
Abstract
Aims/hypothesis
The mechanisms of the protective effects of exendin-4 on NEFA-induced beta cell apoptosis were investigated.
Methods
The effects of exendin-4 and palmitate were evaluated in human and murine islets, rat insulin-secreting INS-1E cells and murine glucagon-secreting alpha-TC1-6 cells. mRNA and protein expression/phosphorylation were measured by real-time RT-PCR and immunoblotting or immunofluorescence, respectively. Small interfering (si)RNAs for Ib1 and Gpr40 were used. Cell apoptosis was quantified by two independent assays. Insulin release was assessed with an insulin ELISA.
Results
Exposure of human and murine primary islets and INS-1E cells, but not alpha-TC1-6 cells, to exendin-4 inhibited phosphorylation of the stress kinases, c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK), and prevented apoptosis in response to palmitate. Exendin-4 increased the protein content of islet-brain 1 (IB1), an endogenous JNK blocker; however, siRNA-mediated reduction of IB1 did not impair the ability of exendin-4 to inhibit JNK and prevent apoptosis. Exendin-4 reduced G-protein-coupled receptor 40 (GPR40) expression and inhibited palmitate-induced phosphorylation of mitogen-activated kinase kinase (MKK)4 and MKK7. The effects of exendin-4 were abrogated in the presence of the protein kinase A (PKA) inhibitors, H89 and KT5720. Knockdown of GPR40, as well as use of a specific GPR40 antagonist, resulted in diminished palmitate-induced JNK and p38 MAPK phosphorylation and apoptosis. Furthermore, inhibition of JNK and p38 MAPK activity prevented palmitate-induced apoptosis.
Conclusions/interpretation
Exendin-4 counteracts the proapoptotic effects of palmitate in beta cells by reducing GPR40 expression and inhibiting MKK7- and MKK4-dependent phosphorylation of the stress kinases, JNK and p38 MAPK, in a PKA-dependent manner.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
NEFAs cooperate with other metabolic factors, as well as with hormonal and genetic abnormalities, to promote beta cell dysfunction leading to hyperglycaemia [1]. If chronically in excess, saturated NEFAs can reduce insulin biosynthesis [2] and secretion [3] and induce beta cell apoptosis [4, 5], a phenomenon termed ‘lipotoxicity’. Therefore, improved knowledge of lipid signalling in the beta cell will allow a better understanding of the mechanisms of beta cell failure in diabetes and of the potential impact of therapeutic options on survival and secretory function of residual beta cells.
Long-chain fatty acids are the natural ligands for the orphan, G-protein-coupled receptor 40 (GPR40), a member of a large family of proteins expressed preferentially in pancreatic beta cells [6]. Several pathways have been proposed to mediate NEFA-induced apoptosis in beta cells, including activation of the stress kinases, c-Jun N-terminal protein kinase (JNK) and p38 mitogen-activated protein kinase (MAPK). Analogous to other MAPKs, JNKs are stimulated following the sequential activation of protein kinases, which include two dual-specificity MAPK kinases, MKK4 and MKK7, and multiple MAPK kinase kinases [7]. While MKK7 is a specific activator of JNKs, MKK4 can also phosphorylate the Thr-Gly-Tyr motif of p38 MAPK [7]. Activation of JNK and p38 MAPK is known to induce beta cell apoptosis [8]; however, the role of NEFA-mediated GPR40 activation in MKK/JNK signalling and beta cell death has not been clarified.
Strategies targeted at enhancing insulin secretion while preserving beta cell mass are currently used or under development for type 2 diabetes treatment [9]. These include glucagon-like peptide 1 (GLP-1) analogues, dipeptidyl peptidase-4 (DPP-4) inhibitors, GPR40 agonists and glucokinase activators [10–12]. DPP-4-resistant GLP-1 analogues, such as exendin-4, are characterised by a relatively long half-life while fully exploiting the insulinotropic effects of GLP-1 [13]. Like other GLP-1 receptor agonists, exendin-4 exerts powerful prosurvival effects on pancreatic beta cells [14], and protects them against apoptosis induced by a variety of stimuli, such as NEFAs. However, the molecular mechanisms underlying the antiapoptotic effect of exendin-4 on NEFA-induced beta cell death are still poorly defined.
Methods
Cell culture
Rat insulin-secreting INS-1E cells (passage 15–30; a gift from C.B. Wollheim, University of Geneva, Geneva, Switzerland) were grown as previously described [15]. Murine glucagon-secreting alpha-TC1-6 cells (passage 35–40; a gift from Professor F. Purrello, University of Catania, Catania, Italy) were grown in monolayer at 37°C in a humidified incubator gassed with 5% CO2, in DMEM containing 4 mmol/l l-glutamine supplemented with 0.02% BSA, 3 g/l glucose, 10% heat-inactivated FBS, 100 IU/ml penicillin, 100 μg/ml streptomycin, 0.1 mmol/l non-essential amino acids (all from Gibco Invitrogen, Paisley, UK) and 15 mmol/l HEPES (pH 7.4).
Animals
Wild-type C57Bl/6 mice were purchased from Charles River Laboratories (Calco, Italy). All animal experimentation respected the regulations and guidelines of Italy and the European Union and the NIH Principles of Laboratory Animal Care (NIH, publication no. 85-23, revised 1985). All the experiments with mice described in this paper have been evaluated and approved (internal ID 0907) by the ethics committee ‘Comitato Etico per la Sperimentazione Animale’ (CESA) of IRSG, Biogem.
Islet isolation and culture
Mouse islets were isolated by bile duct perfusion and collagenase digestion as described [16]. After isolation, they were cultured free floating in RPMI culture medium (Gibco Invitrogen) at 5.5 mmol/l glucose concentration and studied within 3 days of isolation. Cell viability, measured by Trypan Blue exclusion, was 90% after 3 days in culture. Human islets were isolated and cultured as previously described [17], after approval by the local ethics committee of the University of Pisa.
Treatments
INS-1E cells and islets were pretreated with or without 10 nmol/l exendin-4 (exenatide, from Eli Lilly and Co, Indianapolis, IN, USA) for 16 h, followed by stimulation with 0.5 mmol/l palmitate (Sigma-Aldrich, St Louis, MO, USA). Palmitate was dissolved in 0.1 mmol/l NaOH at 70°C for 30 min, and 5 mmol/l palmitate was complexed with 10% essentially fatty acid-free BSA (NEFA/BSA molar ratio of 3.3:1). As indicated, cells were preincubated with SP600125 (10 μmol/l) for 2 h or with the JNK inhibitor, D-JNKi peptide (10 μg/ml), for 16 h. D-JNKi linked to an FITC fluorochrome (D-JNKi-FITC) was used to visualise cell entry and accumulation. Cells were preincubated with SB203580 (40 μmol/l) or SB202190 (10 μmol/l) for 30 min, or with the GPR40 antagonist, GW1100 (1 μmol/l; from Calbiochem, Darmstadt, Germany), for 30 min before treatment with palmitate. Cells were preincubated with H89 (5 μmol/l) for 30 min or KT5720 (1 μmol/l) for 60 min before treatment with exendin-4. In other experiments, cells were stimulated with the GPR40 agonist II (10 μmol/l; from Calbiochem) for 1 h. The JNK inhibitor, JNK-I peptide linked to a 5(6)-carboxyfluorescein (5(6)-FAM) fluorochrome, was from PRIMM (Milan, Italy). SB202190 and KT5720 were from Sigma-Aldrich. All other inhibitors were provided by Calbiochem.
Cell transfection
INS-1E cells were seeded on six-well plates. Upon reaching ∼70% confluence, cells were cultured in growth medium without antibiotics, and after 24 h they were transiently transfected with 60 nmol/l small interfering (si)RNA Gpr40 (also known as Ffar1) (SASI Rn01 00056248 RA NM 153304; Sigma-Aldrich) or siRNA Ib1 (also known as Mapk8ip1) (5′-AACAGCGATGGATTGACCAG-3′) using Lipofectamine 2000 (6 μl/well; Gibco Invitrogen) according to the manufacturer’s instructions. siRNA transfection of murine islets was performed as previously described [18], using dispersed islet cells and 60 nmol/l siRNA Gpr40 (SASI_Mm 01_00102368; Sigma-Aldrich) with Lipofectamine 2000. Lipofectamine 2000 only (Mock) and scrambled siRNA (5′-CGGAAGCGUGUUAACGCAUAU-3′; SCR-siRNA) controls were carried out in parallel.
Immunoblotting
Cells were lysed in buffer containing 50 mmol/l HEPES, pH 7.5, 150 mmol/l NaCl, 1 mmol/l MgCl2, 1 mmol/l CaCl2, 4 mmol/l EDTA, 1% Triton X-100, 10% glycerol, 50 mmol/l NaF and 10 mmol/l sodium pyrophosphate, supplemented with 100 μmol/l phenylmethylsulfonyl fluoride, 5 ng/ml leupeptin, 1 μg/ml aprotinin and 2 mmol/l Na3VO4. Equal protein samples (60 μg) from the cell lysates were separated by SDS-PAGE (7% or 10% gels), as appropriate, and analysed by immunoblotting as previously described [15].
Immunofluorescence
For immunofluorescence studies, equal amounts of size-matched islets (400 islets/each experimental condition) were dissociated with 3 mmol/l EGTA, 20 mg/ml BSA, 2.8 mmol/l glucose and 0.83 μg/ml trypsin to obtain single-cell suspensions. Human beta cells and INS-1E cells were seeded in poly-d-lysine-coated glass coverslips (Esco-Erie Scientific, Portsmouth, NH, USA) and cultured for 24 h in complete medium. To examine JNK phosphorylation, INS-1E cells and human pancreatic beta cells were fixed in ice-cold 4% paraformaldehyde for 15 min. Permeabilisation was achieved in TRIS-buffered saline (TBS)/0.2% Triton X-100 for 10 min; after two washes with TBS/0.1% Triton X-100, cells were exposed to a primary polyclonal antibody against phosphorylated JNK (1:500 dilution) in TBS/3% BSA at 4°C overnight, and then cells were incubated with a secondary Alexa Fluor 546 anti-rabbit goat antibody (1:1,000 dilution; Molecular Probes, Eugene, OR, USA) for 1 h at 25°C. To visualise nuclei, INS-1E cells and human beta cells were stained with TO-PRO-3 (1:5,000 dilution; Molecular Probes). Pictures were acquired on a Leica TCS SP2 laser scanning spectral confocal microscope (Leica Microsystems, Heerbrugg, Switzerland).
Gene expression analysis by quantitative RT-PCR
Total RNA was isolated using RNeasy Mini Kit (Qiagen, Hilden, Germany). Genomic DNA contamination was eliminated by DNase digestion (Qiagen), and 500 ng total RNA was used for cDNA synthesis using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Weiterstadt, Germany). Primers were designed using Primer Express 3.0 (Applied Biosystems) (electronic supplementary material [ESM] Table 1). The PCRs were carried out in an ABI PRISM 7500 System (Applied Biosystems) under the following conditions: 50°C for 2 min, 95°C for 10 min, 40 cycles at 95°C for 15 s, and 60°C for 1 min. Relative RNA levels were determined by analysing the changes in SYBR green fluorescence during PCR using the ΔΔCt method. The mRNA level of each gene was normalised using Gusb.
Measurement of apoptosis
Apoptosis was measured using the Cell Death Detection ELISAPLUS kit (Roche Biochemicals, Indianapolis, IN, USA), according to the manufacturer’s instructions.
Glucose-stimulated insulin secretion (GSIS)
Isolated mouse islets were seeded into 250 ml RPMI-1640 medium with 11.1 mmol/l glucose in 48-well dishes at eight islets/well. After preincubation in KRB containing 3.3 mmol/l glucose for 1 h, the islets were treated for 1 h in KRB containing the indicated reagents with low (3.3 mmol/l) or stimulatory (25 mmol/l) glucose concentrations. The supernatant fractions were then obtained from each reaction well and frozen at −70°C for subsequent determination of insulin concentrations using an insulin ELISA kit (Millipore, Bedford, MA, USA).
Antibodies
Polyclonal antibodies against phosphorylated Thr183/Tyr185-JNK and total JNK, phosphorylated Ser63-c-Jun and total c-Jun, phosphorylated Thr180/Tyr182-p38 MAPK and total p38 MAPK, phosphorylated Ser271/Thr275-MKK7 and total MKK7, phosphorylated Ser257/Thr261-MKK4 and total MKK4, phosphorylated Ser133-cAMP-response-element-binding protein (CREB) and total CREB or cleaved caspase-3 (Asp175) were purchased from Cell Signaling Technology (Beverly, MA, USA). Mouse monoclonal antibody against islet-brain 1/JNK interacting protein 1 (IB1) was from BD Transduction Laboratories (Lexington, KT, USA). Polyclonal GPR40 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Statistical analysis
All data are presented as mean ± SEM and were analysed by Student’s t test or ANOVA, followed by the Tukey multiple comparison test, where appropriate. Statistical significance was set at p < 0.05.
Results
Exendin-4 inhibits palmitate-induced stress kinase activation and apoptosis in beta cells
To investigate NEFA-induced cell death and its potential prevention by the GLP-1 receptor agonist exendin-4, human and murine pancreatic islets were exposed to palmitate for up to 48 h. Apoptosis was found to be increased between 2.5- and 3.5-fold by palmitate, and this was evident after 16–24 h of exposure to the fatty acid (p < 0.05 vs basal; Fig. 1a, b). Challenge of human and murine islets with palmitate also resulted in a two- to three-fold increase in phosphorylation of the stress kinases, JNK (p < 0.05; Fig. 1c–e) and p38 MAPK (p < 0.05; Fig. 1d, e). However, when both human and murine pancreatic islets were pretreated with exendin-4 (10 nmol/l) for 16 h, palmitate-induced apoptosis was markedly reduced (p < 0.05 vs palmitate-exposed islets; Fig. 1a, b). In addition, exendin-4 almost abrogated the increase in JNK (Fig. 1c–e) and p38 MAPK (Fig. 1d, e) phosphorylation in response to palmitate (p < 0.05 vs islets not pretreated with exendin-4). A similar ability of exendin-4 to prevent palmitate-induced apoptosis (evaluated with two independent assays; Fig. 2a, b) and phosphorylation of JNK (Fig. 2c, d) and p38 MAPK (Fig. 2e) was observed in the rat insulin-secreting cell line, INS-1E.

Effects of exendin-4 on palmitate-induced apoptosis and phosphorylation of JNK and p38 MAPK in human and murine pancreatic islets. (a, b) Human and murine pancreatic islets were incubated with or without 10 nmol/l exendin-4 for 16 h, and then exposed to 0.5 mmol/l palmitate for 24 h. Apoptosis was evaluated by measuring cytoplasmic oligosomes with an ELISA in human (a) and murine (b) islets. At least four experiments were performed. (c) Analysis of phosphorylated JNK in beta cells isolated from human islets. The photographs show the phosphorylated form of JNK visualised by immunofluorescence with phospho(P)-JNK antibody (red); an insulin antibody was used to identify beta cells (green), and TO-PRO-3 was used to visualise the nuclei (blue). Scale bars, 13 μm. The bar graph shows the mean fluorescence intensity for each antibody staining the beta cells (white and black bars for insulin and phospho-JNK antibodies, respectively). Data represent units relative to the fluorescence of each antibody in the control condition (i.e. cells not exposed to palmitate nor to exendin-4) set as 100% and are expressed as mean ± SE of three independent experiments. (d, e) Representative immunoblots of JNK phosphorylation and JNK protein content, p38 MAPK phosphorylation and p38 MAPK protein content in human (d) and murine (e) pancreatic islets. At least four independent experiments were performed. *p < 0.05 vs basal (cells not exposed to palmitate); †p < 0.05 vs cells treated with palmitate but not with exendin-4. Ex-4, exendin-4; Palm, palmitate

Effects of exendin-4 on palmitate-induced apoptosis and phosphorylation of JNK and p38 MAPK in rodent insulin-secreting cells. INS-1E cells were incubated with or without 10 nmol/l exendin-4 for 16 h, and then exposed to 0.5 mmol/l palmitate for 24 h. (a) Apoptosis evaluated by measuring cytoplasmic oligosomes with an ELISA (quantification of five experiments). (b) Apoptosis evaluated by immunoblotting of cleaved caspase-3 (representative immunoblot; quantification of five experiments). GAPDH protein content was measured by immunoblotting to assess protein loading. (c) Representative immunoblots of JNK phosphorylation and JNK protein content, and ratio of phosphorylated to total JNK; the p46 and p54 JNK isoforms were quantified separately (white and black bars, respectively; quantification of five experiments). (d) Analysis of phosphorylated JNK in INS-1E cells. The phosphorylated form of JNK was visualised by immunofluorescence with phospho-JNK antibody (red), and TO-PRO-3 was used to visualise the nuclei (blue), as described in the Methods section (four experiments). Scale bars, 18 μm. (e) Representative immunoblots of p38 MAPK phosphorylation and p38 MAPK protein content, and ratio of phosphorylated to total p38 MAPK (five experiments). *p < 0.05 vs control cells; †p < 0.05 vs cells treated with palmitate but not with exendin-4. Ex-4, exendin-4; Palm, palmitate
The effects of non-saturated fatty acids capable of activating GPR40 were also investigated. Apoptosis was found to be increased ∼1.5-fold in oleate-treated INS-1E cells, and pretreatment with exendin-4 (10 nmol/l) for 16 h completely inhibited oleate-induced apoptosis (p < 0.05 vs palmitate-exposed cells; ESM Fig. 1a, b).
A demonstration that activation of both JNK and p38 MAPK is involved in the apoptotic effect of palmitate was achieved in murine pancreatic islets and INS-1 cells, respectively. Inhibition of JNK phosphorylation and activity using the SP600125 inhibitor (10 μmol/l) (ESM Fig. 2a, c) largely prevented palmitate-induced apoptosis (p < 0.05 vs control; Fig. 3a, ESM Fig. 3a). Palmitate-induced apoptosis was also reduced by ∼50% in the presence of SB203580 (40 μmol/l) (p < 0.05 vs control; Fig. 3b, ESM Fig. 3b), a specific p38 MAPK inhibitor (ESM Fig. 2b, d). Similar results were obtained using additional chemical inhibitors of JNK (i.e. the specific JNK inhibitor peptide, JNKi, 10 μg/ml) or p38 MAPK (i.e. SB202190, 10 μmol/l) (ESM Fig. 4a–d). Furthermore, when murine pancreatic islets and INS-1E cells were pretreated with both JNK and p38 MAPK inhibitors, the apoptotic effect of palmitate was completely abrogated (p < 0.05; Fig. 3c, ESM Fig. 3c).

Signalling mechanisms mediating the effects of palmitate on apoptosis of murine pancreatic islets. Murine pancreatic islets were pretreated with 10 μmol/l SP600125 for 2 h and/or 40 μmol/l SB203580 for 30 min, and then exposed to 0.5 mmol/l palmitate for 24 h (a) Effects of the JNK inhibitor, SP600125, on palmitate-induced apoptosis evaluated by measuring cytoplasmic oligosomes in murine islets (four experiments). (b) Effects of the p38 MAPK inhibitor, SB203580, on palmitate-induced apoptosis evaluated by measuring cytoplasmic oligosomes in murine islets (four experiments). (c) Effects of the JNK inhibitor, SP600125, and p38 MAPK inhibitor, SB203580, on palmitate-induced apoptosis in murine islets (four experiments). *p < 0.05 vs control cells not exposed to palmitate; †p < 0.05 vs cells treated with palmitate but not with JNK or p38 MAPK inhibitors. Palm, palmitate
Effects of exendin-4 on palmitate-induced stress kinase activation and apoptosis in alpha cells
To evaluate the possibility that excess saturated fatty acids could also promote apoptosis of alpha cells, similar studies were carried out in the glucagon-secreting alpha-TC1-6 cell line. Exposure of alpha-TC1-6 cells to palmitate for up 48 h resulted in a time-dependent increase in JNK phosphorylation and cellular apoptosis (Fig. 4a, b); however, in contrast with beta cells, exendin-4 did not inhibit these effects of palmitate in pancreatic alpha cells (Fig. 4c–e).

Effects of exendin-4 on palmitate-induced JNK phosphorylation and apoptosis in α-TC1-6 cells. (a, b) Cells were incubated with 0.5 mmol/l palmitate for the indicated times or left untreated. (a) Apoptosis evaluated by measuring cytoplasmic oligosomes with an ELISA. (b) Representative immunoblots of JNK phosphorylation and protein content and relative ratio of phosphorylated to total JNK. (c, d) α-TC1-6 cells were incubated with or without 10 nmol/l exendin-4 for 16 h and then exposed to 0.5 mmol/l palmitate for 24 h. (c) Representative immunoblots of JNK phosphorylation and JNK protein content, and ratio of phosphorylated to total JNK; the p46 and p54 JNK isoforms were quantified separately (grey and black bars, respectively). (d) Analysis of phosphorylated JNK in α-TC1-6 cells. The phosphorylated form of JNK was visualised by immunofluorescence with phospho-JNK antibody (red), and TO-PRO-3 was used to visualise the nuclei (blue), as described in the Methods section. Scale bars, 48 μm. (e) Apoptosis evaluated by measuring cytoplasmic oligosomes with an ELISA. At least four independent experiments were performed for each panel. *p < 0.05 vs control cells not exposed to palmitate. Ex-4, exendin-4; Palm, palmitate
Exendin-4 inhibits GPR40 expression in beta cells
Since exendin-4 reduced palmitate-induced JNK and p38 MAPK phosphorylation, the possibility that GLP-1 receptor activation could regulate the expression levels of GPR40, which serves as a major cell-surface receptor for palmitate [6], was investigated. Exposure of human islets to exendin-4 for up to 24 h reduced GPR40 mRNA and GPR40 protein levels by 50% (p < 0.05 vs basal; Fig. 5a, b). A similar ability of exendin-4 to reduce GPR40 protein expression was observed also in murine islets and INS-1E cells (p < 0.05 vs basal; Fig. 5c–e). In contrast, treatment of INS-1E cells with palmitate for up to 48 h did not affect Gpr40 mRNA levels (data not shown).

Effects of exendin-4 on GPR40 levels in human and murine pancreatic islets and rodent insulin-secreting cells. Cells were incubated with 10 nmol/l exendin-4 for the indicated times or left untreated. (a) GPR40 mRNA levels from human islets were evaluated by quantitative real-time RT-PCR and normalised using GUSB mRNA as internal control. (b) Total GPR40 protein content was evaluated by immunoblotting. GAPDH was measured to assess protein loading. The ratio of GPR40 to GAPDH in four experiments is also shown. (c) Total GPR40 protein content from murine islets was evaluated by immunoblotting. GAPDH was measured to assess protein loading. The ratio of GPR40 to GAPDH in four experiments is also shown. (d) Gpr40 mRNA levels from INS-1E cells were evaluated by quantitative real-time RT-PCR and normalised using Gusb mRNA as internal control. (e) Total GPR40 protein content was evaluated by immunoblotting. GAPDH was measured to assess protein loading. The ratio of GPR40 to GAPDH in five experiments is also shown. *p < 0.05 vs control cells
The physiological significance of exendin-4-induced reduction of GPR40 expression was investigated next using both a GPR40 agonist/antagonist approach and a specific siRNA to reduce GPR40 protein expression. Treatment of murine islets with the GPR40 agonist II (10 μmol/l for 1 h) or exendin-4 (10 nmol/l for 16 h) resulted in enhanced GSIS by ∼30–50% (p < 0.05 vs control; ESM Fig. 5a). Furthermore, pretreatment of islets with exendin-4 for 16 h and subsequent challenge with the GPR40 agonist II for 1 h resulted in a similar augmentation of GSIS (ESM Fig. 5a), indicating that the effects of exendin-4 and the GPR40 agonist were not additive. However, exposure of both murine islets and INS-1E cells to the GPR40 agonist II for longer times (i.e. up to 48 h) provoked an increase in cellular apoptosis by 1.8-fold (p < 0.05 vs basal; Fig. 6a, ESM Fig. 6a, b), albeit the magnitude of this response was lower than the palmitate-induced apoptosis (p < 0.05; Fig. 6a, b). Pretreatment of murine islets with exendin-4 for 4 h did not inhibit the apoptotic response to palmitate or GPR40 agonist II, while this effect was observed when exendin-4 was used for longer times (i.e. for 16 h and 24 h) (Fig. 6a), in parallel with the inhibition of GPR40 expression (Fig. 5c). Furthermore, a specific GPR40 antagonist (GW1100; 1 μmol/l), which neutralised the ability of GPR40 agonist II to enhance GSIS in murine pancreatic islets (ESM Fig. 5b), inhibited GPR40 agonist II- and palmitate-induced apoptosis by >90% and 50%, respectively (Fig. 6b). An RNA interference strategy was also used to selectively suppress Gpr40 expression and determine the resulting effects on palmitate-induced apoptosis. Murine islets and confluent INS-1E cells transfected with an siRNA sequence targeting Gpr40 showed a 65% reduction in the GPR40 protein levels (p < 0.05; ESM Fig. 7a, b). Under these conditions, palmitate-induced apoptosis was inhibited by ∼50% (p < 0.05 vs control; Fig. 6c, ESM Fig. 7c), and so was the activation of the stress kinases, JNK and p38 MAPK (p < 0.05 vs control; Fig. 6d, e; ESM Fig. 7d, e). Altogether, these findings indicate that GPR40 conveys a proapoptotic signal that can be activated when beta cells are exposed to palmitate.

Role of GPR40 in palmitate-induced apoptosis and signalling in murine pancreatic islets. (a) Effects of exendin-4 on GPR40 agonist-induced and palmitate-induced apoptosis. Islets were incubated with 10 nmol/l exendin-4 for 4, 16 or 24 h, and then exposed to 10 μmol/l GPR40 agonist II for 24 h or 0.5 mmol/l palmitate for 24 h. Apoptosis was evaluated by measuring cytoplasmic oligosomes with an ELISA (four experiments). (b) Effects of GPR40 antagonist on palmitate- and GPR40 agonist II-induced apoptosis. Murine pancreatic islets were preincubated with the specific GPR40 antagonist GW1100 (1 μmol/l) for 30 min, and then exposed to 10 μmol/l GPR40 agonist II for 24 h or 0.5 mmol/l palmitate for 24 h. Apoptosis was evaluated by measuring cytoplasmic oligosomes with an ELISA (four experiments). (c–e) Effects of siRNA-mediated knockdown of GPR40 on palmitate-induced apoptosis and signalling in murine pancreatic islets. (c) Apoptosis evaluated by measuring cytoplasmic oligosomes with an ELISA (four experiments). (d) Representative immunoblots of JNK phosphorylation and JNK protein content, and ratio of phosphorylated to total JNK (four experiments). (e) Representative immunoblots of p38 MAPK phosphorylation and protein content, and ratio of phosphorylated to total p38 MAPK (four experiments). *p < 0.05 vs control islets; †p < 0.05 vs palmitate-treated islets; ‡p < 0.05 vs GPR40 agonist II-treated islets. Ex-4, exendin-4; GPR40 Ag, GPR40 agonist II; GPR40 An, GPR40 antagonist GW1100; Palm, palmitate
Signalling mechanisms involved in exendin-4 inhibition of palmitate-mediated apoptosis
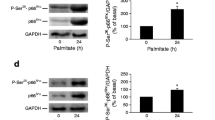
IB1, a scaffold protein that tethers components of the JNK pathway acting as an endogenous blocker, has been suggested to mediate the protective actions of exendin-4 against apoptosis elicited by the cytokine, IL-1β [19]. Therefore, IB1 was next studied in the context of exendin-4 inhibition of NEFA-induced apoptosis. Indeed, when INS-1E cells were exposed to palmitate for up to 48 h, IB1 protein content was initially increased and then reduced starting at 24 h (p < 0.05; ESM Fig. 8; and data not shown); in addition, exendin-4 treatment for 16 h increased IB1 protein content both in the absence and presence of palmitate (p < 0.05; ESM Fig. 8a). However, when a specific siRNA was used to selectively reduce IB1 protein levels (p < 0.05; ESM Fig. 8b), the ability of exendin-4 to increase IB1 expression was prevented (p < 0.05; ESM Fig. 8b), and yet the ability of exendin-4 to inhibit palmitate-stimulated JNK activity, evaluated by measuring c-Jun phosphorylation, was unaltered (p < 0.05; ESM Fig. 8c). It is noteworthy that, when Ib1 was silenced, the ability of exendin-4 to abrogate palmitate-triggered apoptosis of INS-1E cells was also fully preserved (ESM Fig. 8d). Thus, albeit palmitate and exendin-4 exert apparently opposite effects on IB1 protein expression in INS-1E cells, changes in IB1 protein levels do not seem to mediate the ability of exendin-4 to prevent palmitate-induced apoptosis. On the other hand, exendin-4 inhibited palmitate-induced phosphorylation of the kinases, MKK4 and MKK7, which are involved in JNK and p38 MAPK activation (p < 0.05 vs palmitate-treated INS-1E cells; ESM Fig. 9a, b), indicating that the GLP-1 analogue may exert inhibitory effects on the JNK signalling pathway upstream of IB1.
GLP-1 effects on beta cell growth and survival are largely mediated by activation of the cAMP/protein kinase A (PKA)/CREB signalling pathway [20]. We have previously shown that exposure of INS-1E cells to exendin-4 for up to 24 h induced significant increases in intracellular cAMP concentrations and CREB phosphorylation [15]. When cells were pretreated with two independent PKA inhibitors, H89 (5 μmol/l) and KT5720 (1 μmol/l), exendin-4-induced CREB phosphorylation was abrogated (ESM Fig. 10a, b), and the ability of the GLP-1 receptor agonist to reduce Gpr40 mRNA and GPR40 protein levels was also prevented (p < 0.05 vs exendin-4-treated cells; Fig. 7a, b; and data not shown). In addition, inhibition of PKA prevented the ability of exendin-4 to suppress palmitate-stimulated cellular apoptosis (p < 0.05 vs basal; Fig. 7c), as well as MKK4, MKK7, JNK and p38 MAPK phosphorylation (p < 0.05 vs basal; ESM Fig. 11a–d). Thus, exendin-4 counteracts the palmitate effects on beta cell apoptosis by reducing GPR40 protein expression and by inhibiting MKK7- and MKK4-dependent phosphorylation of JNK and p38 MAPK in a PKA-dependent manner.

Involvement of PKA in the effects of exendin-4 on GPR40 expression and on palmitate-induced apoptosis in rodent insulin-secreting cells. (a, b) INS-1E cells were pretreated with 5 μmol/l H89 for 30 min (a) or 1 μmol/l KT5720 for 60 min (b), or left untreated, and then incubated with or without 10 nmol/l exendin-4 for 16 h. Total GPR40 protein content was evaluated by immunoblotting with GAPDH used as control for equal protein loading. The ratio of GPR40 to GAPDH is also shown (five experiments). (c) INS-1E cells were pretreated with 5 μmol/l H89 for 30 min or with 1 μmol/l KT5720 for 60 min, or left untreated, and then incubated with or without 10 nmol/l exendin-4 for 16 h before stimulation with 0.5 mmol/l palmitate for 24 h, as indicated. Apoptosis was evaluated by measuring cytoplasmic oligosomes with an ELISA (five experiments). *p < 0.05 vs control cells; †p < 0.05 vs exendin-4-treated cells in the absence of the PKA inhibitor; ‡p < 0.05 vs cells treated with palmitate alone. Ex-4, exendin-4; Palm, palmitate
Discussion
This study was designed to identify the mechanisms of the protective effects of GLP-1 mimetics on NEFA-induced beta cell death. The degree of saturation of the fatty acids seems to be important for the cytotoxic effect [4, 21, 22], and thus in this study the saturated NEFA, palmitic acid, one of the most abundant fatty acids in plasma, was used. We show that exposure of isolated human and murine pancreatic islets and cultured insulinoma cell lines to palmitate promotes apoptosis by activating JNK and p38 MAPK signalling, and that pretreatment of beta cells with the GLP-1 receptor agonist, exendin-4, prevents the proapoptotic effects of palmitate by multiple mechanisms, including reduction of GPR40 expression and inhibition of MKK4/MKK7-dependent JNK and p38 MAPK activation. The effects of exendin-4 were apparently specific for beta cells, since they were not observed in glucagon-secreting alpha-TC1-6 cells, in which palmitate also induced JNK signalling and apoptosis. These findings may reflect the low level of GLP-1 receptor expression in alpha-TC1-6 cells (A. Natalicchio and F. Giorgino, unpublished results) [23].
GPR40 is highly expressed in pancreatic beta cells and insulin-secreting cell lines, is activated by NEFAs, and plays an important role in the chain of events linking hyperinsulinaemia, obesity and type 2 diabetes [24, 25]. Consistent with its role in glucose-dependent insulin secretion, activation of GPR40 has recently been exploited as a therapeutic target for treatment of type 2 diabetes [11]. However, mice lacking GPR40 display significant protection from obesity-induced hyperglycaemia [25], and overexpression of GPR40 in beta cells results in impaired beta cell function and diabetes [25]. A possible explanation for these apparently divergent effects of GPR40 on beta cell function could be related to the timing of exposure of the receptor to NEFAs, with short-term exposure resulting in increased insulin secretion and long-term exposure leading to beta cell dysfunction, respectively. In this study, exposure of both murine islets and INS-1E cells to a specific GPR40 agonist for several hours resulted in increased apoptosis (ESM Fig. 6), even though the magnitude of this response was lower compared with palmitate-induced cell death (Fig. 6). In contrast, short-term GPR40 stimulation resulted in enhanced GSIS (ESM Fig. 5). Furthermore, results with both a GPR40 antagonist and a specific Gpr40 siRNA indicated that beta cell death induced by long-term exposure to palmitate results, at least in part, from activation of GPR40 (Fig. 6, ESM Fig. 7). In line with these findings, other studies have demonstrated that inhibition of GPR40 protects beta cells from apoptosis [26, 27]. In addition, apoptosis induced by a GPR40-dependent mechanism was also demonstrated in other cellular systems [28]. However, other investigators have recently claimed a protective role of GPR40 in beta cell survival, showing increased beta cell death after treatment with a GPR40 antagonist but unexpectedly no effect when a GPR40 agonist was used [29]. The reasons for these discrepancies are unknown at present and probably reflect different signalling properties of the individual GPR40-binding molecules. Interestingly, the effects of the GPR40 agonist and exendin-4 on insulin release were not additive (ESM Fig. 5), probably because of exendin-4-mediated inhibition of GPR40 expression (Fig. 5).
Exposure of cells to NEFAs results in increased intracellular calcium concentrations and phosphorylation of members of the MAPK family. Two MAPK kinases (MKK4 and MKK7) reportedly phosphorylate and activate the stress kinase, JNK [30, 31]. While MKK7 is a specific activator of JNK, MKK4 can also phosphorylate the Thr-Gly-Tyr motif of p38 MAPK. In this study, the ability of exendin-4 to promote beta cell survival against palmitate was shown to involve an inhibitory effect on palmitate-induced MKK4/MKK7-mediated JNK and p38 MAPK activation. GPR40 knockdown reduced, but did not abolish, palmitate-induced JNK and p38 MAPK phosphorylation and apoptosis; similarly, blocking GPR40 with a specific GPR40 antagonist partially inhibited palmitate-induced apoptosis (Fig. 6, ESM Fig. 7). In contrast, pretreatment with exendin-4 completely prevented JNK and p38 MAPK activation in response to palmitate (Figs 1 and 2). Since GPR40 protein levels were similarly reduced in Gpr40 siRNA- and exendin-4-treated beta cells, it is likely that the effects of exendin-4 on inhibition of the MKK4/MKK7 signalling cascade are, at least in part, independent of the reduction in GPR40 protein expression.
The involvement of other NEFA-dependent signals in the promotion of beta cell dysfunction and death has also been proposed. These include activation of Erk-1/2 [32], NF-κB [33], protein-kinase C (PKC)-δ [34], endoplasmic reticulum (ER) stress [35], calpain-10 [36] and ceramide formation [37], and induction of sterol regulatory element-binding protein [38], macrophage migration inhibitory factor [39], cell death-inducing DNA fragmentation factor a-like effector A [40] and cell death-inducing DFF45-like effector b [41]. NEFA-mediated inhibition of Akt activity [42] has also been implicated. In this study, beta cell apoptosis was fully suppressed when both JNK and p38 MAPK protein activities were inhibited (Fig. 3 and ESM Fig. 3). It is likely that inhibition of JNK/p38 MAPK activities would also reduce serine phosphorylation of IRS-1 and IRS-2, as previously reported in TNF-α-treated beta cells in response to exendin-4 [15]; thus, albeit other pathways may be involved in NEFA-mediated rodent and human beta cell death, activation of the stress kinases, JNK and p38 MAPK, appears to represent a key signalling node in this process.
Administration of GLP-1 was associated with proliferative and antiapoptotic effects in both endocrine and exocrine compartments of the pancreas [43, 44], and significantly reduced apoptotic beta cells in experimental diabetes models [45]. GLP-1 receptor agonists can inhibit the proapoptotic pathways triggered by a variety of stimuli, including IL-1β, TNF-α and rapamycin, by interfering with JNK and possibly p38 MAPK activation [15, 19, 46]. The cAMP/PKA/CREB cascade represents a central mechanism of GLP-1 action, specifically when this relates to promotion of beta cell survival [47], even though other signalling mediators may also be implicated [48, 49]. Impairment of CREB-mediated transcription led to loss of islets by apoptosis and diminished the antiapoptotic action of exendin-4 in human islets [50]. In line with these results, our previous studies demonstrated that inhibition of TNF-α-induced JNK phosphorylation by exendin-4 was abrogated when cells were preincubated with H89, a specific inhibitor of PKA [15]. Also in this study, the antiapoptotic action of exendin-4 was significantly reversed by two specific PKA inhibitors, H89 and KT5720. Furthermore, PKA activation was found to be involved in exendin-4-induced GPR40 inhibition and suppression of MKK4/MKK7 signalling. In future studies, it will be important to decipher the mechanism by which PKA regulates GPR40 expression possibly at the level of gene transcription.
Abbreviations
- CREB:
-
cAMP-response-element-binding protein
- DPP-4:
-
Dipeptidyl peptidase-4
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- GLP-1:
-
Glucagon-like peptide-1
- GPR40:
-
G-protein-coupled receptor 40
- GSIS:
-
Glucose-stimulated insulin secretion
- IB1:
-
Islet-brain 1
- JNK:
-
c-Jun N-terminal kinases
- MAPK:
-
Mitogen-activated protein kinase
- MKK:
-
Mitogen-activated protein kinase kinase
- PKA:
-
Protein kinase A
- siRNA:
-
Small interfering RNA
- TBS:
-
TRIS-buffered saline
References
Marchetti P, Dotta F, Lauro D, Purrello F (2008) An overview of pancreatic beta-cell defects in human type 2 diabetes: implications for treatment. Regul Pept 146:4–11
Poitout V, Hagman D, Stein R, Artner I, Robertson RP, Harmon JS (2006) Regulation of the insulin gene by glucose and fatty acids. J Nutr 136:873–876
Giacca A, Xiao C, Oprescu AI, Carpentier AC, Lewis GF (2011) Lipid-induced pancreatic β-cell dysfunction: focus on in vivo studies. Am J Physiol Endocrinol Metab 300:E255–E262
El-Assaad W, Buteau J, Peyot ML et al (2003) Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology 144:4154–4163
Lupi R, Dotta F, Marselli L et al (2002) Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes 51:1437–1442
Briscoe CP, Tadayyon M, Andrews JL et al (2003) The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem 278:11303–11311
Davis RJ (2000) Signal transduction by the JNK group of MAP kinases. Cell 103:239–252
Widenmaier SB, Ao Z, Kim SJ, Warnock G, McIntosh CH (2009) Suppression of p38 MAPK MAPK and JNK via Akt-mediated inhibition of apoptosis signal-regulating kinase 1 constitutes a core component of the beta-cell pro-survival effects of glucose-dependent insulinotropic polypeptide. J Biol Chem 284:30372–30382
Tahrani AA, Bailey CJ, del Prato S, Narnett AH (2011) Management of type 2 diabetes: new and future developments in treatment. Lancet 378:182–197
Mudaliar S, Henry RR (2012) The incretin hormones: from scientific discovery to practical therapeutics. Diabetologia 55:1865–1868
Burant CF, Viswanathan P, Marcinak J, Cap C et al (2012) TAK-875 versus placebo or glimepiride in type 2 diabetes mellitus: a phase 2, randomized, double-blind, placebo-controlled trial. Lancet 379:1403–1411
Doliba NM, Qin W, Najafi H et al (2012) Glucokinase activation repairs defective bioenergetics of islets of Langerhans isolate from type 2 diabetics. Am J Physiol Endocrinol Metab 302:E87–E102
Giorgino F, Laviola L, Leonardini A, Natalicchio A (2006) GLP-1: a new approach for type 2 diabetes therapy. Diabetes Res Clin Pract 74:S152–S155
Garber AJ (2011) Incretin effects on β-cell function, replication, and mass: the human perspective. Diabetes Care 34:S258–S263
Natalicchio A, de Stefano F, Orlando MR et al (2010) Exendin-4 prevents c-Jun N-terminal protein kinase activation by tumor necrosis factor-alpha (TNFalpha) and inhibits TNFalpha-induced apoptosis in insulin-secreting cells. Endocrinology 151:2019–2029
Li DS, Yuan YH, Tu HJ, Liang QL, Dai LJ (2009) A protocol for islet isolation from mouse pancreas. Nat Protoc 4:1649–1652
Masini M, Bugliani M, Lupi R et al (2009) Autophagy in human type 2 diabetes pancreatic beta-cells. Diabetologia 52:1083–1086
Hagerkvist R, Mokhtari D, Myers JW, Tengholm A, Welsh N (2005) siRNA produced by recombinant dicer mediates efficient gene silencing in islet cells. Ann N Y Acad Sci 1040:114–122
Ferdaoussi M, Abdelli S, Yang JY et al (2008) Exendin-4 protects beta-cells from interleukin-1 beta-induced apoptosis by interfering with the c-Jun NH2-terminal kinase pathway. Diabetes 57:1205–1215
Hui H, Nourparvar A, Zhao X, Perfetti R (2003) Glucagon-like peptide-1 inhibits apoptosis of insulin-secreting cells via a cyclic 5'-adenosine monophosphate-dependent protein kinase A- and a phosphatidylinositol 3-kinase-dependent pathway. Endocrinology 144:1444–1455
Yaney GC, Corkey BE (2003) Fatty acid metabolism and insulin secretion in pancreatic beta-cells. Diabetologia 46:1297–1312
Maedler K, Spinas GA, Dyntar D, Moritz W, Kaiser N, Donath MY (2001) Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 50:69–76
Moens K, Heimberg H, Flamez D et al (1996) Expression and functional activity of glucagon, glucagon-like peptide I, and glucose-dependent insulinotropic peptide receptors in rat pancreatic islet cells. Diabetes 45:257–261
Itoh Y, Kawamata Y, Harada M et al (2003) Free fatty acids regulate insulin secretion from pancreatic beta-cells through GPR40. Nature 422:173–176
Steneberg P, Rubins N, Bartoov-Shifman R, Walker MD, Edlund H (2005) The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metab 1:245–258
Zhang Y, Xu M, Zhang S et al (2007) The role of G protein-coupled receptor 40 in lipoapoptosis in mouse beta-cell line NIT-1. J Mol Endocrinol 38:651–661
Wu J, Sun P, Zhang X et al (2012) Inhibition of GPR40 protects MIN6 β-cells from palmitate-induced ER stress and apoptosis. J Cell Biochem 113:1152–1158
Mieczkowska A, Baslé MF, Chappard D, Mabilleau G (2012) Thiazolidinediones induce osteocyte apoptosis by a G protein-coupled receptor 40-dependent mechanism. J Biol Chem 287:23517–23526
Wagner R, Kaiser G, Gerst F et al (2013) Reevaluation of fatty acid receptor 1 (FFAR1/GPR40) as drug target for the stimulation of insulin secretion in humans. Diabetes 62:2106–2111
Wang X, Destrument A, Tournier C (2007) Physiological roles of MKK4 and MKK7: insights from animal models. Biochim Biophys Acta 1773:1349–1357
Jaeschke A, Davis RJ (2007) Metabolic stress signaling mediated by mixed-lineage kinases. Mol Cell 27:498–508
Plaisance V, Perret V, Favre D et al (2009) Role of the transcriptional factor C/EBPbeta in free fatty acid-elicited beta-cell failure. Mol Cell Endocrinol 305:47–55
Kharroubi I, Ladrière L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL (2004) Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology 145:5087–5096
Eitel K, Staiger H, Rieger J et al (2003) Protein kinase Cδ activation and translocation to the nucleus are required for fatty acid-induced apoptosis of insulin-secreting cells. Diabetes 52:991–999
Cnop M, Ladrière L, Igoillo-Esteve M, Moura RF, Cunha DA (2010) Causes and cures for endoplasmic reticulum stress in lipotoxic β-cell dysfunction. Diabetes Obes Metab 2:76–82
Johnson JD, Han Z, Otani K et al (2004) RyR2 and calpain-10 delineate a novel apoptosis pathway in pancreatic islets. J Biol Chem 279:24794–24802
Veluthakal R, Palanivel R, Zhao Y, McDonald P, Gruber S, Kowluru A (2005) Ceramide induces mitochondrial abnormalities in insulin-secreting INS-1 cells: potential mechanisms underlying ceramide-mediated metabolic dysfunction of the β cell. Apoptosis 10:841–850
Li J, Liu X, Ran X et al (2010) Sterol regulatory element-binding protein-1c knockdown protected INS-1E cells from lipotoxicity. Diabetes Obes Metab 12:35–46
Saksida T, Stosic-Grujicic S, Timotijevic G, Sandler S, Stojanovic I (2011) Macrophage migration inhibitory factor deficiency protects pancreatic islets from palmitic acid-induced apoptosis. Immunol Cell Biol 90:688–698
Omae N, Ito M, Hase S et al (2012) Suppression of FoxO1/cell death-inducing DNA fragmentation factor α-like effector A (Cidea) axis protects mouse β-cells against palmitic acid-induced apoptosis. Mol Cell Endocrinol 348:297–304
Li H, Song Y, Zhang LJ et al (2012) Cell death-inducing DFF45-like effector b (Cideb) is present in pancreatic beta-cells and involved in palmitate induced beta-cell apoptosis. Diabetes Metab Res Rev 28:145–155
Wrede CE, Dickson LM, Lingohr MK, Briaud I, Rhodes CJ (2002) Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic β-cells (INS-1). J Biol Chem 277:49676–49684
Bulotta A, Farilla L, Hui H, Perfetti R (2004) The role of GLP-1 in the regulation of islet cell mass. Cell Biochem Biophys 40:65–78
Farilla L, Hui H, Bertolotto C et al (2002) Glucagon-like peptide-1 promotes islet cell growth and inhibits apoptosis in Zucker diabetic rats. Endocrinology 143:4397–4408
Li Y, Hansotia T, Yusta B, Ris F, Halban PA, Drucker DJ (2003) Glucagon-like peptide-1 receptor signaling modulates beta-cell apoptosis. J Biol Chem 278:471–478
Kawasaki Y, Harashima S, Sasaki M et al (2010) Exendin-4 protects pancreatic beta-cells from the cytotoxic effect of rapamycin by inhibiting JNK and p38 MAPK phosphorylation. Horm Metab Res 42:311–317
Kwon G, Pappan KL, Marshall CA, Schaffer JE, McDaniel ML (2004) cAMP dose-dependently prevents palmitate-induced apoptosis by both protein kinase A- and cAMP-guanine nucleotide exchange factor-dependent pathways in beta-cells. J Biol Chem 279:8938–8945
Buteau J, Foisy S, Rhodes CJ, Carpenter L, Biden TJ, Prentki M (2001) Protein kinase Czeta activation mediates glucagon-like peptide-1-induced pancreatic beta-cell proliferation. Diabetes 50:2237–2243
Yusta B, Baggio LL, Estall JL et al (2006) GLP-1 receptor activation improves beta-cell function and survival following induction of endoplasmic reticulum stress. Cell Metab 4:391–406
Sarkar SA, Gunter J, Bouchard R et al (2007) Dominant negative mutant forms of the cAMP response element binding protein induce apoptosis and decrease the anti-apoptotic action of growth factors in human islets. Diabetologia 50:1649–1659
Funding
This work was supported by a grant from the Ministero dell’Università e della Ricerca, Italy, to FG.
Duality of interest
FG is a consultant for and has received lecture fees from Eli Lilly & Co, AstraZeneca and Bristol-Myers Squibb. FG has received grant support from Eli Lilly, AstraZeneca and Bristol-Myers Squibb. All other authors declare that there is no duality of interest associated with their contribution to this manuscript.
Contribution statement
AN, RL and FG designed the study. FT, GB, EC and PM acquired data. RL, NM and GB performed experiments and data analysis. AN, RL, LL, SP and FG performed data analysis and interpretation. AC, AP, MRO and AL participated in interpretation and discussion of the data. AN, RL and FG wrote the manuscript. PM, LL, SP, FT, GB, NM, AP, EC, MRO, AL and AC critically reviewed the manuscript for intellectual content. All authors approved the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Annalisa Natalicchio and Rossella Labarbuta contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
Effects of exendin-4 on oleate-induced apoptosis in rodent insulin-secreting cells. Apoptosis was evaluated by measuring cytoplasmic oligosomes with an ELISA assay a. INS-1E cells were incubated with 0.5 mmol/l oleate for the indicated times or left untreated (four experiments). b. INS-1E cells were incubated with or without 10 nmol/l exendin-4 for 16 h and then exposed to 0.5 mmol/l oleate for 24 h (four experiments). *P < 0.05 vs. control cells; †P < 0.05 vs. cells exposed to oleate but not to exendin-4. Ex-4, exendin-4. (PDF 76 kb)
ESM Fig. 2
Validation of the use of JNK and p38 MAPK inhibitors in murine pancreatic islets and INS-1E cells. a, c. Effects of the JNK inhibitor SP600125 on palmitate-induced JNK phosphorylation in murine pancreatic islets (a) and INS1E cells (c). Cells were pre-treated with 10 μmol/l SP600125 for 2 h and then exposed to 0.5 mmol/l palmitate. Representative immunoblots of JNK phosphorylation and protein content, and ratio of phosphorylated to total JNK isoforms (p46 JNK, white bars; p54 JNK, black bars) (four experiments). b, d. Effects of the p38 MAPK inhibitor SB203580 on palmitate-induced p38 MAPK phosphorylation in murine pancreatic islets (b) and INS1E cells (d). Cells were pre-treated with 40 μmol/l SB203580 for 30 min and then exposed 0.5 mmol/l palmitate. Representative immunoblots of p38 MAPK phosphorylation and protein content, and ratio of phosphorylated to total p38 MAPK (four experiments). *P < 0.05 vs. control cells; †P < 0.05 vs. palmitate-treated cells. Palm, palmitate. (PDF 122 kb)
ESM Fig. 3
Signaling mechanisms mediating the effects of palmitate on apoptosis of INS-1E cells. a. Effects of the JNK inhibitor SP600125 on palmitate-induced apoptosis evaluated by measuring cytoplasmic oligosomes in INS-1E cells (five experiments). b. Effects of the p38 MAPK inhibitor SB203580 on palmitate-induced apoptosis evaluated by measuring cytoplasmic oligosomes in INS-1E cells (five experiments). c. Effects of the JNK inhibitor SP600125 and p38 MAPK inhibitor SB203580 on palmitate-induced apoptosis in INS-1E cells (five experiments). *p < 0.05 vs. control cells; †p < 0.05 vs. cells treated with palmitate but not with exendin-4. Palm, palmitate. (PDF 79 kb)
ESM Fig. 4
Effects of the JNK inhibitor JNKi and the p38 MAPK inhibitor SB202190 on palmitate-induced apoptosis in rodent insulin-secreting cells. INS-1E cells were pre-treated with the JNKi peptide (10 μg/ml for 16 h) or SB202190 (10 μmol/l for 30 min) and then exposed to 0.5 mmol/l palmitate for 24 h. a. Representative immunoblots of c-Jun phosphorylation and protein content, and ratio of phosphorylated to total c-Jun. b. Palmitate-induced apoptosis, evaluated by measuring the release of cytoplasmic oligosomes with an ELISA assay, in the absence or presence of JNKi (four experiments). c. Representative immunoblots of p38 MAPK phosphorylation and protein content, and ratio of phosphorylated to total p38 MAPK. d. Palmitate-induced apoptosis, evaluated by measuring the release of cytoplasmic oligosomes with an ELISA assay, in the absence or presence of SB202190 (four experiments). *P < 0.05 vs. control cells; †P < 0.05 vs. palmitate-treated cells in the absence of the indicated kinase inhibitor. Palm, palmitate. (PDF 89 kb)
ESM Fig. 5
a. Role of GPR40 in glucose-stimulated insulin secretion (GSIS) and effects of exendin-4 on GPR40-mediated potentiation of GSIS in murine pancreatic islets. Islets were pretreated with 10 nmol/l exendin-4 for 16 h or left untreated, then incubated in the presence or absence of 10 μmol/l GPR40 Agonist II for 1 h, and finally incubated in KRBH buffer containing 3.3 or 25 mmol/l glucose (3.3 mmol/l glucose, white bars; 25 mmol/l glucose, grey bars) and the reagents indicated above for 1 h. Insulin secretion was evaluated by measuring insulin concentrations in the conditioned medium with an ELISA assay (four experiments). b. Effect of GPR40 Antagonist (GW1100) on GPR40 Agonist II-mediated potentiation of GSIS in murine pancreatic islets. Islets, pretreated with 1 μmol/l GW1100 for 30 min or left untreated, were exposed to 10 μmol/l GPR40 Agonist II for 1 h and then incubated in KRBH buffer containing 3.3 or 25 mmol/l glucose for 1 h (3.3 mmol/l glucose, white bars; 25 mmol/l glucose, grey bars). Insulin secretion was evaluated by measuring insulin concentrations in the conditioned medium with an ELISA assay (four experiments). *p < 0.05 vs. islets incubated in 3.3 mmol/l glucose; †p < 0.05 vs. control islets incubated in 25 mmol/l glucose; Ex-4, exendin-4; GPR40 Ag, GPR40 Agonist II; GPR40 An, GPR40 Antagonist GW1100. (PDF 89 kb)
ESM Fig. 6
Effects of long-term stimulation of GPR40 on apoptosis in murine pancreatic islets and rodent insulin-secreting cells. Apoptosis was evaluated by measuring cytoplasmic oligosomes with an ELISA assay. a. Murine pancreatic islets were incubated with 10 μmol/l GPR40 Agonist II for the indicated times or left untreated. b. INS-1E cells were incubated with 10 μmol/l GPR40 Agonist II for the indicated times or left untreated. At least four independent experiments were performed. *p < 0.05 vs. control cells; GPR40 Ag, GPR40 Agonist II. (PDF 32 kb)
ESM Fig. 7
siRNA-mediated knock-down of Gpr40. Murine pancreatic islets and INS-1E cells were transfected with a siRNA sequence targeting Gpr40 (siGpr40, 60 nmol/l), with a control scrambled siRNA sequence (60 nmol/l, SCR), or with transfection reagents only (mock), as described under Materials and Methods. Representative immunoblots showing GPR40 protein expression in murine islets (a) and INS-1E cells (b). Glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) was analyzed as control for equal protein loading (four experiments). c, e. Effects of siRNA-mediated knock-down of Gpr40 on palmitate-induced signaling and apoptosis in INS-1E cells. c. Apoptosis evaluated by measuring cytoplasmic oligosomes with an ELISA assay in INS-1E cells (five experiments). d. Representative immunoblots of JNK phosphorylation and JNK protein content, and ratio of phosphorylated to total JNK (five experiments) in INS-1E cells. e. Representative immunoblots of p38 MAPK phosphorylation and protein content, and ratio of phosphorylated to total p38 MAPK in INS-1E cells (five experiments). *p < 0.05 vs. control cells; †p < 0.05 vs. palmitate-treated cells. Palm, palmitate. (PDF 105 kb)
ESM Fig. 8
Role of IB1 in the protective effects of exendin-4 in rodent insulin-secreting cells. a. INS-1E cells were incubated with or without 10 nmol/l exendin-4 for 16 h, and then exposed to 0.5 mmol/l palmitate for 24 h. Total IB1 protein levels were determined by immunoblotting with specific antibodies. Glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) was measured to assess protein loading. The ratio of IB1 to GAPDH in five experiments is also shown. b. siRNA-mediated knock-down of Ib1. INS-1E cells were transfected with a siRNA sequence targeting Ib1 (siIb1, 60 nmol/l), with a control scrambled siRNA sequence (60 nmol/l, SCR), or with transfection reagents only (mock), as described under Materials and Methods. Representative immunoblot showing IB1 protein levels with GAPDH used as control for equal protein loading. Data are presented after normalization to the GAPDH protein levels in five experiments. c. Representative immunoblots of c-Jun Ser63 phosphorylation and protein content detected by immunoblotting, and ratio of phosphorylated to total c-Jun (five experiments). d. Apoptosis evaluated by measuring cytoplasmic oligosomes with an ELISA assay (five experiments). *p < 0.05 vs. control cells; †p < 0.05 vs. palmitate-treated cells. Palm, palmitate; Ex-4, exendin-4. (PDF 105 kb)
ESM Fig. 9
Effects of exendin-4 on palmitate-induced phosphorylation of MKK4 and MKK7 in rodent insulin-secreting cells. INS-1E cells were incubated with or without 10 nmol/l exendin-4 for 16 h and then exposed to 0.5 mmol/l palmitate. a. Representative immunoblots of MKK4 Ser257/Thr261 phosphorylation and protein content detected, and ratio of phosphorylated to total MKK4. b. Representative immunoblots of MKK7 Ser271/Thr275 phosphorylation and protein content, and ratio of phosphorylated to total MKK7. At least four independent experiments were performed.*p < 0.05 vs. control cells; † p < 0.05 vs. palmitate-treated cells. Palm, palmitate; Ex-4, exendin-4. (PDF 99 kb)
ESM Fig. 10
Effects of the PKA inhibitors H89 (a) and KT5720 (b) on exendin-4-induced CREB phosphorylation in rodent insulin-secreting cells. INS-1E cells were treated with 5 μmol/l H89 or 1 μmol/l KT 5720 for 60 min, or left untreated, and then incubated with or without 10 nmol/l exendin-4 for 30 min. In each panel, representative immunoblots of CREB phosphorylation and protein content, and the ratio of phosphorylated to total CREB are shown. At least four independent experiments were performed.*p < 0.05 vs. control cells; †p < 0.05 vs. exendin-4-treated cells in the absence of the PKA inhibitor. Ex-4, exendin-4. (PDF 91 kb)
ESM Fig. 11
Involvement of PKA in the effects of exendin-4 on palmitate-induced MKK4, MKK7, JNK and p38 MAPK phosphorylation in rodent insulin-secreting cells. INS-1E cells were pre-treated with 5 μmol/l H89 for 30 min, or left untreated, and then incubated with or without 10 nmol/l exendin-4 for 16 h prior to stimulation with 0.5 mmol/l palmitate, as indicated. a. Representative immunoblots of MKK4 phosphorylation and protein content and ratio of phosphorylated to total MKK4. b. Representative immunoblots of MKK7 phosphorylation and protein content and relative ratio of phosphorylated to total MKK7. c. Representative immunoblots of JNK phosphorylation and protein content and relative ratio of phosphorylated to total JNK; p46 JNK, white bars; p54 JNK, black bars. d. Representative immunoblots of p38 MAPK phosphorylation and protein content and relative ratio of phosphorylated to total p38 MAPK. At least five independent experiments were performed.*p < 0.05 vs. control cells; †p < 0.05 vs. palmitate-treated cells. Palm, palmitate; Ex-4, exendin-4. (PDF 164 kb)
ESM Table 1
(PDF 949 kb)
Rights and permissions
About this article
Cite this article
Natalicchio, A., Labarbuta, R., Tortosa, F. et al. Exendin-4 protects pancreatic beta cells from palmitate-induced apoptosis by interfering with GPR40 and the MKK4/7 stress kinase signalling pathway. Diabetologia 56, 2456–2466 (2013). https://doi.org/10.1007/s00125-013-3028-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-3028-4