
Abstract
Aims/hypothesis
Pro-opiomelanocortin (POMC) neurons in the arcuate nucleus (ARC) regulate energy homeostasis by secreting α-melanocyte-stimulating hormone (α-MSH), derived from POMC precursor, in response to leptin signalling. Expression of Pomc is subject to multiple modes of regulation, including epigenetic regulation. Methyl-CpG-binding protein 2 (MeCP2), a nuclear protein essential for neuronal function, interacts with promoters to influence gene expression. We aim to address whether MeCP2 regulates hypothalamic Pomc expression and to investigate the role of epigenetics, particularly DNA methylation, in this process.
Methods
We generated a mouse line with MeCP2 specifically deleted in POMC neurons (Mecp2 flox/y/Pomc-Cre [PKO]) and characterised its metabolic phenotypes. We examined the DNA methylation pattern of the Pomc promoter and its impact on hypothalamic gene expression. We also studied the requirement of MeCP2 for, and the effects of, DNA methylation on Pomc promoter activity using luciferase assays.
Results
PKO mice are overweight, with increased fat mass resulting from increased food intake and respiratory exchange ratio. PKO mice also exhibit elevated plasma leptin. Deletion of MeCP2 in POMC neurons leads to increased DNA methylation of the hypothalamic Pomc promoter and reduced Pomc expression. Furthermore, in vitro studies show that hypermethylation of the Pomc promoter reduces its transcriptional activity and reveal a functional synergy between MeCP2 and cAMP responsive element binding protein 1 (CREB1) in positively regulating the Pomc promoter.
Conclusions/interpretation
Our results demonstrate that MeCP2 positively regulates Pomc expression in the hypothalamus. Absence of MeCP2 in POMC neurons leads to increased DNA methylation of the Pomc promoter, which, in turn, downregulates Pomc expression, leading to obesity in mice with an accentuating degree of leptin resistance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The fundamental cause of overweight and obesity is an energy imbalance, i.e. increased food intake and/or reduced energy expenditure. The leptin–melanocortin pathway is a key intrinsic regulator in the maintenance of energy homeostasis [1]. Leptin is secreted from adipose tissue and acts on at least two groups of neurons: POMC neurons, which are activated by leptin to produce anorexigenic POMC; and neuropeptide Y (NPY)/Agouti-related peptide (AgRP) neurons, which produce orexigenic NPY and AgRP and are inhibited by leptin signalling [2, 3]. Regulation of Pomc expression is critical for energy homeostasis, as impaired Pomc regulation leads to leptin resistance and obesity [4–6]. Pomc transcription is mainly activated by leptin through the JAK2/STAT3 pathway [7–10]. Deficits in JAK2/STAT3 signalling in POMC neurons are associated with reduced Pomc expression, leptin resistance and obesity [11, 12].
The expression of Pomc is subject to multiple levels of control. In addition to upstream signalling, such as leptin signalling, epigenetic modification of the Pomc promoter may also play a significant role in the regulation of its expression. The role and mechanism of leptin signalling in regulating Pomc expression is relatively well understood [13], while the evidence for epigenetic regulation in hypothalamic Pomc expression has just begun to emerge. Embedded in a CpG island, the 5′ promoter of human POMC is methylated in normal non-expressing tissues, and is specifically unmethylated in expressing tissues, tumours and some cell lines [14]. Changes in peri-conceptional or perinatal environment can change DNA methylation levels of the Pomc promoter, which is associated with obesity in adulthood. Pomc promoter was found to be hypomethylated after peri-conceptional undernutrition in sheep [15]. Early postnatal overfeeding predisposed the rats to obesity, and hypothalamic Pomc promoter was found to be hypermethylated and its expression was downregulated [16]. These studies all indicate that the hypothalamic Pomc promoter is a key target of perinatal epigenetic programming that leads to permanent changes in gene expression and increased risk of obesity.
Methylated DNA is able to recruit methyl-CpG-binding protein 2 (MeCP2), which regulates gene expression [17]. Mutation of Mecp2 is associated with Rett syndrome (RTT), a neuropsychiatric disorder [18]. MeCP2 has long been recognised as a transcriptional repressor that acts by binding to methylated DNA and recruiting a repressor complex [19, 20]. Only recently has it been recognised that MeCP2 in the hypothalamus may function as a transcriptional activator through its interaction with cAMP responsive element binding protein 1 (CREB1) in a DNA-methylation-independent manner [21]. Many mouse models with mutant or deleted MeCP2 have been established to study RTT [18, 22–26]. Besides RTT-like phenotypes, some of these mouse models share a common obesity phenotype. Deletion of MeCP2 in postmitotic neurons, in postnatal CNS, or in Sim1-expressing neurons all result in an overweight or obese phenotype [22, 25, 26], indicating that MeCP2 in the CNS may play a role in regulating energy homeostasis.
We investigated whether and how MeCP2 could play a role in the epigenetic regulation of Pomc expression in the hypothalamus, and whether and how the epigenetic regulation, especially DNA methylation, could contribute to obesity and leptin resistance, based on knowledge that: (1) α-melanocyte-stimulating hormone (α-MSH), a POMC product, is a key regulator of energy homeostasis; (2) the Pomc promoter is a target of epigenetic modification and the changes in DNA methylation on Pomc promoter are associated with obesity; and (3) MeCP2 is an important player in epigenetic regulation and plays an important role in the hypothalamus as a transcriptional regulator.
Methods
Animal welfare
All experiments involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of the Agency for Science, Technology and Research (A*STAR). All mice in this study were housed and bred in the Biological Resource Center (A*STAR) on a 12 h/12 h light/dark cycle with free access to food and water.
Generation of a mouse line with POMC-neuron-specific deletion of MeCP2
Two mouse lines B6.129P2-Mecp2tm1Bird/J and Tg(Pomc1-Cre)16Lowl/J were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). The two lines were crossed to generate Mecp2 flox/y (control) and Mecp2 flox/y/Pomc-Cre (PKO). Genotyping primer sequences are shown in electronic supplementary material (ESM) Table 1.
Body weight and body composition
Age-matched littermates were weighed weekly or biweekly after weaning. Body composition was measured using EchoMRI-100 (Echo Medical Systems, Houston, TX, USA) as previously described [27].
Animal monitoring
Oxymax/Comprehensive Lab Animal Monitoring System (CLAMS; Columbus Instruments, Columbus, OH, USA) was used to measure oxygen consumption (\( \overset{\cdot }{V}{\mathrm{O}}_2 \)), carbon dioxide production (\( \overset{\cdot }{V}{\mathrm{CO}}_2 \)), food intake and activity [28]. Paired mice were individually housed in the chambers that were maintained at 24 ± 1°C, and provided with free access to chow diet and water. All measurements were taken every 15 min for 3 consecutive days after the mice were acclimatised for 2 days. Respiratory exchange ratio (RER) was calculated as the ratio of \( \overset{\cdot }{V}{\mathrm{CO}}_2 \) to \( \overset{\cdot }{V}{\mathrm{O}}_2 \). Basal metabolic rate (BMR) was determined by averaging the lowest plateau region of oxygen consumption curve corresponding to resting periods. The X-total or Z-total movement was the total number of times the motion sensors detected for horizontal or vertical movement, respectively. All data collected from monitoring over 3 days were used in relevant analyses.
Plasma leptin measurements
Mice were fasted for 2 h before blood collection. Blood samples were collected into Microvette blood collection tubes (Kent Scientific Cooperation, Torrington, CT, USA) and cleared by centrifugation at 10,000 g for 2 min at 4°C. Plasma was collected and used for mouse leptin measurement (mouse leptin ELISA, Millipore, Billerica, MA, USA) according to the manufacturer’s instructions.
Immunohistochemistry
Immunohistochemistry was carried out as previously described with slight modifications [29]. Mice were perfused with 4% paraformaldehyde and brains were dissected and post-fixed at 4°C overnight. After cryopreservation in 30% sucrose, coronal cryosections (16 μm) containing hypothalamus were cut from the brain blocks and probed with anti-MeCP2 antibody (1:500; Upstate; catalogue number ABE171, Billerica, MA, USA) and anti-POMC precursor (1:500, Phoenix Pharmaceuticals, Burlingame, CA, USA) in blocking buffer (3% goat serum, 3% BSA and 0.01% Triton X-100 in PBS) for 48 h at 4°C, followed by incubation with Alexa Fluor 488 goat anti-chicken antibody (1:1,000, Invitrogen, Carlsbad, CA, USA) and Alexa Fluor 594 goat anti-rabbit (1:1,000, Invitrogen) for 1 h at room temperature. Samples were then subjected to confocal imaging using a Nikon A1R+si confocal microscope.
RNA extraction and quantitative PCR
Mouse hypothalamus was dissected into RNAlater (Invitrogen), kept overnight at 4°C and then stored at −80°C until required. RNA was extracted from the hypothalami with RNeasy Mini Kit (Qiagen, Duesseldorf, Germany) according to the manufacturer’s instructions, treated with DNase I (Invitrogen) and reverse-transcribed with RevertAid (Fermentas, Ottawa, ON, Canada). Quantitative PCR was performed using SYBR Green PCR master mix (Invitrogen) and primers (see ESM Table 1) on StepOnePlus real-time PCR system (Invitrogen). Samples were analysed in triplicate and normalised against Gapdh as an internal control. Relative changes in gene expression were calculated by the ΔΔCt method.
DNA extraction and bisulfite sequencing
Mouse hypothalamus was dissected and snap-frozen in liquid nitrogen. Genomic DNA was extracted from mouse hypothalami using DNeasy Blood & Tissue Kit (Qiagen). Sodium bisulfite conversion was carried out using 1–2 μg genomic DNA with Epitect Bisulfite Kits (Qiagen). Sequences for the bisulfite sequencing PCR primers are shown in the ESM. The PCR products were purified by gel extraction kit (Qiagen) and ligated into pGEM-T Easy vector (Promega, Madison, WI, USA). PCR products were sequenced for the Pomc promoter region and at least 15 positive clones were analysed.
DNA plasmids
The Pomc promoter–luciferase construct (pGL3-Pomc) was a generous gift from D. Accili (Columbia University, New York, NY, USA). pCMV-SPORT6-CREB1 was purchased from Open Biosystem (catalogue number MMM1013-64862). Myc-tagged CREB1 was generated by PCR and subcloned into pCMV5-Myc at EcoRI and BamHI sites. The primer sequences are shown in ESM Table 1. To generate glutathione S-transferase (GST)-CREB1, pCMV-Myc-CREB1 was digested with EcoRI and BamHI, and subcloned into the EcoRI and XhoI sites of pGEX-KG.
Methyltransferase treatment
Plasmid pGL3-Pomc was treated with different methyltransferases targeting CpGs of varying sequences, including MSssI (CG), HpaII (CCGG) and HhaI (GCGC) (New England BioLabs, Ipswich, MA, USA), according to the manufacturer’s instructions. Methyltransferase-treated DNA preparations were then purified using a PCR purification kit (Qiagen) and used for transfection.
Luciferase assay
The luciferase assay was done as previously described [30]. Briefly, HEK293T cells were transfected with relevant plasmids using FuGENE6 (Roche, Basel, Switzerland). After 2 days, transfected cells were washed with PBS, lysed in 200 μl passive lysis buffer (Promega) and subjected to luciferase activity measurement on a luminometer (Molecular Devices, SpectraMax L, Sunnyvale, CA, USA). The firefly luciferase activity was normalised against Renilla luciferase activity.
Co-immunoprecipitation
HEK293T cells were transfected with MeCP2 (wild type [WT] or R106W) or Myc-CREB1 using the calcium phosphate transfection method. At 2 days after transfection, whole-cell lysates were prepared as previously described [30] and used for co-immunoprecipitation (Co-IP). From the mixture 1 mg was incubated with anti-Flag M2 affinity gel (Sigma-Aldrich, Munich, Germany), EZview Red c-Myc-Agarose (Sigma-Aldrich) or control IgG, respectively, overnight at 4°C. The immunoprecipitates were washed extensively, resuspended in ×2 sample buffer and subjected to SDS-PAGE followed by immunoblotting with Flag (Sigma-Aldrich) or Myc (Santa Cruz, CA, USA) antibodies. From the total cell volume, 2% was used as input.
GST-CREB1 protein expression, purification and pull down
BL21 bacterial cells were transformed to express GST-CREB1 or GST. Fusion proteins were purified using glutathione–Sepharose-4B beads (Amersham, Buckinghamshire, UK) according to the manufacturer’s instructions. As for GST-pull-down assay, protein-coupled glutathione beads were incubated with 500 μg respective total cell lysate of Flag-MeCP2-transfected HEK293 cells for 1 h at 4°C in TNET buffer (50 mmol/l Tris-Cl, pH 7.4, 150 mmol/l NaCl, 1 mmol/l EDTA, 1% TritonX-100). The beads were washed in TNET buffer, resolved by SDS-PAGE and immunoblotted with Flag (Sigma-Aldrich) or CREB1 (Novus biological, Littleton, CO, USA) antibodies.
Statistical analysis
Data were presented as means ± SEM. Statistics was performed by using two-tailed Student’s t test or one-way ANOVA followed by Tukey’s post hoc test. The significance limit was set at p < 0.05.
Results
Specific deletion of MeCP2 in POMC neurons
MeCP2 was deleted specifically in POMC neurons by crossing the mice in which exons 3 and 4 of Mecp2 were flanked by two loxP sites [18] and mice with Cre driven by Pomc1 [31–34] (Fig. 1a). Male Mecp2 flox/y (control) and Mecp2 flox/y/Pomc-Cre (PKO) mice were used for experiments (Fig. 1b). Selective deletion of MeCP2 in POMC neurons was validated by immunohistochemistry for both MeCP2 and POMC precursor. As shown in Fig. 1c, at the ARC region where POMC neurons were concentrated, POMC precursor was distributed in the cytoplasm while MeCP2 was restricted to the nucleus. MeCP2 was present in the nuclei of POMC neurons of the control mouse but not in those of the PKO mouse, confirming the specific deletion of MeCP2 in POMC neurons.

Generation of PKO mice. (a) Homologous recombination and Cre excision strategy for generating PKO mice. Control (fl/fl) showing targeted exon 3 and 4 and flanking loxP sites, PKO showing Mecp2 gene structure after Cre excision. (b) Representative genotyping patterns for Mecp2 +/y, Mecp2 fl/y/Pomc-Cre (PKO), Mecp2 +/y/Pomc-Cre and Mecp2 fl/y (control). (c) Confocal images of double immunofluorescence for the detection of MeCP2 (green) and POMC precursor (red) in coronal brain sections of control and PKO mice. Arrows point to POMC neurons (red) with clear MeCP2 signal (green) in control (third row), but not in PKO (fourth row), indicating specific deletion of MeCP2 in POMC neurons of PKO mice. Scale bars: 100 μm (first and second rows) and 20 μm (third and fourth rows). Ctrl, control
Increased body weight and fat mass and development of leptin resistance in PKO mice
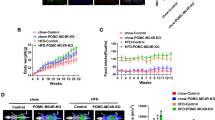
Given that POMC neurons regulate food intake and energy expenditure, we first assessed the roles of MeCP2 in these processes by monitoring the body weight of the mice. The PKO mice had higher body weights starting from the age of 4 months (Fig. 2a). Body composition analysis showed that the PKO mice had higher fat mass compared with controls from the age of 4 months. At 8 months of age, the PKO mice continued to show increased fat mass; however, they exhibited decreased lean mass as a percentage of body weight (Fig. 2b, c), suggesting that body weight gain in the PKO mice was mainly due to increased adiposity. To examine the relative contribution of increased food intake and decreased energy expenditure to the observed body weight gain, we performed indirect calorimetry on the PKO and control mice by using the CLAMS metabolic chambers. The PKO mice had higher food intake and RER when compared with controls (Fig. 2d, e), though no difference was observed in oxygen consumption, BMR or physical activities between PKO mice and controls (Fig. 2f–i). Increased RER in PKO mice indicated lower usage of fat as an energy source, consistent with increased fat mass in these mice. Compared with controls, PKO mice had higher circulating leptin levels at the ages of 4 and 8 months (Fig. 3a). Because leptin is secreted proportionally from adipose tissue, we normalised leptin level to fat mass. After fat mass normalisation, the leptin level of PKO mice was still higher than that of controls at the age of 8 months (Fig. 3b). The findings of higher leptin levels and increased food intake in the PKO mice suggest that these mice developed leptin resistance. Although the increases in food intake and RER in PKO mice were marginal and did not reach statistical significance at the age of 5 months, and became significant only at the age of 9 months (Fig. 2d, e), the subtle energy surplus contributed to significant body weight difference over time (Fig. 2a). Collectively, these data indicate that increased food intake and reduced consumption of fat as an energy source account for the increased body weight and adiposity in the PKO mice.

Increased body weight and fat mass, and higher food intake and RER in PKO mice. (a) Increased body weight in PKO mice from the age of 16 weeks (n = 9–15 per genotype). (b) Higher fat and lean mass in PKO mice by the age of 8 months (n = 6–7 per genotype for 4-month-old groups and 18–20 per genotype for 8-month-old groups). (c) Higher fat content and lower lean mass in PKO mice at 8 months of age. (d, e) Increased food intake (d) and RER (e) in 9-month-old PKO mice. (f–i) Similar O2 consumption (f), activity level (g, h) and BMR (i) between PKO and control mice. n = 9–12 per genotype. White squares (a) or bars (b–i), control; black squares (a) or bars (b-i), PKO. Data are presented as mean ± SEM. PKO vs control mice of the same group: * p < 0.05 and ** p < 0.01. m, months

Increased leptin level in PKO mice. (a) Elevated leptin level in PKO mice at the age of 4 and 8 months. n = 21–28 mice per genotype for 4-month-old groups and n = 14–17 mice per genotype for 8-month-old groups. (b) Higher fat-mass-normalised leptin level in PKO mice at the age of 8 months. n = 8–10 mice per genotype for 4-month-old groups and n = 11–14 mice per genotype for 8-month-old groups. White bars, control; black bars, PKO. Data are presented as mean ± SEM. PKO vs control mice of the same group: * p < 0.05 and ** p < 0.01. Ctrl, control
Reduced hypothalamic Pomc expression in the PKO mice
To understand the mechanism underlying the increased food intake, body weight and fat mass, we performed real-time quantitative PCR to examine Pomc expression in the hypothalamus. As expected, only the PKO mice – not the controls – expressed Cre in the hypothalami (Fig. 4a). At the age of 4 months, the hypothalamic Pomc mRNA level was already lower in PKO mice (Fig. 4b). As Pomc expression is positively regulated by leptin, we normalised the Pomc expression to leptin level. As the PKO mice had higher leptin levels, the normalised Pomc expression was even lower in the PKO mice when compared with the controls (Fig. 4c). Npy mRNA level was unaltered, while Agrp mRNA level was lower in PKO mice, possibly to compensate for reduced Pomc levels (Fig. 4d, e). At the age of 9 months, the hypothalamic Pomc mRNA level remained lower in PKO mice, but no difference was found in Npy and Agrp levels (Fig. 4f–h). These results suggest that an absence of MeCP2 in POMC neurons contributes to reduced Pomc expression, and that MeCP2 in POMC neurons plays a role in regulating Pomc expression.

Reduced Pomc expression in the hypothalamus of PKO mice. (a). Real-time quantitative PCR measurements using hypothalamic samples showed that Cre was only expressed in PKO mice. (b) Hypothalamic Pomc expression was reduced in 4-month-old PKO mice. (c) Leptin-level-normalised Pomc expression was lower in 4-month-old PKO hypothalamus. (d) Npy expression in the PKO mice was similar to that in the control mice at 4 months of age. n = 3 per genotype. (e) Agrp expression was reduced in 4-month-old PKO mice. (f) Hypothalamic Pomc expression was reduced in 9-month-old PKO mice. Npy (g) and Agrp (h) expression in the PKO mice was similar to that in the control mice at the age of 9 months. n = 5–12 mice per genotype for 4-month-old groups and n = 5–6 per genotype for 9-month-old groups unless otherwise indicated. White bars, control; black bars, PKO. All gene expression was normalised to Gapdh. Data are presented as mean ± SEM. PKO vs control mice of the same group: * p < 0.05, ** p < 0.01 and *** p < 0.001
Increased DNA methylation of the Pomc promoter in PKO mice
We tested whether the DNA methylation status of Pomc promoter was changed, as this may explain the altered Pomc expression in the PKO hypothalamus. We examined the methylation status of 18 CpGs between −209 to +13 on the Pomc promoter using hypothalamus samples from PKO and control mice. Overall, DNA methylation levels at different CpG sites on the Pomc promoter showed an upward trend in the PKO mice compared with controls when the mice were 4 month olds, with eight out of 18 CpGs showing significantly higher methylation levels in the PKO mice (Fig. 5a). DNA methylation level increased considerably in control mice, and by 15 months of age there was no difference in the methylation level between the PKO and control mice (Fig. 5b). At the age of 4 months, the average percentage of methylated cytosines of all 18 CpGs was lower in the control than in the PKO mice (35.2 ± 2.5% vs 52.0 ± 3.6%; Fig. 5c). By the age of 15 months, the percentage of DNA methylation was no longer different between the two genotypes (Fig. 5c). Notably, reduced hypothalamic Pomc expression was observed at the age of 4 months (Fig. 4b, c), but not at the age of 15 months (0.31 ± 0.017 vs 0.32 ± 0.075, n = 3 per genotype), suggesting a negative correlation between DNA methylation level and Pomc expression.

Increased DNA methylation of the Pomc promoter in the PKO hypothalamus. (a) At the age of 4 months, DNA methylation at eight out of 18 CpG sites on the Pomc promoter was upregulated in PKO hypothalamus compared with that of controls. (b) At the age of 15 months, no difference was observed in the levels of DNA methylation of the Pomc promoter in the hypothalamuses of control and PKO mice. (c) The average percentage of methylated cytosines on the Pomc promoter was higher in PKO mice at the age of 4 months, but similar to controls at the age of 15 months. The percentage of methylated cytosines at different CpG sites between −209 and +13 on the Pomc promoter was measured by bisulfite sequencing from at least 15 positive clones per mouse (n = 4–5 mice per genotype). White bars, control (a–c); black bars, PKO (a–c). Data are presented as mean ± SEM. PKO vs control mice of the same group: * p < 0.05. (d) Effects of DNA methylation on Pomc promoter activity by luciferase assay. Reduced Pomc promoter activity was observed in HEK293 cells that were treated with different methyltransferases (MSssI, HpaII and HhaI). Firefly luciferase activity was normalised to that of Renilla luciferase. Methyltransferase-treated luciferase activity is expressed as a percentage of control. Data are presented as mean ± SEM of three independent experiments, each performed in triplicate: ** p < 0.01 and *** p < 0.001
Hypermethylation of the Pomc promoter decreases its activity
We further examined the relationship between Pomc promoter methylation and its expression by luciferase assay. A plasmid (pGL3-Pomc) containing the luciferase gene driven by the Pomc promoter was treated with different methyltransferases targeting CpGs of varying sequences, including MSssI (targeting CG), HpaII (CCGG) and HhaI (GCGC). The plasmid pGL3-Pomc contained −646 to +65 of the Pomc gene, which covered the 480 bp upstream of the transcription start site that was required for the full promoter activity [35, 36]. Within the promoter region, there were 20 CpG sites targeted by MSssI, one CpG site by HpaII and four CpG sites by HhaI. All three methyltransferases significantly reduced Pomc promoter activity, with lower promoter activity corresponding to more methylated CpG sites (Fig. 5d), suggesting that hypermethylation of Pomc promoter decreases its promoter activity.
Functional synergy of MeCP2 and CREB1 in regulating Pomc promoter activity
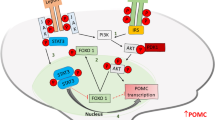
Deletion of MeCP2 in POMC neurons led to reduced Pomc expression, suggesting that MeCP2 may be a positive regulator of Pomc promoter activity. We examined this possibility and also tested whether CREB1 was a co-activator by using the luciferase assay. Co-transfection of WT MeCP2 and CREB1 resulted in a significant activation of Pomc promoter activity compared with transfection of MeCP2 or CREB1 alone (Fig. 6a). Expression of a mutant MeCP2 containing the point mutation R106W failed to enhance Pomc promoter activity (Fig. 6a). Co-IP experiments showed that MeCP2 and CREB1 interacted with each other, and that MeCP2 R106W mutant showed weaker interaction with CREB1 (Fig. 6b). The GST pull-down assay confirmed the direct interaction between WT MeCP2 and CREB1, which was abolished by an R106W mutation in MeCP2 (Fig. 6c), suggesting that MeCP2-CREB1 interaction may be required for the synergistic effect on Pomc promoter activity. However, as the R106W mutation lies within the methyl-CpG binding domain of MeCP2, methyl-CpG binding activity of MeCP2 may also be required for the regulation. These experiments suggest that MeCP2 functions as an activator of Pomc promoter, and the activation requires its interaction with CREB1.

Functional synergy of MeCP2 and CREB1 in regulating the Pomc promoter. (a) pGL3-Pomc reporter plasmid was transfected with MeCP2 (WT or R106W), CREB1 or both into HEK293 cells and luciferase activity was measured. Co-expression of WT MeCP2 and CREB1 significantly potentiated Pomc promoter activity, which was abolished when WT MeCP2 was replaced with MeCP2 R106W mutant. Firefly luciferase activity was normalised to that of Renilla luciferase. Luciferase activity of different treatment was normalised to luciferase activity of pGL3-Pomc alone. Data are presented as mean ± SEM of three independent experiments, each measured in triplicate: ** p < 0.01. (b) Co-IP experiments showing Flag-MeCP2 and Myc-CREB1 interaction. (c) GST pull-down assay showing that GST-CREB1 could pull down WT Flag-MeCP2 but not R106W mutant. IP, immunoprecipitation; IB, immunoblot
Discussion
In this study, we demonstrate that mice with specific deletion of MeCP2 in POMC neurons exhibit increased body weight and fat mass, and leptin resistance. PKO mice show increased Pomc promoter methylation and reduced hypothalamic Pomc mRNA, consistent with a regulatory role of MeCP2 in POMC neurons. Moreover, MeCP2 and CREB1 synergistically promote Pomc expression in vitro.
MeCP2 is highly expressed in the CNS. It has been proposed that different RTT phenotypes are caused by loss of MeCP2 function in specific neurons [26]. There are numerous mouse models with mutation or deletion of MeCP2 that give rise to an array of RTT-like phenotypes [18, 22–26]. However, most of these mouse models have altered MeCP2 in all or most of the CNS, making it difficult to delineate the specific role of MeCP2 in different neurons. Overweight or obesity is a common phenotype in mouse models with deletion of MeCP2 in postmitotic neurons, in postnatal CNS and in Sim1-expressing neurons [22, 25, 26], as well as in some patients with atypical RTT [37–40]. A plausible mechanism for the overweight or obesity phenotype may be the reduced levels of brain-derived neuropeptide (BDNF), an established regulator of energy balance [41] and target of MeCP2 regulation [42]. In our study, we deleted MeCP2 in a single functional neuron type (POMC neurons), which plays a key role in the regulation of energy homeostasis. As Bdnf expression and secretion is secondary to POMC neuron activation [41], we did not measure Bdnf expression and chose to focus on Pomc expression in the PKO mice. The findings that PKO mice show dysregulated energy homeostasis indicate that MeCP2 in POMC neurons is essential in energy homeostasis regulation.
The signalling cascades involved in the regulation of the functions of POMC neuron have been extensively studied; they include the Janus kinase 2 (JAK2)-signal transducer and activator of transcription 3 (STAT3) and phosphoinositide-3-kinase (PI3K) pathways activated by leptin and insulin, respectively [32, 43]. As many of these signalling pathways in POMC neurons converge at the regulation of Pomc expression, defects at different steps along the signalling pathways lead to reduced Pomc expression and leptin resistance. However, changes on Pomc promoter itself, such as epigenetic modifications, directly affect Pomc expression regardless of upstream signalling [14], which may in turn contribute to leptin resistance. The Pomc promoter fragment in this study is within the tissue-specific CpG island that is differentially methylated in different tissue types [44]. Consistent with the finding that Pomc expression is closely related to its promoter methylation [45], we observed an increased DNA methylation of the Pomc promoter and decreased Pomc mRNA expression in vitro and in the PKO hypothalamus. The molecular mechanism underlying the increased Pomc promoter methylation in the absence of MeCP2 is unknown. A possible model based on a study of the epigenetic regulation of Pdx1 in intrauterine growth retardation [46] is that the absence of MeCP2 causes reduced Pomc expression that results in a transition from open active to less active chromatin conformation, which features lower histone acetylation and higher dimethylated H3K9. As histone methylation and DNA methylation reciprocally influence each other [47], increased dimethylated H3K9 may lead to a higher level of DNA methylation, resulting in a closed inactive chromatin conformation and further reduction of Pomc expression.
According to this model, MeCP2 functions as an activator of Pomc expression. This notion is supported by the luciferase reporter assay experiments, which show that MeCP2 regulates Pomc promoter activity positively and synergistically with CREB1 (Fig. 6a). This is consistent with a previous finding showing a synergistic effect between MeCP2 and CREB1 [21]. It appears that binding between MeCP2 and CREB1 is required for their cooperation, as mutant MeCP2 that does not bind to CREB1 fails to upregulate Pomc promoter activity. Further studies, such as sequential chromatin IP (ChIP), will help to confirm the simultaneous occupation of MeCP2 and CREB1 on the Pomc promoter and the functional synergy between MeCP2 and CREB1 on Pomc expression.
Two recent studies report that MeCP2 binds to both 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) with similar high affinities [48, 49]. Highly expressed in CNS, 5hmC is enriched in actively transcribed genes, whereas 5mC is enriched in less active genes. Moreover, MeCP2 is the major 5hmC-binding protein in the brain [49]. The bisulfite conversion method used in our study does not differentiate 5mC and 5hmC. Technologies are being developed to allow analysis of 5hmC at single-base resolution. Future studies may be directed to examine whether 5hmC exists on the Pomc promoter and whether MeCP2 binds to 5hmC to regulate Pomc transcription. MeCP2 is also critical during neuron development as MeCP2 deletion in mouse brain results in reduced neuron size [22]. We did not observe a significant reduction in POMC neuron size or number in PKO mice. However, a more detailed analysis is needed to determine the effect of MeCP2 deletion on POMC neuron development in PKO mice.
Our results show that lack of MeCP2 in POMC neurons is associated with increased DNA methylation of the Pomc promoter and decreased Pomc expression in the hypothalamus, which accounts for the observed obesity and leptin resistance phenotypes, including increased food intake, less burning of fat and higher body weight. This study demonstrates that MeCP2 is a positive regulator of Pomc expression in the hypothalamus; and hypermethylation of the Pomc promoter reduces Pomc expression in the hypothalamus and contributes to leptin resistance.
Abbreviations
- AgRP:
-
Agouti-related peptide
- ARC:
-
Arcuate nucleus
- A*STAR:
-
Agency for Science, Technology and Research
- BMR:
-
Basal metabolic rate
- CLAMS:
-
Oxymax/Comprehensive Lab Animal Monitoring System
- Co-IP:
-
Co-immunoprecipitation
- CREB1:
-
cAMP responsive element binding protein 1
- 5hmC:
-
5-Hydroxymethylcytosine
- 5mC:
-
5-Methylcytosine
- MeCP2:
-
Methyl-CpG-binding protein 2
- α-MSH:
-
α-Melanocyte-stimulating hormone
- NPY:
-
Neuropeptide Y
- POMC:
-
Pro-opiomelanocortin
- PKO:
-
Mecp2 flox/y/Pomc-Cre
- RER:
-
Respiratory exchange ratio
- RTT:
-
Rett syndrome
- WT:
-
Wild type
References
Cone RD (1999) The central melanocortin system and energy homeostasis. Trends Endocrinol Metab 10:211–216
Cowley MA, Smart JL, Rubinstein M et al (2001) Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411:480–484
Gong L, Yao F, Hockman K et al (2008) Signal transducer and activator of transcription-3 is required in hypothalamic agouti-related protein/neuropeptide Y neurons for normal energy homeostasis. Endocrinology 149:3346–3354
Leal-Cerro A, Considine RV, Peino R et al (1996) Serum immunoreactive-leptin levels are increased in patients with Cushing's syndrome. Horm Metab Res 28:711–713
Farooqi IS, O'Rahilly S (2005) Monogenic obesity in humans. Annu Rev Med 56:443–458
Maffei M, Stoffel M, Barone M et al (1996) Absence of mutations in the human OB gene in obese/diabetic subjects. Diabetes 45:679–682
Banks AS, Davis SM, Bates SH, Myers MG (2000) Activation of downstream signals by the long form of the leptin receptor. J Biol Chem 275:14563–14572
White DW, Kuropatwinski KK, Devos R, Baumann H, Tartaglia LA (1997) Leptin receptor (OB-R) signaling. Cytoplasmic domain mutational analysis and evidence for receptor homo-oligomerization. J Biol Chem 272:4065–4071
Vaisse C, Halaas JL, Horvath CM, Darnell JE, Stoffel M, Friedman JM (1996) Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet 14:95–97
McCowen KC, Chow JC, Smith RJ (1998) Leptin signaling in the hypothalamus of normal rats in vivo. Endocrinology 139:4442–4447
Bates SH, Stearns WH, Dundon TA et al (2003) STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature 421:856–859
Gao Q, Wolfgang MJ, Neschen S et al (2004) Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc Natl Acad Sci U S A 101:4661–4666
Morris DL, Rui L (2009) Recent advances in understanding leptin signaling and leptin resistance. Am J Physiol Endocrinol Metab 297:E1247–E1259
Newell-Price J (2003) Proopiomelanocortin gene expression and DNA methylation: implications for Cushing's syndrome and beyond. J Endocrinol 177:365–372
Stevens A, Begum G, White A (2011) Epigenetic changes in the hypothalamic pro-opiomelanocortin gene: a mechanism linking maternal undernutrition to obesity in the offspring? Eur J Pharmacol 660:194–201
Plagemann A, Harder T, Brunn M et al (2009) Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: an epigenetic model of obesity and the metabolic syndrome. J Physiol 587:4963–4976
Lewis JD, Meehan RR, Henzel WJ et al (1992) Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 69:905–914
Guy J, Hendrich B, Holmes M, Martin JE, Bird A (2001) A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet 27:322–326
Nan X, Ng HH, Johnson CA et al (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393:386–389
Jones PL, Veenstra GJ, Wade PA et al (1998) Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 19:187–191
Chahrour M, Jung SY, Shaw C et al (2008) MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320:1224–1229
Chen RZ, Akbarian S, Tudor M, Jaenisch R (2001) Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet 27:327–331
Samaco RC, Fryer JD, Ren J et al (2008) A partial loss of function allele of methyl-CpG-binding protein 2 predicts a human neurodevelopmental syndrome. Hum Mol Genet 17:1718–1727
Shahbazian M, Young J, Yuva-Paylor L et al (2002) Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 35:243–254
Gemelli T, Berton O, Nelson ED, Perrotti LI, Jaenisch R, Monteggia LM (2006) Postnatal loss of methyl-CpG binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of Rett syndrome in mice. Biol Psychiatry 59:468–476
Fyffe SL, Neul JL, Samaco RC et al (2008) Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron 59:947–958
Gustavsson N, Lao Y, Maximov A et al (2008) Impaired insulin secretion and glucose intolerance in synaptotagmin-7 null mutant mice. Proc Natl Acad Sci U S A 105:3992–3997
Lou PH, Gustavsson N, Wang Y, Radda GK, Han W (2011) Increased lipolysis and energy expenditure in a mouse model with severely impaired glucagon secretion. PLoS One 6:e26671
Piper ML, Unger EK, Myers MG, Xu AW (2008) Specific physiological roles for signal transducer and activator of transcription 3 in leptin receptor-expressing neurons. Mol Endocrinol 22:751–759
Yang G, Lim C, Li C et al (2009) FoxO1 inhibits leptin regulation of pro-opiomelanocortin promoter activity by blocking STAT3 interaction with specificity protein 1. J Biol Chem 284:3719–3727
Balthasar N, Coppari R, McMinn J et al (2004) Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron 42:983–991
Xu AW, Ste-Marie L, Kaelin CB, Barsh GS (2007) Inactivation of signal transducer and activator of transcription 3 in proopiomelanocortin (Pomc) neurons causes decreased pomc expression, mild obesity, and defects in compensatory refeeding. Endocrinology 148:72–80
van de Wall E, Leshan R, Xu AW et al (2008) Collective and individual functions of leptin receptor modulated neurons controlling metabolism and ingestion. Endocrinology 149:1773–1785
Vong L, Ye C, Yang Z, Choi B, Chua S, Lowell BB (2011) Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71:142–154
Kitamura T, Feng Y, Kitamura YI et al (2006) Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med 12:534–540
Jeannotte L, Trifiro MA, Plante RK, Chamberland M, Drouin J (1987) Tissue-specific activity of the pro-opiomelanocortin gene promoter. Mol Cell Biol 7:4058–4064
Couvert P, Bienvenu T, Aquaviva C et al (2001) MECP2 is highly mutated in X-linked mental retardation. Hum Mol Genet 10:941–946
Zappella M, Meloni I, Longo I, Hayek G, Renieri A (2001) Preserved speech variants of the Rett syndrome: molecular and clinical analysis. Am J Med Genet 104:14–22
Kleefstra T, Yntema HG, Oudakker AR et al (2002) De novo MECP2 frameshift mutation in a boy with moderate mental retardation, obesity and gynaecomastia. Clin Genet 61:359–362
Adegbola AA, Gonzales ML, Chess A, LaSalle JM, Cox GF (2009) A novel hypomorphic MECP2 point mutation is associated with a neuropsychiatric phenotype. Hum Genet 124:615–623
Xu B, Goulding EH, Zang K et al (2003) Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci 6:736–742
Zhou Z, Hong EJ, Cohen S et al (2006) Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 52:255–269
Kim MS, Pak YK, Jang PG et al (2006) Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat Neurosci 9:901–906
Newell-Price J, King P, Clark AJ (2001) The CpG island promoter of the human proopiomelanocortin gene is methylated in nonexpressing normal tissue and tumors and represses expression. Mol Endocrinol 15:338–348
Plagemann A (2005) Perinatal programming and functional teratogenesis: impact on body weight regulation and obesity. Physiol Behav 86:661–668
Pinney SE, Simmons RA (2010) Epigenetic mechanisms in the development of type 2 diabetes. Trends Endocrinol Metab 21:223–229
Cedar H, Bergman Y (2009) Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 10:295–304
Szulwach KE, Li X, Li Y et al (2011) 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci 14:1607–1616
Mellén M, Ayata P, Dewell S, Kriaucionis S, Heintz N (2012) MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 151:1417–1430
Funding
Research in the laboratories of WH was supported by an intramural funding from A*STAR Biomedical Research Council.
Declaration of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author contributions
WH and YES designed the research. XW performed the experiments. XW and WH wrote the manuscript; all authors were involved in analysing and interpreting the data, and revising the article. All authors approved the final manuscript to be published.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
(PDF 66 kb)
Rights and permissions
About this article
Cite this article
Wang, X., Lacza, Z., Sun, Y.E. et al. Leptin resistance and obesity in mice with deletion of methyl-CpG-binding protein 2 (MeCP2) in hypothalamic pro-opiomelanocortin (POMC) neurons. Diabetologia 57, 236–245 (2014). https://doi.org/10.1007/s00125-013-3072-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-3072-0