with a small-molecule inhibitor or small interfering RNA attenuated the differentiation of 3T3-L1 adipocytes [11]. JAK2 has also been shown to be required for growth hormone (GH)-dependent differentiation of pre-adipocytes via activation of STAT5 [12], a critical regulator of adipocyte development [13–15].
In mature adipocytes, JAK2 can be activated by several cytokines and hormones, most notably leptin, GH, prolactin, IL-6, leukaemia inhibitory factor (LIF), oncostatin M (OSM), ciliary neurotrophic factor (CNTF) and IFN-γ. These cytokines and hormones regulate many aspects of adipose tissue biology. For example, leptin [16], IL-6 [17, 18], IFN-γ [19], GH [20] and prolactin [21] have all been shown to exert lipolytic effects on isolated adipocytes and adipose tissue. Furthermore, leptin, CNTF and LIF can inhibit fatty acid synthesis in white adipose tissue either via a central mechanism [22] or by inhibiting the expression of lipogenic enzymes [23, 24]. In addition to regulating adipose expansion, CNTF enhances the expression of uncoupling protein 1 induced by β3-adrenergic stimulation in brown adipose tissue [25]. OSM, a member of the IL-6 family of cytokines, suppresses adiponectin expression and induces dedifferentiation of adipocytes [26]. Whether all these effects are mediated by JAK2 and to what extent JAK2 activation is required for them to occur has not been established.
At the cellular level, JAK2 functions primarily by transducing signals from cytokines and activating downstream STAT proteins. There is also evidence of STAT-independent functions of JAK2 in adipocytes. It has been shown that fatty acid binding protein 4/adipocyte protein 2 (AP2), a highly-expressed lipid-binding protein in adipocytes, associated with the unphosphorylated form of JAK2 and attenuated its signalling [27]. Nevertheless, the cell-specific functions of adipocyte JAK2 are not well understood.
Recently, adipocyte-specific deletion of Jak2 driven by the adiponectin promoter has been shown to result in reduced lipolysis and increased body fat [28]. The molecular mechanisms behind this and the metabolic consequences are not clear. In this study, we studied the metabolic effects of adipose JAK2 deficiency. We show that mice with impaired adipose JAK2 signalling driven by the Ap2 (also known as Fabp4) promoter develop profound adiposity when on a regular chow diet, and that this is associated with reduced energy expenditure. Thus, while glucose metabolism in young mice is normal, these mice develop whole-body insulin resistance as they age.
Methods
Animals
Adipocyte-specific Jak2 knockout (A-Jak2 KO) mice were generated by breeding mice with the Jak2 gene flanked by loxP sites (Jak2 fl/fl) [29, 30] (kindly provided by Kay-Uwe Wagner, University of Nebraska Medical Center, Omaha, NE, USA) with mice expressing Cre recombinase under the control of the Ap2 promoter (aP2Cre +; purchased from Jackson Laboratory, Bar Harbor, ME, USA). The resulting aP2Cre + Jak2 +/fl mice were intercrossed to generate aP2Cre + Jak2 +/+, aP2Cre + Jak2 +/fl and aP2Cre + Jak2 fl/fl (herein referred to as A-Jak2 KO) mice. Mice were maintained on a mixed 129Sv and C57BL/6 background, and housed in a pathogen-free facility at the Toronto Medical Discovery Tower (Toronto, ON, Canada) with a 12 h light–dark cycle and free access to standard irradiated rodent chow (5% energy from fat; Harlan Teklad, Indianapolis, IN, USA). All animal experimental protocols were approved by the Toronto General Research Institute Animal Care Committee.
In vivo metabolic analyses
Blood glucose measurements, the glucose tolerance test (GTT) and insulin tolerance test (ITT), and quantification of glucose-stimulated insulin secretion were done as previously described [6]. For insulin signalling experiments, mice fasted overnight were injected i.p. with human regular insulin (5 U/kg) or PBS. Tissues were removed 10 min later and snap-frozen in liquid nitrogen. Rectal temperature was measured in fed mice between 10:00 and 11:00 hours. To measure energy expenditure, mice were individually housed in a comprehensive laboratory animal monitoring system (Columbus Instruments, Columbus, OH, USA) with free access to food and water. After 24 h acclimatisaton to the apparatus, data for 24 h were collected.
Body composition
The body composition of 5-month-old male mice with free access to chow was assessed using nuclear magnetic resonance spectroscopy (Biospec 70/30; Bruker, Ettlingen, Germany). Fat volume was quantified using a combination of semi-automated and manual segmentation tools in MIPAV software (Version 7.0.1; National Institute of Health, Bethesda, MD, USA).
Analyses of serum variables
Overnight-fasted mice were anaesthetised and blood was collected by cardiac puncture. Serum insulin levels were measured by a mouse insulin ELISA kit (Crystal Chem, Downers Grove, IL, USA). Serum GH and IGF-1 levels were determined by radioimmunoassay, and serum adiponectin, leptin, TNF-α and IL-6 levels by the Luminex 100 System (Luminex, Austin, TX, USA) at the Mouse Metabolic Phenotyping Centre (Vanderbilt University, Nashville, TN, USA). Serum total cholesterol, triacylglycerol, HDL-cholesterol and NEFA were also assayed at the Mouse Metabolic Phenotyping Centre (Vanderbilt University).
Histological analysis
Liver, pancreas and adipose tissue were removed, fixed and processed to paraffin blocks. Tissue sections were stained with haematoxylin and eosin. Adipocyte size was measured using cellSens software (Olympus, Tokyo, Japan). Adipocyte number per fat pad was calculated from the total fat pad volume: average adipocyte volume ratio, using the method developed by Lemonnier [31]. Immunohistochemistry analysis was performed on pancreatic sections using anti-insulin antibody (Dako, Carpinteria, CA, USA) as previously described [32]. Scanned sections were analysed with ImageScope version 11.0.2.716 software (Aperio Technologies, Vista, CA, USA).
Ex vivo lipolysis
Perigonadal fat pads were surgically removed from 6-month-old mice, cut into 50 mg pieces and incubated in Krebs–Ringer bicarbonate buffer containing 2% wt/vol. fatty acid-free BSA. After a preincubation period of 1 h at 37°C, isoprenaline (Sigma, St Louis, MO, USA), recombinant murine leptin (PeproTech, Rocky Hill, NJ, USA) or recombinant mouse GH (National Hormone and Peptide Program, Torrance, CA, USA) was added directly to the incubation medium. After 2 h of incubation, glycerol release into the medium was determined using a free glycerol reagent (Sigma).
RNA isolation and quantitative RT-PCR
Total RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA). RNA was reverse-transcribed with random primers using M-MLV enzyme (Invitrogen), and quantitative real-time PCR was performed using SYBR Green master mix on a 7900HT Fast-Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA). Each sample was run in triplicate.
Immunoblotting
Tissues were mechanically homogenised in ice-cold lysis buffer and centrifuged for 10 min at 14,000 g and 4°C. The resulting supernatant fraction was separated by SDS-PAGE and immunoblotted with antibodies to phospho-Akt (S473), total Akt, total JAK2 and GAPDH (Cell Signaling Technology, Danvers, MA, USA).
Statistical analysis
Data are presented as mean ± SEM. Values were analysed by two-tailed independent-sample Student’s t test or one-way ANOVA, as appropriate, using GraphPad Prism version 5 (GraphPad Software, La Jolla, CA, USA). A value of p < 0.05 was considered statistically significant.
Results
Disruption of adipocyte JAK2 increases body weight
A-Jak2 KO mice were viable and fertile with no gross abnormalities compared with littermate controls. Quantitative RT-PCR and western blot analyses showed a significant reduction of Jak2 mRNA and protein abundance in the inguinal, perigonadal and brown adipose tissue (Fig. 1a, b). JAK2 abundance in other tissues, including liver, skeletal muscle, brain and macrophages was not affected (Fig. 1c). Selective deficiency of JAK2 in adipose tissue did not affect circulating levels of GH (which requires JAK2 for signal transduction) and its downstream target IGF-1 (electronic supplementary material [ESM] Fig. 1a, b).

Increased body weight in A-Jak2 KO mice. (a) mRNA expression of Jak2 in inguinal (Ing.), perigonadal (Peri.) and interscapular brown adipose tissue (BAT) from aP2Cre + Jak2 +/+ (black bars) and A-Jak2 KO (white bars) mice at 5–6 months of age. Values are normalised to 18S mRNA levels and expressed as fold changes relative to the aP2Cre + Jak2 +/+ group; n ≥ 3. (b) Lysates from perigonadal white adipose tissue (WAT) and BAT from aP2Cre + Jak2 +/+ and A-Jak2 KO mice were prepared and processed for immunoblotting for JAK2. Protein band intensity was quantified by ImageJ software and normalised to the aP2Cre + Jak2 +/+ group; n = 4–6. (c) Tissue lysates from 5- to 6-month-old mice were prepared and processed for immunoblotting for JAK2. MΦ, macrophages. (d) Growth curves of male and (e) female aP2Cre + Jak2 +/+ (black squares), aP2Cre + Jak2 +/fl (grey squares) and A-Jak2 KO (black diamonds) mice and littermate controls; n ≥ 8. (f) A representative photograph of female aP2Cre + Jak2 +/+ and A-Jak2 KO littermates at 6 months of age. (g–l) Body weight, body length (measured from snout to anus) and BMI at 5–6 months of age in (g, i, k) male (n ≥ 10) and (h, j, l) female (n ≥ 7) control (black bars) and A-Jak2 KO (white bars) mice. All results are mean ± SEM; *p < 0.05, **p < 0.01 and ***p < 0.001
To study the role of adipocyte JAK2 in body weight regulation, we followed A-Jak2 KO mice and littermate controls on a standard chow diet and monitored their body weight monthly. As shown in Fig. 1d, e, starting from 3 months of age for males and 2 months of age for females, A-Jak2 KO mice progressively gained more body weight than aP2Cre + Jak2 +/+ and aP2Cre + Jak2 +/fl littermate controls. This was particularly evident in female mice (Fig. 1f). Interestingly, heterozygous aP2Cre + Jak2 +/fl mice exhibited a similar growth curve to aP2Cre + Jak2 +/+ mice, suggesting that gene dosage had no effect on body weight. For subsequent analyses, we combined data from aP2Cre + Jak2 +/+ and aP2Cre + Jak2 +/fl mice, collectively referring to them as controls. By 6 months of age, male and female A-Jak2 KO mice weighed approximately 40.3% and 65.8% more than their control littermates, respectively (Fig. 1g, h). Body length was slightly greater (Fig. 1i, j), while BMI was significantly higher in A-Jak2 KO mice (Fig. 1k, l).
Disruption of adipocyte JAK2 leads to increased adiposity
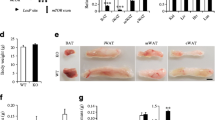
To determine the underlying basis for the increased body weight, we measured body composition by nuclear magnetic resonance spectroscopy. Lean body mass was similar in A-Jak2 KO mice and control littermates (Fig. 2a, b). However, fat mass and body fat content were significantly increased in A-Jak2 KO mice at 5 months of age (Fig. 2a–c). Similarly, when we isolated and weighed fat pads from mice at 5–6 months of age, all fat pads examined from A-Jak2 KO mice weighed more than those examined from control littermates. The differences reached statistical significance for absolute fat pad weight (Fig. 2d, g) and per cent total body weight (Fig. 2e, h). Notably, greater differences between genotypes were observed in both sexes for the inguinal depot compared with the perigonadal depot. On the other hand, the absolute weight of other organs was comparable between the genotype groups, with the exception of the liver, which weighed more in female A-Jak2 KO mice (Fig. 2f, i). Together, these results suggest that adipocyte JAK2 deficiency leads to higher body weight due to increased adiposity.

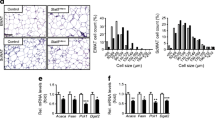
Increased adiposity in A-Jak2 KO mice. (a) Body composition of male mice at 5 months of age as measured by nuclear magnetic resonance spectroscopy, with (b, c) quantification as indicated; n = 5. Black bars, control; white bars, A-Jak2 KO. (d, e) Inguinal (Ing.), perigonadal (Peri.), retroperitoneal (Retro.), mesenteric (Mes.) and interscapular brown adipose tissue (BAT) fat pads were removed from 5- to 6-month-old male (n ≥ 11) and (g, h) female (n ≥ 10) mice and weighed. Results are expressed as absolute fat pad weight or per cent total body weight. (f) Liver, pancreas, spleen, heart and kidneys were removed from 5- to 6-month-old male (n ≥ 10) and (i) female (n ≥ 6) mice and weighed. Results are expressed as absolute organ weight. (j) Representative micrographs of haematoxylin and eosin staining of inguinal, perigonadal and interscapular BAT sections from 5- to 6-month-old female mice. Scale bar, 40 μm. (k) Quantification of adipocyte size distribution, (l) average adipocyte size and (m) cell number in perigonadal fat pads from tissue sections as above (j); n = 3. All results are mean ± SEM; *p < 0.05, **p < 0.01 and ***p < 0.001
Haematoxylin and eosin staining revealed the presence of enlarged adipocytes in inguinal, perigonadal and interscapular brown adipose tissue in A-Jak2 KO mice (Fig. 2j). Analysis of adipocyte size, distribution and number suggested a threefold increase in the average size of perigonadal adipocytes, with no change in adipocyte number (Fig. 2k–m). Similar changes were observed for adipocytes from the inguinal depot (data not shown). These results indicate that the adipose tissue expansion observed in A-Jak2 KO mice is due to adipocyte hypertrophy, not to an increase in cell number.
A-Jak2 KO mice have normal energy metabolism at 1 month of age, but display reduced energy expenditure as they age
To delineate the physiological mechanisms that would account for the apparent positive energy balance in A-Jak2 KO mice, we measured their food intake and energy expenditure by indirect calorimetry. At 1 month of age when A-Jak2 KO mice had a similar body weight to control littermates, there were no significant differences in absolute food or water intake, volume of oxygen consumption (\( \overset{\cdotp }{V}{\mathrm{O}}_2 \)), respiratory exchange ratio (RER), physical activity or body temperature between the genotype groups in both sexes (ESM Fig. 2a-l). These data suggest that adipose JAK2 deficiency had no direct effect on energy balance.
By 5–6 months of age, A-Jak2 KO mice still had normal absolute daily food and water intake, suggesting no change in energy intake (Fig. 3a, b, g, h). A-Jak2 KO mice were also comparable to control littermates in terms of the energy source utilised, as indicated by a similar RER (Fig. 3c, i). On the other hand, \( \overset{\cdotp }{V}{\mathrm{O}}_2 \) was significantly lower in A-Jak2 KO mice of both sexes (Fig. 3d, j), consistent with their reduced energy expenditure. This was associated with significantly reduced physical activity in female A-Jak2 KO mice (Fig. 3k). Male A-Jak2 KO mice also showed a non-significant decrease in physical activity compared with control littermates (Fig. 3e). In contrast, body temperature was not altered by adipocyte JAK2 deficiency (Fig. 3f, l). Together, these results suggest that the progressive increase in body weight in A-Jak2 KO mice may be due, at least in part, to decreased energy expenditure.

Reduced energy expenditure in A-Jak2 KO mice at 5–6 months of age. (a–e) Male (n = 6) and (g–k) female (n ≥ 6) control (black bars) and A-Jak2 KO (white bars) mice aged 5–6 months were housed individually in metabolic chambers with free access to food and water, and energy balance data were collected for 24 h. (a, g) Daily food and (b, h) water intake were determined by weighing the chow and measuring the volume of drinking water, respectively, before and after the measurement period. (c, i) RER, calculated as the ratio of volume of carbon dioxide production (\( \overset{\cdotp }{V}{\mathrm{CO}}_2 \)):\( \overset{\cdotp }{V}{\mathrm{O}}_2 \). (d, j) \( \overset{\cdotp }{V}{\mathrm{O}}_2 \) and (e, k) physical activity expressed as average number of infra-red beam breaks during one measurement interval. (f) Rectal temperature of male and (l) female control and A-Jak2 KO mice; n ≥ 3. All results are mean ± SEM; *p < 0.05, **p < 0.01 and ***p < 0.001
Adipose JAK2 deficiency leads to impaired lipolysis
To elucidate molecular changes in adipose tissue that could account for the observed phenotype in A-Jak2 KO mice, we analysed the expression of genes involved in lipid and glucose metabolism in perigonadal adipose tissue. At 1 month of age, gene expression was not altered by JAK2 deficiency (ESM Fig. 3a, b). The genes studied included known STAT target genes, e.g. Fabp4, fatty acid synthase (Fas, also known as Fasn), acyl-CoA oxidase 1 (Acox1) and pyruvate dehydrogenase kinase (Pdk4). By 5–6 months of age, genes implicated in lipid accumulation became differentially regulated. An overall upregulation of genes implicated in adipogenesis was observed (Fig. 4a). On the other hand, enzymes regulating fatty acid synthesis showed decreased expression. mRNA levels of hormone sensitive lipase (Lipe) were significantly increased, whereas expression of adipose triacylglycerol lipase (Pnpla2) was reduced in A-Jak2 KO mice (Fig. 4a). In addition, the mRNA abundance of genes involved in β-oxidation was elevated, suggesting a disruption in adipose lipid homeostasis. In contrast, the expression of genes regulating glucose metabolism was not altered by JAK2 deficiency (Fig. 4b).

Impaired lipolysis and disrupted lipid metabolism in A-Jak2 KO mice. (a) mRNA expression of genes involved in lipid metabolism in perigonadal adipose tissue from mice at 5–6 months of age; n ≥ 6. Values are normalised to 18S mRNA levels and expressed as fold changes relative to the control group. Cebpa encodes CCAAT/enhancer binding protein, alpha; Srebf1, sterol regulatory element-binding protein 1c; Fabp4, fatty acid binding protein 4; Pparg, peroxisome proliferator-activated receptor gamma; Acc, acetyl-CoA carboxylase; Fas, fatty acid synthase; Lipe, hormone sensitive lipase; Pnpla2, adipose triacylglycerol lipase; Cpt-1 (also known as Cpt1a), carnitine palmitoyltransferase 1; Acox1, acyl-CoA oxidase 1, palmitoyl. Black bars, control; white bars, A-Jak2 KO. (b) mRNA expression of genes implicated in glucose metabolism in tissue as above (a). Irs1 encodes insulin receptor substrate 1; Slc2a4, solute carrier family 2 (facilitated glucose transporter), member 4; Pdk4, pyruvate dehydrogenase kinase isozyme 4. (c) Glycerol release from perigonadal fat explants of 6-month-old mice stimulated with isoprenaline, recombinant leptin or GH. Results are from triplicate samples repeated three times. (d) Serum levels of NEFA, (e) triacylglycerol and (f) total cholesterol from overnight-fasted mice at 5–6 months of age; n ≥ 4. All results are mean ± SEM; *p < 0.05, **p < 0.01 and † p = 0.06
Next, given the well-known lipolytic effects of JAK2-activating cytokines and hormones, we measured ex vivo lipolysis in adipose explants from 5- to 6-month-old mice. As shown in Fig. 4c, baseline glycerol release was not affected by adipose JAK2 deficiency. On the other hand, while isoprenaline induced a robust lipolytic response in controls, this effect was significantly attenuated in A-Jak2 KO mice. Furthermore, glycerol release in response to leptin and GH was completely abolished in A-Jak2 KO adipose tissue, indicating impaired lipolysis.
Consistent with disrupted lipid homeostasis in adipose tissue, A-Jak2 KO mice showed higher NEFA levels in the circulation at 5–6 months of age (Fig. 4d), whereas levels of triacylglycerol (Fig. 4e), total cholesterol (Fig. 4f) and cholesterol lipoproteins (ESM Fig. 3c) were not changed. Finally, despite the presence of massive adiposity and increased circulating NEFA concentration, liver tissue from A-Jak2 KO mice did not accumulate a significant amount of lipid, as shown by histological staining (ESM Fig. 3d). Together, these results suggest that adipose JAK2 deficiency results in defective lipolysis, leading to a disruption of whole-body lipid homeostasis.
A-Jak2 KO mice have disrupted adipokine secretion
To investigate the effects of JAK2 deficiency on adipose function, we also examined expression patterns of adipokines. At 1 month of age, mRNA levels of leptin, adiponectin and Tnf-a (also known as Tnf) were similar, whereas expression of Il6 was downregulated in perigonadal adipose tissue from A-Jak2 KO mice (ESM Fig. 4). As A-Jak2 KO mice accumulated more adipose mass with age, their perigonadal adipose tissue upregulated the mRNA transcription of leptin and Tnf-a (Fig. 5a). Consistent with this, levels of these adipokines were also elevated in the circulation (Fig. 5b–e). In addition, while gene expression of adiponectin was not changed (Fig. 5a), circulating adiponectin levels were significantly reduced in A-Jak2 KO mice (Fig. 5c).

Disrupted adipokine profile in A-Jak2 KO mice. (a) mRNA expression of genes encoding adipokines, determined in perigonadal adipose tissue from mice at 5–6 months of age; n ≥ 8. Values are normalised to 18S mRNA levels and expressed as fold changes relative to the control group. Lep encodes leptin; Adipoq, adiponectin. Black bars, control; white bars, A-Jak2 KO. (b–e) Serum levels of adipokines as indicated from overnight-fasted mice at 5–6 months of age; n ≥ 4. All results are mean ± SEM; *p < 0.05, **p < 0.01 and ***p < 0.001; † p = 0.07
A-Jak2 KO mice show impaired insulin sensitivity as they age
To determine the metabolic consequences of adipocyte JAK2 disruption, we assessed glucose metabolism in A-Jak2 KO mice. At 2 months of age, A-Jak2 KO mice displayed no changes in random or fasting blood glucose levels (ESM Fig. 5a, b, e, f). Responses of blood glucose to exogenous glucose and insulin administration were also similar in A-Jak2 KO compared with control mice (ESM Fig. 5c, d, g, h). This normal glucose metabolism was present despite the significant increase in adiposity in female A-Jak2 KO mice.
By 5–6 months of age, male and female A-Jak2 KO mice maintained normal levels of random and fasting blood glucose (Fig. 6a, b, f, g). An i.p. GTT suggested the presence of glucose intolerance, especially in female A-Jak2 KO mice (Fig. 6c, h). However, when we performed an OGTT using a fixed dose of glucose (50 mg) to eliminate the difference in dosing between genotype groups, glucose tolerance in A-Jak2 KO mice was comparable to that in control littermates (ESM Fig. 6a, b), suggesting that A-Jak2 KO mice may be able to maintain glucose homeostasis at this age. Nevertheless, while male A-Jak2 KO mice showed no significant change in insulin sensitivity (Fig. 6d, e), female A-Jak2 KO mice showed impaired insulin sensitivity during an ITT (Fig. 6i) and higher fasting serum insulin levels (Fig. 6j). Next, to assess organ-specific insulin sensitivity, female A-Jak2 KO mice and control littermates were challenged with insulin, and tissues removed and processed for analysis of insulin signalling by western blotting. As shown in Fig. 6k, insulin-stimulated Akt phosphorylation was significantly attenuated in perigonadal adipose tissue, liver and skeletal muscle of A-Jak2 KO mice, suggesting the presence of whole-body insulin resistance. This was associated with increased beta cell area, probably as a compensatory response to increased insulin demand (ESM Fig. 6c, d). However, glucose-stimulated insulin secretion was normal in A-Jak2 KO mice (ESM Fig. 6e). In summary, selective JAK2 deficiency in adipocytes results in whole-body insulin resistance in association with disrupted lipid homeostasis and adipokine secretion.

Impaired insulin sensitivity in A-Jak2 KO mice at 5–6 months of age. (a, f) Random blood glucose, (b, g) fasting blood glucose, (c, h) i.p. GTT (1 g/kg), (d, i) i.p. ITT (1.0 U/kg) and (e, j) fasting serum insulin levels at 5–6 months of age in male (n ≥ 5) and female (n ≥ 5) mice. Black, control; white or grey, A-Jak2 KO. (k) Perigonadal white adipose tissue (WAT), liver and skeletal muscle lysates were prepared from 6-month-old female mice that had been injected with insulin (5 U/kg) or PBS, and resolved by SDS-PAGE. Lysates were immunoblotted for phospho-Akt (S473), total Akt and GAPDH. Protein band intensity was quantified by ImageJ software and normalised to the PBS-injected control group; n = 3. All results are mean ± SEM; *p < 0.05 and **p < 0.01
Discussion
In this study, we investigated the role of JAK2, a key mediator of cytokine signalling, in adipose tissue biology and whole-body metabolism. Using mice lacking Jak2 in adipocytes, we show that JAK2 plays an essential role in maintaining adipose mass, such that its deficiency resulted in extensive adipose tissue expansion even on a regular chow diet. This was associated with reduced energy expenditure and impaired insulin sensitivity with age. Notably, the phenotypic differences between A-Jak2 KO mice and control littermates were more profound in females. The mechanisms underlying this sex-related difference are not clear, but may reflect sexual dimorphic actions of JAK2-activating cytokines or compensatory pathways activated in response to JAK2 deficiency in male mice.
To disrupt JAK2 specifically in adipocytes, we used Ap2-Cre transgenic mice, a widely used model to study the adipocyte-specific function of genes of interest [33, 34]. However, the Ap2 promoter/enhancer has been shown to drive transgene expression during embryonic development [35]. Therefore, we cannot rule out a contribution of JAK2 deficiency in other tissues to the observed phenotype of A-Jak2 KO mice. Nevertheless, our results are in agreement with a recent report characterising mice with adipose-specific Jak2 deletion driven by the adiponectin promoter [28]. Similarly to our mice, male knockout mice in this model had normal body weight, but significantly increased body fat at 8–10 weeks of age.
The marked adiposity observed with adipocyte JAK2 deficiency is consistent with previous studies in which various components of the JAK2–STAT pathway were disrupted in adipocytes. In particular, disruption of the GH receptor (GHR) [36] and STAT3 [37] using the Ap2 promoter, and knockdown of the leptin receptor using antisense RNA [38] all resulted in increased body weight and adiposity. Of all the models, A-Jak2 KO mice had the most striking increase in body weight. This is probably due to the combined disruption of signalling from multiple cytokines as a consequence of JAK2 deficiency. Interestingly, the perigonadal fat depot had the smallest increase in mass, especially in male A-Jak2 KO mice. This is in line with observations in adipocyte-specific Ghr knockout mice [36] and may be due to different responses of various fat depots to the action of GH [39].
Previous studies in vitro have suggested a critical role of JAK2 in adipogenesis [11, 12]. Although direct in vivo evidence is lacking for JAK2, studies in different mouse models support the importance of its downstream protein, STAT5, in adipocyte differentiation [40, 41]. Therefore, JAK2 deficiency may have an impact on adipocyte development in our model. However, A-Jak2 KO mice probably did not have impaired adipogenesis because they maintained a similar number of adipocytes compared with control littermates. This lack of an effect on adipogenesis may be due to the late deletion of Jak2 induced by the Ap2 promoter, as AP2 is commonly regarded as a marker of terminal adipocyte differentiation [42].
With no change in adipogenesis and no direct effect on energy balance, adipocyte-specific JAK2 deficiency might alter body composition by modulating lipid turnover in the adipose tissue. Indeed, a number of JAK2-activating cytokines and hormones including leptin [16], IL-6 [17, 18], IFN-γ [19], GH [20] and prolactin [21] have well-documented lipolytic effects. In agreement with this, adiponectin promoter-driven Jak2 deletion resulted in impaired lipolysis in white adipose tissue [28]. In this work, we further showed that adipocyte JAK2 is required for leptin- and GH-stimulated lipolysis. Our results are consistent with previous work showing defective leptin- and GH-stimulated lipolysis in adipocytes from mice lacking STAT3, and STAT5A and STAT5B, respectively [37, 43]. However, in contrast to these models, adipose JAK2 deficiency also significantly attenuated catecholamine-induced lipolysis, albeit to a lesser degree, indicating that adipose JAK2 directly modulates the lipolytic program.
We postulate that whole-body metabolic changes later in life in A-Jak2 KO mice are likely to be secondary to increased adiposity and the ensuing insulin resistance, as A-Jak2 KO mice were metabolically normal at a younger age despite profound obesity. Specifically, with blunted insulin action, insulin-stimulated expression of lipogenic genes including acetyl-CoA carboxylase (Acc, also known as Acaca) and Fas would be diminished [44]. The inhibition of lipolysis via suppression of Lipe would also be attenuated [45]. This might explain the higher circulating levels of NEFA in older A-Jak2 KO mice.
In line with insulin resistance resulting from disrupted adipose tissue homeostasis, we observed higher circulating levels of leptin and TNF-α, and lower levels of adiponectin, which could all contribute to the metabolic disturbances in older A-Jak2 KO mice. While histological analysis of the liver did not suggest excessive lipid deposition, it is possible that with age and more severe insulin resistance, fatty liver may eventually develop in A-Jak2 KO mice. Importantly, our knockout animals were phenotypically distinct in terms of whole-body metabolism compared with other models of disrupted JAK–STAT signalling. The disruption of GHR in adipocytes did not affect glucose homeostasis or hepatic lipid content up to 20 weeks of age [36], whereas mice with diminished adipose leptin signalling displayed glucose intolerance by as early as 6–7 weeks of age, and liver steatosis by 19–20 weeks [38]. In contrast, although glucose homeostasis was relatively normal in mice with adipose-specific STAT3 deficiency, fatty liver was present by 20 weeks of age [37]. These differences in whole-body metabolism are present despite the same primary defect in body weight homeostasis, demonstrating the complexity of metabolic regulation and the diverse functions of the JAK–STAT pathway in adipose physiology.
Results from this work and our previous study on hepatocyte-specific Jak2 deletion suggest that JAK2 acts to keep lipid content in check in metabolic tissues with high lipid turnover. We propose that disruption of this important regulator leads to excessive lipid accumulation locally. This promotes neutral lipid storage and prevents spillover into the circulation and other organs. Consequently, despite massive liver steatosis, hepatocyte-specific Jak2 knockout mice did not develop inflammatory liver damage and were in fact protected from high-fat diet-induced obesity and glucose intolerance [6]. In the case of adipose JAK2 deficiency, lipid accumulation in adipose tissue was initially benign with no adverse effects on metabolic variables. However, the progressive deposition of lipid eventually exceeded the storage capacity of existing adipocytes, leading to the release of NEFA into the circulation and the ensuing metabolic consequences as mice grew older.
In summary, A-Jak2 KO mice develop increased body mass and adiposity due to defective lipolysis. With age, this increased adiposity leads to dyslipidaemia, insulin resistance and dysregulation of adipocyte gene expression. Therefore, JAK2 in adipocytes is required for body weight homeostasis and prevents excessive lipid accumulation in adipose tissue. Targeting the JAK–STAT pathway may provide a new therapeutic option for the treatment of obesity and associated type 2 diabetes.
Abbreviations
- A-Jak2 KO:
-
Adipocyte-specific Jak2 knockout
- AP2:
-
Adipocyte protein 2
- CNTF:
-
Ciliary neurotrophic factor
- GH:
-
Growth hormone
- GHR:
-
GH receptor
- GTT:
-
Glucose tolerance test
- ITT:
-
Insulin tolerance test
- JAK:
-
Janus kinase
- LIF:
-
Leukaemia inhibitory factor
- OSM:
-
Oncostatin M
- RER:
-
Respiratory exchange ratio
- STAT:
-
Signal transducer and activator of transcription
- \( \overset{\cdotp }{V}{\mathrm{O}}_2 \) :
-
Volume of oxygen consumption
References
Sun K, Kusminski CM, Scherer PE (2011) Adipose tissue remodeling and obesity. J Clin Invest 121:2094–2101
Guilherme A, Virbasius JV, Puri V, Czech MP (2008) Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol 9:367–377
Kahn BB, Flier JS (2000) Obesity and insulin resistance. J Clin Invest 106:473–481
Swinburn BA, Sacks G, Hall KD et al (2011) The global obesity pandemic: shaped by global drivers and local environments. Lancet 378:804–814
Richard AJ, Stephens JM (2011) Emerging roles of JAK–STAT signaling pathways in adipocytes. Trends Endocrinol Metab 22:325–332
Shi SY, Garcia Martin R, Duncan RE et al (2012) Hepatocyte-specific deletion of Janus kinase 2 (JAK2) protects against diet-induced steatohepatitis and glucose intolerance. J Biol Chem 287:10277–10288
Sos BC, Harris C, Nordstrom SM et al (2011) Abrogation of growth hormone secretion rescues fatty liver in mice with hepatocyte-specific deletion of JAK2. J Clin Invest 121:1412–1423
Stewart WC, Morrison RF, Young SL, Stephens JM (1999) Regulation of signal transducers and activators of transcription (STATs) by effectors of adipogenesis: coordinate regulation of STATs 1, 5A, and 5B with peroxisome proliferator-activated receptor-gamma and C/AAAT enhancer binding protein-alpha. Biochim Biophys Acta 1452:188–196
Hellgren G, Albertsson-Wikland K, Billig H, Carlsson LM, Carlsson B (2001) Growth hormone receptor interaction with Jak proteins differs between tissues. J Interferon Cytokine Res 21:75–83
Gomez-Ambrosi J, Catalan V, Diez-Caballero A et al (2004) Gene expression profile of omental adipose tissue in human obesity. FASEB J 18:215–217
Zhang K, Guo W, Yang Y, Wu J (2011) JAK2/STAT3 pathway is involved in the early stage of adipogenesis through regulating C/EBPbeta transcription. J Cell Biochem 112:488–497
Yarwood SJ, Sale EM, Sale GJ, Houslay MD, Kilgour E, Anderson NG (1999) Growth hormone-dependent differentiation of 3T3-F442A preadipocytes requires Janus kinase/signal transducer and activator of transcription but not mitogen-activated protein kinase or p70 S6 kinase signaling. J Biol Chem 274:8662–8668
Floyd ZE, Stephens JM (2003) STAT5A promotes adipogenesis in nonprecursor cells and associates with the glucocorticoid receptor during adipocyte differentiation. Diabetes 52:308–314
Shang CA, Waters MJ (2003) Constitutively active signal transducer and activator of transcription 5 can replace the requirement for growth hormone in adipogenesis of 3T3-F442A preadipocytes. Mol Endocrinol 17:2494–2508
Nanbu-Wakao R, Morikawa Y, Matsumura I et al (2002) Stimulation of 3T3-L1 adipogenesis by signal transducer and activator of transcription 5. Mol Endocrinol 16:1565–1576
Siegrist-Kaiser CA, Pauli V, Juge-Aubry CE et al (1997) Direct effects of leptin on brown and white adipose tissue. J Clin Invest 100:2858–2864
Trujillo ME, Sullivan S, Harten I, Schneider SH, Greenberg AS, Fried SK (2004) Interleukin-6 regulates human adipose tissue lipid metabolism and leptin production in vitro. J Clin Endocrinol Metab 89:5577–5582
van Hall G, Steensberg A, Sacchetti M et al (2003) Interleukin-6 stimulates lipolysis and fat oxidation in humans. J Clin Endocrinol Metab 88:3005–3010
Memon RA, Feingold KR, Moser AH, Doerrler W, Grunfeld C (1992) In vivo effects of interferon-alpha and interferon-gamma on lipolysis and ketogenesis. Endocrinology 131:1695–1702
Goodman HM (1968) Multiple effects of growth hormone on lipolysis. Endocrinology 83:300–308
Fielder PJ, Talamantes F (1987) The lipolytic effects of mouse placental lactogen II, mouse prolactin, and mouse growth hormone on adipose tissue from virgin and pregnant mice. Endocrinology 121:493–497
Buettner C, Muse ED, Cheng A et al (2008) Leptin controls adipose tissue lipogenesis via central, STAT3-independent mechanisms. Nat Med 14:667–675
Zvonic S, Cornelius P, Stewart WC, Mynatt RL, Stephens JM (2003) The regulation and activation of ciliary neurotrophic factor signaling proteins in adipocytes. J Biol Chem 278:2228–2235
Hogan JC, Stephens JM (2005) Effects of leukemia inhibitory factor on 3T3-L1 adipocytes. J Endocrinol 185:485–496
Ott V, Fasshauer M, Dalski A, Klein HH, Klein J (2002) Direct effects of ciliary neurotrophic factor on brown adipocytes: evidence for a role in peripheral regulation of energy homeostasis. J Endocrinol 173:R1–R8
Song HY, Kim MR, Lee MJ et al (2007) Oncostatin M decreases adiponectin expression and induces dedifferentiation of adipocytes by JAK3- and MEK-dependent pathways. Int J Biochem Cell Biol 39:439–449
Thompson BR, Mazurkiewicz-Munoz AM, Suttles J, Carter-Su C, Bernlohr DA (2009) Interaction of adipocyte fatty acid-binding protein (AFABP) and JAK2: AFABP/aP2 as a regulator of JAK2 signaling. J Biol Chem 284:13473–13480
Nordstrom SM, Tran JL, Sos BC, Wagner KU, Weiss EJ (2013) Disruption of JAK2 in adipocytes impairs lipolysis and improves fatty liver in mice with elevated GH. Mol Endocrinol 27:1333–1342
Krempler A, Qi Y, Triplett AA, Zhu J, Rui H, Wagner KU (2004) Generation of a conditional knockout allele for the Janus kinase 2 (Jak2) gene in mice. Genesis 40:52–57
Wagner KU, Krempler A, Triplett AA et al (2004) Impaired alveologenesis and maintenance of secretory mammary epithelial cells in Jak2 conditional knockout mice. Mol Cell Biol 24:5510–5520
Lemonnier D (1972) Effect of age, sex, and sites on the cellularity of the adipose tissue in mice and rats rendered obese by a high-fat diet. J Clin Invest 51:2907–2915
Choi D, Schroer SA, Lu SY et al (2010) Erythropoietin protects against diabetes through direct effects on pancreatic beta cells. J Exp Med 207:2831–2842
Bluher M, Michael MD, Peroni OD et al (2002) Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev Cell 3:25–38
He W, Barak Y, Hevener A et al (2003) Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A 100:15712–15717
Urs S, Harrington A, Liaw L, Small D (2006) Selective expression of an aP2/fatty acid binding protein 4-Cre transgene in non-adipogenic tissues during embryonic development. Transgenic Res 15:647–653
List EO, Berryman DE, Funk K et al (2013) The role of GH in adipose tissue: lessons from adipose-specific GH receptor gene-disrupted mice. Mol Endocrinol 27:524–535
Cernkovich ER, Deng J, Bond MC, Combs TP, Harp JB (2008) Adipose-specific disruption of signal transducer and activator of transcription 3 increases body weight and adiposity. Endocrinology 149:1581–1590
Huan JN, Li J, Han Y, Chen K, Wu N, Zhao AZ (2003) Adipocyte-selective reduction of the leptin receptors induced by antisense RNA leads to increased adiposity, dyslipidemia, and insulin resistance. J Biol Chem 278:45638–45650
List EO, Palmer AJ, Berryman DE, Bower B, Kelder B, Kopchick JJ (2009) Growth hormone improves body composition, fasting blood glucose, glucose tolerance and liver triacylglycerol in a mouse model of diet-induced obesity and type 2 diabetes. Diabetologia 52:1647–1655
Stewart WC, Pearcy LA, Floyd ZE, Stephens JM (2011) STAT5A expression in Swiss 3T3 cells promotes adipogenesis in vivo in an athymic mice model system. Obesity (Silver Spring) 19:1731–1734
Teglund S, McKay C, Schuetz E et al (1998) Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell 93:841–850
Hunt CR, Ro JH, Dobson DE, Min HY, Spiegelman BM (1986) Adipocyte P2 gene: developmental expression and homology of 5'-flanking sequences among fat cell-specific genes. Proc Natl Acad Sci U S A 83:3786–3790
Fain JN, Ihle JH, Bahouth SW (1999) Stimulation of lipolysis but not of leptin release by growth hormone is abolished in adipose tissue from Stat5a and b knockout mice. Biochem Biophys Res Commun 263:201–205
Girard J, Perdereau D, Foufelle F, Prip-Buus C, Ferre P (1994) Regulation of lipogenic enzyme gene expression by nutrients and hormones. FASEB J 8:36–42
Saltiel AR, Kahn CR (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414:799–806
Acknowledgements
We would like to thank K.-U. Wagner (University of Nebraska Medical Center, Omaha, NE, USA) for generously providing the Jak2 floxed mice and for the genotyping protocols. We would also like to thank J. Chung (Toronto General Research Institute, Toronto, ON, Canada) for help with the OGTT studies.
Funding
This work was supported by operating grants from the Canadian Institute of Health Research MOP-81148 and MOP-93707, and by a Canadian Diabetes Association Grant-in-aid (to MW). MW is supported by the Canada Research Chair in Signal Transduction in Diabetes Pathogenesis. SYS is supported by a Canadian Institutes of Health Research Doctoral Research Award, a Canadian Diabetes Association Doctoral Student Research Award and a Canadian Liver Foundation Graduate Studentship. CTL is supported by the Eliot Phillipson Clinician Scientist Training Program, and a Banting and Best Diabetes Centre Post-doctoral Fellowship. TS is supported by a Canadian Diabetes Association Doctoral Student Research Award and by a Banting and Best Diabetes Centre Novo Nordisk Studentship.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
SYS designed and performed experiments, interpreted and analysed data, and wrote and edited the manuscript. CTL, JJB, TS, S-YL and SAS performed experiments, analysed data and edited the manuscript. MW designed experiments, interpreted and analysed data, and critically edited the manuscript. All authors have read and approved the final version of the manuscript. MW is responsible for the integrity of the work as a whole.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
(PDF 175 kb)
ESM Fig. 2
(PDF 340 kb)
ESM Fig. 3
(PDF 1759 kb)
ESM Fig. 4
(PDF 198 kb)
ESM Fig. 5
(PDF 262 kb)
ESM Fig. 6
(PDF 1505 kb)
Rights and permissions
About this article
Cite this article
Shi, S.Y., Luk, C.T., Brunt, J.J. et al. Adipocyte-specific deficiency of Janus kinase (JAK) 2 in mice impairs lipolysis and increases body weight, and leads to insulin resistance with ageing. Diabetologia 57, 1016–1026 (2014). https://doi.org/10.1007/s00125-014-3185-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-014-3185-0