
Abstract
Aims/hypothesis
Enhanced vascular inflammation, immune cell infiltration and elevated production of reactive oxygen species (ROS) contribute significantly to pro-atherogenic responses in diabetes. We assessed the immunomodulatory role of NADPH oxidase (NOX)-derived ROS in diabetes-accelerated atherosclerosis.
Methods
Diabetes was induced in male Apoe −/− mice with five daily doses of streptozotocin (55 mg kg−1 day−1). Atherosclerotic plaque size, markers of ROS and immune cell accumulation were assessed in addition to flow cytometric analyses of cells isolated from the adjacent mediastinal lymph nodes (meLNs). The role of NOX-derived ROS was investigated using the NOX inhibitor, GKT137831 (60 mg/kg per day; gavage) administered to diabetic and non-diabetic Apoe −/− mice for 10 weeks.
Results
Diabetes increased atherosclerotic plaque development in the aortic sinus and this correlated with increased lesional accumulation of T cells and CD11c+ cells and altered T cell activation in the adjacent meLNs. Diabetic Apoe −/− mice demonstrated an elevation in vascular ROS production and expression of the proinflammatory markers monocyte chemoattractant protein 1, vascular adhesion molecule 1 and IFNγ. Blockade of NOX-derived ROS using GKT137831 prevented the diabetes-mediated increase in atherosclerotic plaque area and associated vascular T cell infiltration and also significantly reduced vascular ROS as well as markers of inflammation and plaque necrotic core area.
Conclusions/interpretation
Diabetes promotes pro-inflammatory immune responses in the aortic sinus and its associated lymphoid tissue. These changes are associated with increased ROS production by NOX. Blockade of NOX-derived ROS using the NOX inhibitor GKT137831 is associated with attenuation of these changes in the immune response and reduces the diabetes-accelerated development of atherosclerotic plaques in Apoe −/− mice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetes increases the risk of developing cardiovascular disease and its serious sequelae, such as stroke and myocardial infarction [1, 2]. Elevated immune-mediated inflammation and oxidative stress are known to play key roles in the initiation and progression of atherosclerosis [3–5]. However, the underlying pathological mechanisms that link the immune response and reactive oxygen species (ROS) to accelerated atherosclerosis development in diabetes remain to be fully elucidated.
An enhanced inflammatory state in diabetes promotes the activation of pro-atherogenic mechanisms in vascular cells and immune cells [6, 7]. Previous studies have identified a marked increase in T cells in aortas from diabetic mice [8] and in human atherosclerotic plaques [9], the majority of which are proinflammatory, IFNγ-producing CD4+ Th1 cells [10]. Furthermore, abundant infiltration of activated T lymphocytes and macrophages are key features of plaque instability and vulnerability to rupture [11].
Diabetic patients suffer from increased oxidative stress, particularly those patients with complications [12]. NADPH oxidase (NOX) enzymes are a major source of vascular ROS [4, 13], with isoforms NOX1 and NOX4 playing key roles in vasculopathies [14–16]. Elevated ROS production in the vessels of diabetic patients and rodents has been shown in parallel with increased NOX expression and activity [17–19]. Furthermore, we recently demonstrated that genetic deletion of Nox1 attenuates atherosclerosis development in diabetic Apoe −/− mice [20]. Oxidative stress induces the expression of pro-inflammatory adhesion and chemotactic molecules, such as vascular adhesion molecule 1 (VCAM-1) and monocyte chemoattractant protein 1 (MCP-1) [20–22], which promote the recruitment and accumulation of inflammatory cells to the developing atherosclerotic lesion [23, 24]. Hyperglycaemia augments the production of proinflammatory cytokines, including IFNγ [25, 26], which further stimulate ROS production by NOX enzymes [4], highlighting another link between oxidative stress and the immune response leading to inflammation in diabetes.
Previous studies have evaluated the effect of diabetes-mediated oxidative stress and inflammation on atherosclerosis development and progression [20], although changes in the immune response in diabetes-accelerated atherosclerosis have not been investigated in detail. We propose that NOX-derived ROS play an important role in the immune–inflammatory responses that contribute to accelerated atherosclerosis development in diabetes. The study had the following purposes: (1) to investigate the effects of diabetes on immune–inflammatory responses known to contribute to atherosclerosis and (2) to use a pharmacological approach to elucidate the link between diabetes-induced ROS production by NOX enzymes and key immune mechanisms that drive atherosclerosis using the novel NOX1/NOX4 inhibitor, GKT137831.
Methods
Animal studies
All experimental protocols were approved by the Alfred Medical Research and Education Precinct (AMREP) Animal Ethics Committee according to the National Health and Medical Research Council of Australia guidelines.
The mice used in this study had unrestricted access to water and standard chow (Specialty Feeds, Glen Forrest, WA, Australia) and were maintained on a 12 h light/dark cycle under pathogen-free conditions in the Precinct Animal Centre of the Baker IDI Heart and Diabetes Institute. Six-week-old male Apoe −/− mice (ARC, Canning Vale, WA, Australia) were rendered diabetic by five daily i.p. injections of streptozotocin (Sigma-Aldrich, St Louis, MO, USA) at a dose of 55 mg/kg in citrate buffer [27, 28]. Only mice with a blood glucose level >15 mmol/l at 1 week after the induction of diabetes were studied. Subgroups of diabetic and non-diabetic Apoe −/− mice were administered the NOX inhibitor, GKT137831 by gavage at a dose of 60 mg/kg per day for 10 weeks. Systolic BP was assessed by a non-invasive tail cuff system in conscious mice [29]. At the conclusion of the study, the mice were killed by an i.p. injection of sodium pentobarbital (100 mg/kg; Euthatal, Sigma-Aldrich, Castle Hill, NSW, Australia).
Tissue collection and preparation
The heart and aorta were dissected, with the aortic sinus cleaned of surrounding cardiac tissue and snap frozen in liquid nitrogen. Hearts, cut in half at a plane parallel to the tips of the atria, were snap frozen in liquid nitrogen and then embedded in optimal cutting temperature compound (OCT) for cryosectioning. The aortic sinus and mediastinal lymph nodes (meLNs) from 8-week-old mice (1 week after the stabilisation of diabetes), for gene expression analyses, were isolated under dissecting microscope (Olympus SZX10; Olympus, Tokyo, Japan). Between four and six meLNs were collected per mouse (n = 4–6 mice/group) at the end of the study period for flow cytometry.
NOX inhibitor
GKT137831, a member of the pyrazolopyridine dione family, is a specific inhibitor of NADPH isoforms NOX1 and NOX4 (NOX1 K i = 110 nmol/l and Nox4 K i = 140 nmol/l) and has been used previously by our group and others to inhibit NOX1- and NOX4-derived ROS in vivo and in vitro [20, 30].
Plasma lipid analysis
Blood was collected in heparin via cardiac puncture and total cholesterol, HDL and LDL concentrations were measured in plasma with a standard commercial enzymatic assay using a Beckman Coulter LX20PRO Analyser (Cat. No. 467825; Beckman Coulter Diagnostics, Gladsville, NSW, Australia).
Atherosclerotic lesion assessment
Aortic sinus lesions were stained with Oil Red O as described previously [31]. Briefly, aortic sinus cryosections (6 μm) were collected starting from the appearance of the first complete leaflet and stained with Oil Red O (Sigma-Aldrich, St Louis, MO, USA). Images were visualised under light microscopy at 40× magnification (Olympus BX50; Olympus Optical, Tokyo, Japan), digitised with a JVC high resolution camera (Olympus BX50; Olympus Optical) and quantified using Image Pro-Plus 6.0 (Media Cybernetics, Silver Spring, MD, USA). For each mouse, three cross-sections, 60 μm apart, were analysed and the results of each group were expressed as lesion area (μm2) ± SEM. Plaque necrosis was quantified by measuring the area of hematoxylin and eosin-negative acellular areas in the intima, as described previously [32], and the results were expressed as percentage of necrotic core per lesion area.
Cell immunostaining and flow cytometry
meLNs were isolated from at least four mice per group (four to six lymph nodes per mouse) into chilled RPMI 1640 medium supplemented with 10% (v/v) heat-inactivated FCS, 100 U/ml penicillin, 100 μg/ml streptomycin and 100 μg/ml l-glutamine. Single-cell suspensions (approximately 1 × 106 cells/ml) were stained for CD3, CD4, CD8, CD19, CD44, CD62L, CD11c and CD11b in combination with Fc block (BD Pharmingen, North Ryde, NSW, Australia) to minimise background staining. Antibody details and gating strategy are outlined in electronic supplementary material (ESM) Table 1 and ESM Fig. 1, respectively. A minimum of 10,000 events were collected on a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, USA) using CellQuest Pro software (BD Biosciences, San Jose, CA, USA) and analysed with GateLogic software (Inivai, Mentone, VIC, Australia). Results were expressed as the ratio of cells expressing specific marker(s) within the given parent gate.
Immunochemistry of atherosclerotic lesions
Frozen sinus sections were stained with antibodies specific for murine nitrotyrosine (NT), MCP-1, VCAM-1, CD4, CD8, CD11c and IFNγ. Antibody details and dilutions are outlined in ESM Table 2. Sections were fixed with acetone and quenched in 3% H2O2 in TRIS-buffered saline (pH 7.6) to inhibit endogenous peroxidase activity. Sections were incubated with 2.5% Normal Horse Serum (1:10; Vector Laboratories, Burlingame, CA, USA) for 30–60 min at room temperature. Primary antibodies were applied for 1 h at room temperature, although MCP-1 and CD11c required overnight incubation at 4°C in a humidified chamber. Sections were incubated with sequential blocks to avidin and biotin (Vector Laboratories) followed by the appropriate biotinylated secondary antibody (Vector Laboratories) for 30 min. All sections were incubated with horseradish peroxidase-conjugated streptavidin (Vectastain Elite ABC Staining Kit; Vector Laboratories) visualised with 3,3′-diamino-benzidine tetrahydrochloride/H2O2 (DAB; Sigma-Aldrich, St Louis, MO, USA) and counterstained with Mayer’s haematoxylin before mounting with DePex (Sigma-Aldrich, St Louis, MO, USA). Cells staining positive for CD4, CD8 and CD11c were expressed as cell number per unit area of lesion. All other results expressed positively stained area as a percentage of the total lesion area.
Quantitative RT-PCR
Total RNA was extracted from homogenised aortic sinuses and one or two meLNs in Trizol reagent (Invitrogen Australia, Mt Waverly, VIC, Australia) using a bullet blender system (Next Advance, Averill Park, NY, USA). Contaminating DNA was removed after treatment with DNA-free DNAse according to the manufacturer’s specifications (Ambion, Austin, TX, USA). The resulting DNA-free RNA (0.35 μg of sinus RNA and 0.25 μg of meLN RNA) was reverse transcribed into cDNA using the Superscript First Strand Synthesis System (Life Technologies BRL, Grand Island, NY, USA). Gene expression was analysed by quantitative RT-PCR using a Taqman system (ABI Prism 7500; Perkin-Elmer, Poster City, CA, USA) except for IFNγ gene analysis, which used SYBR Green PCR Master Mix (Applied Biosystems, Darmstadt, Germany). Fluorescence for each cycle was analysed quantitatively and gene expression normalised relative to the expression of the housekeeping gene for 18S ribosomal RNA (18S rRNA Taqman Control Reagent kit), which was multiplexed with the gene of interest. For the Syber system, the 18S rRNA control was run separately from the gene of interest but on the same plate. Probes and primers were designed using a Primer Express program (sequences outlined in ESM Table 3) and were purchased from Applied Biosystems (Foster City, CA, USA). Amplifications were performed with the following time course: 50°C for 2 min, 95°C for 10 min and 50 cycles of 94°C for 20 s and 60°C for 1 min. The data were normalised to 18S rRNA and were expressed as fold inductions relative to control non-diabetic Apoe −/− mice (arbitrarily assigned a value of 1).
Measurement of superoxide production in aortic tissue in situ
Superoxide generation was evaluated with the oxidative fluorescent dye dihydroethidium (DHE), as described previously [33]. In brief, cryosections (6 μm) were incubated with 2 μmol/l DHE (Molecular Probes, Eugene, OR, USA) in DMSO, at 37°C for 30 min, rinsed with cold PBS and mounted in AquaMount media for immediate imaging (Olympus BX61; Olympus Optical, Tokyo Japan) using an excitation wavelength of 488 nm and an emission fluorescence filter at 590 nm. Images were taken at a low exposure time to minimise any background fluorescence. Fluorescent ethidium signal was quantified using NIH Image J software (http://rsbweb.nih.gov/ij/) and expressed as the mean fluorescence intensity ± SEM as a percentage of lesion area (n = 5–7).
Statistical analysis
All data are presented as means ± SEM, unless otherwise indicated. Groups were analysed by one-way and two-way ANOVA with an LSD post hoc test for multiple comparison of the means using SPSS Statistics version 20 (IBM, St Leonards, NSW, Australia). A p value of <0.05 was considered statistically significant.
Results
Metabolic variables
Metabolic characteristics for each experimental group are outlined in ESM Table 4. Induction of diabetes significantly increased blood glucose (F = 44.2, p < 0.001) and HbA1c levels (by 37%; F = 44.7, p < 0.001) compared with controls, which did not change following treatment. As expected, diabetic Apoe −/− mice had significantly lower body weights (F = 16.9, p < 0.001), higher plasma concentrations of LDL (F = 12.1, p < 0.001), total cholesterol (F = 12.9, p < 0.001) and triacylglycerols (F = 6.46, p < 0.014) when compared with the controls. Body weight reduction and dyslipidaemia were largely unaffected by treatment with GKT137831, although triacylglycerols were significantly reduced compared with untreated diabetic mice (diabetes–treatment interaction: F = 4.28, p < 0.044).
Assessment of atherosclerotic lesions in the aortic sinus
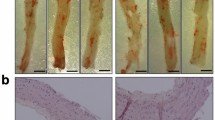
After 10 weeks, diabetic mice showed a significant increase in atherosclerotic plaques within the aortic sinus compared with non-diabetic controls, as assessed by Oil Red O staining (Fig. 1a, b; F = 10.5, p < 0.002). Treatment of diabetic Apoe −/− mice with GKT137831 significantly reduced lesion area in the aortic sinus by 65% in comparison with untreated diabetic Apoe −/− mice (GKT137831 effect: F = 12.9, p < 0.001; diabetes–treatment interaction: F = 41.6, p < 0.001). This effect was not seen in non-diabetic Apoe −/− mice treated with GKT137831.

Diabetes-induced atherosclerosis and enhanced plaque necrotic core area in the aortic sinus is attenuated by NOX inhibition with GKT137831 in mice. Plaque area was measured at 10 weeks after induction of diabetes as a percentage of total aortic sinus area. Oil Red O stained aortic sinuses, magnification ×40 (a) and quantification (b). Representative photomicrographs of haematoxylin and eosin staining in aortic sinus plaques (c) in addition to quantitative analysis of necrotic core area as a percentage of total plaque area (d). Control (Cont) and diabetic (Diab) Apoe −/− mice treated with or without GKT137831 (GKT) for 10 weeks (60 mg/kg per day). Data are presented as means ± SEM (n = 4–6/group). Groups were analysed using one-way ANOVA with an LSD post hoc test. *p < 0.05, Apoe −/− Cont vs Diab; † p < 0.05, Apoe −/− Diab vs treated Diab
Atherosclerotic necrotic core area
To determine the effect of diabetes on accelerated atherosclerosis development in the aortic sinus, we examined plaque necrotic core area. We observed a significant increase in the percentage of necrotic area in plaques from diabetic Apoe −/− mice, which was decreased by GKT137831 (Fig. 1c, d; GKT137831 effect independent of diabetic status: F = 11.3, p < 0.003).
The diabetes-induced effects on lymphocytes in meLNs are attenuated by NOX inhibition
To evaluate the effect of diabetes on the phenotype and activation status of T lymphocytes we examined the draining lymphoid tissue of the aorta, the meLNs, using flow cytometry. The meLNs from diabetic mice showed a significant increase in the proportion of CD3+ lymphocytes, which was reduced by treatment with GKT137831 (Fig. 2a; GKT137831 effect: F = 6.79, p < 0.024; diabetes–treatment interaction: F = 9.60, p < 0.010). Further analysis of this CD3+ subset revealed a significant increase in the proportion of CD8+ lymphocytes in diabetic meLNs, which was attenuated by GKT137831 treatment (Fig. 2b; F = 7.97, p < 0.017). Similarly, there was a significant reduction in CD4+ lymphocytes with response to GKT137831 treatment (Fig. 2c). Diabetic mice also displayed a higher fraction of activated (CD44+CD62L−) CD4+ cells compared with non-diabetic mice (F = 6.93, p < 0.023); this was normalised by GKT137831 treatment (Fig. 2d; GKT137831 effect: F = 5.22, p < 0.043; diabetes–treatment interaction: F = 5.57, p < 0.038). These phenotypic changes were not observed in the CD8+ T lymphocytes (ESM Fig. 2). In addition, diabetes was associated with a decrease in the proportion of CD11c+CD11b+ leucocytes (F = 8.30, p < 0.015) and an increase in CD11c+CD11b− and CD11c−CD11b+ subsets. Treatment with GKT137831 prevented the diabetes-mediated increase in CD11c+CD11b− cells (Fig. 2e, GKT137831 effect: F = 7.12, p < 0.022; diabetes–treatment interaction: F = 10.5, p < 0.002). Diabetes status also influenced the effect of GKT137831 on CD11c−CD11b+ leucocytes (interaction: F = 5.53, p < 0.038), although this did not result in a significant difference compared with untreated diabetic mice.

Effect of diabetes and NOX inhibition on mouse meLN-derived leucocytes. Proportion of CD3+ (a), CD8+ (b) and CD4+ (c) lymphocytes isolated from the meLNs of control (Cont) and diabetic (Diab) Apoe −/− mice treated with or without GKT137831 (GKT) for 10 weeks (60 mg/kg per day). *p < 0.05, Apoe −/− Cont vs Diab; † p < 0.05, Apoe −/− Diab vs treated Diab. Analysis of CD4+ lymphocyte phenotypes based on CD44 and CD62L expression profile (d): activated (CD44+CD62L−); memory (CD44+CD62+) and naive (CD44−CD62L+). Analysis of monocyte-granulocyte subsets based on CD11c and CD11b expression (e): plasmacytoid dendritic cells (pDC; CD11c+CD11b−); monocytoid DC (mDC; CD11c+CD11b+) and macrophages (MFs; CD11c−CD11b+). Data are presented as means ± SEM (n = 4–6/group). Groups were analysed using one-way ANOVA with an LSD post hoc test. *p < 0.05, Apoe −/− control (black bars) vs diabetic (white bars); † p < 0.05, Apoe −/− diabetic vs treated diabetic (hatched bars); and ‡ p < 0.05, control vs treated control (grey bars)
Markers of immune cells in atherosclerotic plaques
Analysis of sinuses stained for cell-specific antigens revealed a significant increase in the number of CD4+ (Fig. 3a, b) and CD8+ (Fig. 3c, d) cells in the plaques from diabetic Apoe −/− mice compared with non-diabetic Apoe −/− mice. In addition, plaques from diabetic Apoe −/− mice showed a marked increase in CD11c+ cells (Fig. 3e, f). Inhibition of NOX with GKT137831 reduced lesional staining for CD4 (diabetes–treatment interaction: F = 11.4, p < 0.005), CD8 (GKT137831 effect: F = 7.74, p < 0.016; diabetes–treatment interaction: F = 11.5, p < 0.005) and CD11c (interaction: F = 5.18, p < 0.040) in diabetic sinus lesions.

Increased infiltration of inflammatory cells to the aortic sinus plaques of diabetic mice is prevented by GKT137831. Representative sections of aortic sinus immunostained for the presence of CD4 (a), CD8 (c) and CD11c (e); original magnification ×100. Control (Cont) and diabetic (Diab) Apoe −/− mice were treated with or without GKT137831 (GKT) for 10 weeks (60 mg kg−1 day−1). Quantitative analysis of the number of cells positive for CD4 (b), CD8 (d), CD11c (f) per lesion area. Data are presented as means ± SEM (n = 5–7/group). Groups were analysed using one-way ANOVA with an LSD post hoc test. *p < 0.05, Apoe −/− Cont vs Diab and † p < 0.05, Apoe −/− Diab vs treated Diab
Proinflammatory gene and protein expression in vascular and lymphoid tissue
Examination of the inflammation marker MCP-1 identified a significant increase in expression of the gene encoding MCP-1 (F = 5.77, p < 0.028) and also in MCP-1 protein (F = 13.4, p < 0.002) expression in the plaques of diabetic Apoe −/− mice; these increases were attenuated with GKT137831 treatment in diabetic Apoe −/− mice alone (Fig. 4a, b; GKT137831 effect: F = 26.2, p < 0.001; diabetes–treatment interaction: F = 11.1, p < 0.001). Similarly, immunohistochemical analysis of VCAM-1 protein also revealed elevated expression in the plaques from diabetic Apoe −/− mice compared with non-diabetic Apoe −/− mice. VCAM-1 expression was significantly reduced in diabetic Apoe −/− mice when treated with GKT137831 (Fig. 4c, diabetes–treatment interaction: F = 8.39, p < 0.010). Aortic sinuses isolated from diabetic Apoe −/− mice showed a significant increase in Ifng mRNA, which was prevented by treatment with GKT137831 (Fig. 4d, GKT137831 effect: F = 4.31, p < 0.05; diabetes–treatment interaction: F = 5.11, p < 0.035). Similarly, leucocytes in the meLNs from diabetic mice expressed significantly higher levels of Ifng mRNA when compared with non-diabetic mice, although this was unaffected by treatment (Fig. 4e, diabetes effect: F = 17.36, p < 0.001). Staining for IFNγ in the sinus showed that the majority of the IFNγ-positive cells were located at the outer endothelial membrane of the leaflet, suggestive of cells entering via the circulation (ESM Fig. 3). In contrast, genes associated with anti-inflammatory responses, Il10 and Foxp3 (encoding transcription factor for T regulatory cells), were unchanged in the meLNs of diabetic mice compared with non-diabetic mice (ESM Fig. 4).

The diabetes-mediated upregulation of immune–inflammatory markers in mouse aortic sinus and its associated lymphoid tissue is prevented by NOX inhibition with GKT137831. RT-PCR gene expression analysis of recruitment molecules: Mcp-1 (also known as Ccl2) (a) and proinflammatory cytokine Ifng mRNA levels in the aortic sinus (d) and meLNs (e) relative to 18S of control (Cont) and diabetic (Diab) Apoe −/− mice treated with or without GKT137831 (GKT) for 10 weeks (60 mg/kg per day). Quantitative analysis of MCP-1 (b) and VCAM-1 (c) positive staining as a percentage of lesion area. Data are presented as means ± SEM (n = 6/group). Groups were analysed using one-way ANOVA with an LSD post hoc test. *p < 0.05, Apoe −/− Cont vs Diab; † p < 0.05, Apoe −/− Diab vs treated Diab; and § p < 0.05, treated Diab vs treated Cont
Oxidative stress in the diabetic atherosclerotic lesion is reduced by NOX inhibition
Immunostaining for NT was analysed to measure the formation of peroxynitrite, a product of superoxide and nitric oxide interaction. In addition, vascular superoxide in diabetes was assessed using the oxidative fluorescent dye DHE. In diabetic mice, NT staining was significantly increased in atherosclerotic plaques when compared with their non-diabetic counterparts (Fig. 5a, b; F = 6.83, p < 0.021). Aortic sinus plaques from Apoe −/− mice with diabetes showed significantly increased ethidium fluorescence compared with non-diabetic controls (Fig. 5c, d; F = 9.95, p < 0.005). The fluorescence appeared to be more intense from cells within a plaque than from cells in the surrounding tissue. Treatment of diabetic mice with GKT137831 reduced both oxidative stress markers, NT (diabetes–treatment interaction: F = 4.87, p < 0.046) and DHE (GKT137831 effect: F = 9.19, p < 0.006; diabetes–treatment interaction: F = 6.88, p < 0.015) in comparison with untreated diabetic mice. Additionally, gene expression analyses revealed a trend towards an increase in Nox1 (p = 0.118) and a significant increase in Nox4 (p = 0.040) mRNA in the aortic sinus of untreated diabetic mice (ESM Table 5). Treatment of diabetic mice with GKT137831 significantly reduced expression of both Nox1 (GKT137831 effect: F = 6.706, p < 0.020) and Nox4 (diabetes–treatment interaction: F = 6.745, p < 0.020). There was no change in the expression of genes encoding other NOX isoforms, Nox2 and p47-phox (also known as Ncf1), or other potential sources of ROS, including endothelial nitric oxide synthase, inducible NOS and xanthine oxidase.

Increased ROS formation in sinus plaques of diabetic mice is attenuated by NOX inhibition. NT immunohistochemistry (a) and quantification (b) in the plaque, as percentage positive staining per total plaque area. Representative photomicrographs of DHE staining in aortic sinus plaques, magnification ×100 (c) and quantification of fluorescence intensity as a percentage of lesion area (d). AU, arbitrary units. Control (Cont) and diabetic (Diab) Apoe −/− mice treated with or without GKT137831 (GKT) for 10 weeks (60 mg/kg per day). Data are presented as means ± SEM (n = 5–7/group). Groups were analysed using one-way ANOVA with an LSD post hoc test. *p < 0.05 Apoe −/− Cont vs Diab; † p < 0.05, Apoe −/− Diab vs treated Diab
Discussion
These studies have demonstrated that diabetes-accelerated atherosclerosis is associated with increased immune cell infiltration into the aortic sinus lesion and altered T cell activation in the adjacent meLNs, and that these pro-atherogenic processes are associated with elevated NOX-derived ROS. The important contributions of immune cell-mediated inflammation and oxidative stress to atherosclerosis have been well established [12, 34, 35]. However, the interactions between key ROS-producing enzymes and inflammatory/immune cell activation in this process remain elusive in the setting of diabetes. To investigate the role of two well-characterised vascular isoforms of the NADPH enzyme, NOX1 and NOX4, in the development of diabetes-associated atherosclerosis, we administered the NOX1/NOX4 dual inhibitor GKT137831 to diabetic Apoe −/− mice.
Diabetes was associated with a significant increase in atherosclerotic plaque area in the aortic sinus of Apoe −/− mice, accompanied by elevated levels of oxidative stress as reflected by enhanced NT and DHE staining in the plaque. Treatment with GKT137831 prevented the diabetes-associated increase in ROS production and attenuated the accelerated development of atherosclerosis in the aortic sinus. Recent data published by our group demonstrated that treatment with GKT137831 significantly reduced vascular ROS production and atherosclerotic plaque formation in the aortic arch and total aorta of diabetic Apoe −/− mice, and this was also associated with a reduction in diabetes-induced leucocyte adhesion to the vascular wall [20].
A key finding of this study in Apoe −/− mice is the impact of diabetes on adaptive immune cell phenotypes in the draining lymph nodes for the aorta. Diabetes increased the proportion of CD8+ T cells and activated CD4+ T cells in the meLNs, which was associated with an increased accumulation of T cells into the atherosclerotic lesion. Previous studies have shown that hyperglycaemia induces activation of pro-inflammatory T lymphocytes and decreases the suppressor function of T regulatory cell subsets [36, 37]. In this study, the increase in activated CD4+ T cells in the meLNs of diabetic Apoe −/− mice did not appear to be associated with aberrant immune suppression, as we did not observe differences in expression of genes encoding Treg and Th2 cell markers (Foxp3 and Il10, respectively). Of note, Kolbus et al showed that T cells in the draining lymph nodes of the aorta are also exposed to antigens acquired in the atherosclerotic plaque, which results in their activation to Th1 cells [38].
The diabetes-associated alterations in T cell functional phenotypes correlated with an increase in Ifng expression in the meLNs and aortic sinus plaque. The proinflammatory cytokine IFNγ has been shown to play an important role in atherosclerosis [39] through recruitment of monocytes and T lymphocytes to the plaque via the induction of chemokines, such as MCP-1 [24]. Diabetes-induced expression of IFNγ, MCP-1 and VCAM-1 in aortic sinus lesions was coupled with a significant increase in the infiltration of CD4+ and CD8+ cells into the atherosclerotic lesion. Previous work by Zhou et al demonstrated a predominance of IFNγ-producing CD4+ Th1 cells in atherosclerotic lesions, indicative of the proinflammatory Th1-CD4+ subset [40]. In line with these findings, we observed more than twice as many CD4+ cells than CD8+ cells in the aortic sinuses of atherosclerosis prone diabetic Apoe −/− mice. Furthermore, we showed localisation of IFNγ protein in the aortic sinus to cells at the luminal face of the leaflets, indicative of infiltrating proinflammatory immune cells from the circulation. Collectively these data suggest a possible link between the inflamed vasculature and its associated lymphoid tissue, which warrants further investigation. In this study, we show for the first time that inhibition of NOX-generated ROS with GKT137831 prevents the diabetes-induced increase in lesional accumulation of CD4+, CD8+ and CD11c+ cells, with an associated reduction in plaque area. Additionally, GKT137831 attenuates the diabetes-induced increases in CD8+ T cells and activated CD4+ T cells in the meLNs in conjunction with reduced Ifng expression in the aortic sinus of diabetic mice. Importantly, many of the changes in immune cells in the meLNs and the aortic sinus of mice treated with the NOX inhibitor were only observed in the setting of diabetes. Collectively, our data provides evidence that a T cell-driven type 1 cytokine response, characterised by IFNγ [41], may predominate in the aortic sinus of diabetic mice under the modulation of NOX-derived ROS.
IFNγ elicits pro-apoptotic actions on macrophage-foam cells and vascular smooth muscle cells in advanced atherosclerotic plaques [42]. Activated CD8+ T lymphocytes secrete large amounts of IFNγ in addition to granzymes and perforins, which elicit apoptosis and necrosis of cells in plaques [43]. In this study, the increase in CD8+ T cells in the aortic sinus lesions of diabetic Apoe −/− mice could signify an important role for T lymphocyte-mediated cytotoxicity in diabetic atherosclerosis. Furthermore, IFNγ secreted by activated T lymphocytes in the plaque can impede collagen synthesis by smooth muscle cells, which reinforces the plaque [44]. Consistent with this notion, plaques from diabetic Apoe −/− mice had significantly larger necrotic cores compared with plaques from non-diabetic mice. This finding is of particular clinical relevance, given the strong link between plaque inflammation, necrotic core size and the risk of plaque rupture [45, 46]. In particular, human diabetic lesions are characterised by very large necrotic cores [47]. Our study reveals that treatment of diabetic mice with GKT137831 prevents the diabetes-mediated increases in lesional CD8+ T cells and necrotic core area. Furthermore, peroxynitrite has been shown to prime T cells for apoptosis [48], suggesting that the increase in NT staining (secondary to peroxynitrite formation) and larger necrotic core that we observed in the plaques of diabetic mice may be associated with enhanced ROS-mediated T cell apoptosis. These results propose an important role for NOX-derived ROS in processes that underscore the progression of atherosclerotic plaques to more unstable, rupture-prone plaques.
Dendritic cells (DCs) play a central role in antigen presentation and activation of T cells in atherosclerosis [49]. A number of cell surface markers are expressed by DCs, although the CD11c+ DCs classically migrate to secondary lymphoid tissues to activate antigen-specific T cells [5]. In this study, we identified a significant increase in CD11c+ cells recruited to the aortic sinus of diabetic mice, which was attenuated by GKT137831 treatment. Furthermore, we found that the proportion of CD11c+CD11b− leucocytes was increased in the meLNs of diabetic mice and this was normalised on treatment with GKT137831. The role of DCs can be atherogenic or atheroprotective depending on priming environment [38]. Interestingly, the induction of ROS correlates with inflammatory DC functionality and expansion [50]. These results implicate NOX1/NOX4-derived ROS in the modulation of DC recruitment to the atherosclerotic vasculature, as well as DC function in the draining lymph nodes, which would increase their exposure to plaque antigens and facilitate interactions between DCs and CD4+ T cells ultimately leading to their activation.
In summary, we show that diabetes promotes atherosclerosis, inflammation and oxidative stress in the aortic sinus of diabetic Apoe −/− mice. Specifically, diabetes was associated with increased expression of genes encoding the proinflammatory molecules IFNγ, MCP-1 and VCAM-1, with evidence of enhanced immune responses in both the vasculature and its associated lymphoid tissue. Our study clearly demonstrated that inhibition of NOX-derived ROS is associated with reduced accumulation and decreased activation of T lymphocytes and CD11c+ cells in diabetes-induced atherosclerotic plaques. In addition, our study has demonstrated a role for NOX-derived ROS in the activation of T lymphocytes in the draining lymph nodes of the aorta, which is likely to contribute to inflammatory responses in the aorta. Treatment of diabetic Apoe −/− mice with the pharmacological NOX inhibitor GKT137831 attenuated many of these pro-atherogenic changes, and this may indicate promotion of a more stable plaque phenotype. These data provide new evidence for the involvement of NOX-derived ROS in fuelling immune-mediated inflammation in aortic sinus lesions of diabetic Apoe −/− mice.
Abbreviations
- DC:
-
Dendritic cell
- DHE:
-
Dihydroethidium
- MCP-1:
-
Monocyte chemoattractant protein 1
- meLN:
-
Mediastinal lymph node
- NT:
-
Nitrotyrosine
- NOX:
-
NADPH oxidase
- ROS:
-
Reactive oxygen species
- VCAM-1:
-
Vascular adhesion molecule 1
References
Kannel WB, McGee DL (1979) Diabetes and cardiovascular disease. The Framingham study. J Am Med Assoc 241:2035–2038
Mäkinen VP, Forsblom C, Thorn LM et al (2009) Network of vascular diseases, death and biochemical characteristics in a set of 4,197 patients with type 1 diabetes (The FinnDiane Study). Cardiovasc Diabetol 8:54
Ross R (1999) Atherosclerosis - an inflammatory disease. N Engl J Med 340:115–126
Lassègue B, Griendling KK (2010) NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol 30:653–661
Hansson GK, Hermansson A (2011) The immune system in atherosclerosis. Nat Immunol 12:204–212
Piga R, Naito Y, Kokura S, Handa O, Yoshikawa T (2007) Short-term high glucose exposure induces monocyte-endothelial cells adhesion and transmigration by increasing VCAM-1 and MCP-1 expression in human aortic endothelial cells. Atherosclerosis 193:328–334
Kanter JE, Bornfeldt KE (2013) Inflammation and diabetes-accelerated atherosclerosis: myeloid cell mediators. Trends Endocrinol Metab 24:137–144
Soro-Paavonen A, Watson AMD, Li J et al (2008) Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes 57:2461–2469
Hansson GK, Holm J, Jonasson L (1989) Detection of activated T lymphocytes in the human atherosclerotic plaque. Am J Pathol 135:169–175
Frostegård J, Ulfgren AK, Nyberg P et al (1999) Cytokine expression in advanced human atherosclerotic plaques: dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis 145:33–43
Van Der Wal AC, Becker AE, van Der Loos CM, Das PK (1994) Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation 89:36–44
Baynes JW (1991) Role of oxidative stress in development of complications in diabetes. Diabetes 40:405–412
Griendling KK, Ushio-Fukai M (1997) NADH/NADPH oxidase and vascular function. Trends Cardiovasc Med 7:301–307
Hink U, Li H, Mollnau H et al (2001) Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res 88:E14–E22
Sedeek M, Callera G, Montezano A et al (2010) Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. Am J Physiol Ren Physiol 299:F1348–F1358
Guzik TJ, Sadowski J, Guzik B et al (2006) Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arterioscler Thromb Vasc Biol 26:333–339
Ding H, Hashem M, Triggle C (2007) Increased oxidative stress in the streptozotocin-induced diabetic apoE-deficient mouse: changes in expression of NADPH oxidase subunits and eNOS. Eur J Pharmacol 561:121–128
Guzik TJ, Mussa S, Gastaldi D et al (2002) Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 105:1656–1662
Wendt MC, Daiber A, Kleschyov AL et al (2005) Differential effects of diabetes on the expression of the gp91 phox homologues nox1 and nox4. Free Radic Biol Med 39:381–391
Gray SP, Di Marco E, Okabe J et al (2013) NADPH Oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 127:1888–1902
Marumo T, Schini-Kerth VB, Fisslthaler B, Busse R (1997) Platelet-derived growth factor-stimulated superoxide anion production modulates activation of transcription factor NF-κB and expression of monocyte chemoattractant protein 1 in human aortic smooth muscle cells. Circulation 96:2361–2367
Kunsch C, Medford RM (1999) Oxidative stress as a regulator of gene expression in the vasculature. Circ Res 85:753–766
Cybulsky MI, Iiyama K, Li H et al (2001) A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Investig 107:1255–1262
Mach F, Sauty A, Iarossi AS et al (1999) Differential expression of three t lymphocyte-activating CXC chemokines by human atheroma-associated cells. J Clin Investig 104:1041–1050
Nareika A, Sundararaj KP, Im YB, Game BA, Lopes-Virella MF, Huang Y (2009) High glucose and interferon gamma synergistically stimulate MMP-1 expression in U937 macrophages by increasing transcription factor STAT1 activity. Atherosclerosis 202:363–371
Esposito K, Nappo F, Marfella R et al (2002) Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress. Circulation 106:2067–2072
Hsueh W, Abel ED, Breslow JL et al (2007) Recipes for creating animal models of diabetic cardiovascular disease. Circ Res 100:1415–1427
Candido R, Jandeleit-Dahm KA, Cao Z et al (2002) Prevention of accelerated atherosclerosis by angiotensin-converting enzyme inhibition in diabetic apolipoprotein E-deficient mice. Circulation 106:246–253
Krege JH, Hodgin JB, Hagaman JR, Smithies O (1995) A noninvasive computerized tail-cuff system for measuring blood pressure in mice. Hypertension 25:1111–1115
Aoyama T, Paik YH, Watanabe S et al (2012) Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology 56:2316–2327
Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA (1987) Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis 68:231–240
Feng B, Zhang D, Kuriakose G, Devlin CM, Kockx M, Tabas I (2003) Niemann-Pick C heterozygosity confers resistance to lesional necrosis and macrophage apoptosis in murine atherosclerosis. Proc Natl Acad Sci U S A 100:10423–10428
Elmarakby AA, Loomis ED, Pollock JS, Pollock DM (2005) NADPH oxidase inhibition attenuates oxidative stress but not hypertension produced by chronic ET-1. Hypertension 45:283–287
Sheehan AL, Carrell S, Johnson B, Stanic B, Banfi B, Miller FJ (2011) Role for Nox1 NADPH oxidase in atherosclerosis. Atherosclerosis 216:321–326
Kanter JE, Averill MM, Leboeuf RC, Bornfeldt KE (2008) Diabetes-accelerated atherosclerosis and inflammation. Circ Res 103:e116–e117
Stentz FB, Kitabchi AE (2005) Hyperglycemia-induced activation of human T-lymphocytes with de novo emergence of insulin receptors and generation of reactive oxygen species. Biochem Biophys Res Commun 335:491–495
Lindley S, Dayan CM, Bishop A, Roep BO, Peatman M, Tree TIM (2005) Defective suppressor function in CD4+CD25+ T cells from patients with type 1 diabetes. Diabetes 54:92–99
Kolbus D, Ramos OH, Berg KE et al (2010) CD8+T cell activation predominate early immune responses to hypercholesterolemia in Apoe−(/)− mice. BMC Immunol 11:58
Gupta S, Pablo AM, Jiang XC, Wang N, Tall AR, Schindler C (1997) IFN-γ, potentiates atherosclerosis in ApoE knock-out mice. J Clin Investig 99:2752–2761
Zhou X, Nicoletti A, Elhage R, Hansson GK (2000) Transfer of CD4+ T cells aggravates atherosclerosis in immunodeficient apolipoprotein E knockout mice. Circulation 102:2919–2922
Uyemura K, Demer LL, Castle SC et al (1996) Cross-regulatory roles of interleukin (IL)-12 and IL-10 in atherosclerosis. J Clin Investig 97:2130–2138
McLaren JE, Ramji DP (2009) Interferon gamma: a master regulator of atherosclerosis. Cytokine Growth Factor Rev 20:125–135
Kyaw T, Winship A, Tay C et al (2013) Cytotoxic and proinflammatory CD8+ T lymphocytes promote development of vulnerable atherosclerotic plaques in apoE-deficient mice. Circulation 127:1028–1039
Libby P, Ridker PM, Maseri A (2002) Inflammation and atherosclerosis. Circulation 105:1135–1143
Falk E, Shah PK, Fuster V (1995) Coronary plaque disruption. Circulation 92:657–671
Thim T, Hagensen MK, Bentzon JF, Falk E (2008) From vulnerable plaque to atherothrombosis. J Intern Med 263:506–516
Burke AP, Kolodgie FD, Zieske A et al (2004) Morphologic findings of coronary atherosclerotic plaques in diabetics: a postmortem study. Arterioscler Thromb Vasc Biol 24:1266–1271
Brito C, Naviliat M, Tiscornia AC et al (1999) Peroxynitrite inhibits T lymphocyte activation and proliferation by promoting impairment of tyrosine phosphorylation and peroxynitrite-driven apoptotic death. J Immunol 162:3356–3366
Bobryshev YV (2010) Dendritic cells and their role in atherogenesis. Lab Investig 90:970–984
Sheng KC, Pietersz GA, Tang CK, Ramsland PA, Apostolopoulos V (2010) Reactive oxygen species level defines two functionally distinctive stages of inflammatory dendritic cell development from mouse bone marrow. J Immunol 184:2863–2872
Acknowledgements
The authors would like to thank E. Grixti for his expert assistance in measuring metabolic variables, M. Arnstein for assistance with general tissue pathology/immunohistochemistry and K. Gilbert for maintenance of the mice (all affiliated with Baker IDI Heart and Diabetes Institute, Melbourne, VIC, Australia). Appreciation extends to G. Paukovic (Burnet Institute, Melbourne, VIC, Australia) for his expertise in flow cytometry. Kind thanks are also given to A. Bobik (Department of Vascular Biology and Atherosclerosis, Baker IDI Heart and Diabetes Institute) for intellectual contribution to discussions.
Funding
This work was supported by the National Health and Medical Research Council project grant of Australia. KAJD is supported by an NHMRC senior Research Fellowship and MEC is an Australian Fellow for the NHMRC and a JDRF scholar. EDM is supported by a Post-graduate Fellowship (PB 12M 6943) from the National Heart Foundation of Australia.
Duality of interest
CS is a paid employee and owns shares in Genkyotex SA, Geneva, Switzerland. All other authors declare that there is no duality of interest associated with their contribution to this manuscript.
Contribution statement
EDM contributed to the direction of the study, conducted data collection and subsequent analyses and wrote and edited the manuscript. SPG directed the study, conducted data analysis and wrote and edited the manuscript. PC performed and analysed some of the immunohistochemical stains and reviewed the manuscript. CK contributed to collection of meLNs, analysis of some immunohistochemistical stains and reviewed the manuscript. AZ assisted with flow cytometry data collection and analysis and reviewed the manuscript. CS was involved in the conception of the study design, contributed to the protocol for GKT137831 preparation and administration and reviewed and edited the manuscript. RMT and HHHWS assisted with data analysis of oxidative stress variables and also reviewed the manuscript. MEC contributed to data analysis and critical revisions of intellectual content of the manuscript. RS assisted with flow cytometry analysis and edited and reviewed the manuscript. KAJD contributed to the design and supervision of the study, provided intellectual input to discussions and edited and reviewed the manuscript. All authors approved the manuscript for publication.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
(PDF 50 kb)
ESM Table 2
(PDF 93 kb)
ESM Table 3
(PDF 73 kb)
ESM Table 4
(PDF 73 kb)
ESM Table 5
(PDF 74 kb)
ESM Fig. 1
(PDF 228 kb)
ESM Fig. 2
(PDF 99 kb)
ESM Fig. 3
(PDF 141 kb)
ESM Fig. 4
(PDF 113 kb)
Rights and permissions
About this article
Cite this article
Di Marco, E., Gray, S.P., Chew, P. et al. Pharmacological inhibition of NOX reduces atherosclerotic lesions, vascular ROS and immune–inflammatory responses in diabetic Apoe −/− mice. Diabetologia 57, 633–642 (2014). https://doi.org/10.1007/s00125-013-3118-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-3118-3