
Abstract
Aims/hypothesis
The small, highly conserved vasoactive peptide urotensin II (UII) is upregulated in atherosclerosis. However, its effects in diabetes-associated atherosclerosis have not been assessed.
Methods
Endothelial cells were grown in normal- and high-glucose (5 and 25 mmol/l) media with and without UII (10−8 mol/l) and/or the UII receptor antagonist, SB-657510 (10−8 mol/l). Apoe knockout (KO) mice with or without streptozotocin-induced diabetes were treated with or without SB-657510 (30 mg kg−1 day−1; n = 20 per group) and followed for 20 weeks. Carotid endarterectomy specimens from diabetic and non-diabetic humans were also evaluated.
Results
In high (but not normal) glucose medium, UII significantly increased CCL2 (encodes macrophage chemoattractant protein 1 [MCP-1]) gene expression (human aortic endothelial cells) and increased monocyte adhesion (HUVECs). UII receptor antagonism in diabetic Apoe KO mice significantly attenuated diabetes-associated atherosclerosis and aortic staining for MCP-1, F4/80 (macrophage marker), cyclooxygenase-2, nitrotyrosine and UII. UII staining was significantly increased in carotid endarterectomies from diabetic compared with non-diabetic individuals, as was staining for MCP-1.
Conclusions/interpretation
This is the first report to demonstrate that UII is increased in diabetes-associated atherosclerosis in humans and rodents. Diabetes-associated plaque development was attenuated by UII receptor antagonism in the experimental setting. Thus UII may represent a novel therapeutic target in the treatment of diabetes-associated atherosclerosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The small vasoactive peptide urotensin II (UII) was originally characterised in fish [1]. The UII receptor (UT) was identified as the previously orphaned receptor G-protein coupled receptor 14 (GPR14) [2–5] or sensory epithelium neuropeptide-like receptor (SENR) [6] and is now named UT [7]. UII and the UT are widely distributed in the vasculature [8], with UII being a highly potent vasoactive hormone [2].
UII and its receptor are known to be upregulated in atherosclerotic plaque from non-diabetic animals and human patients [9–11]. Additionally, chronic UII infusion has been shown to enhance macrophage foam cell formation [12] and atherosclerosis in high-fat-fed Apoe knockout (KO) mice [13]. While UT antagonism has been investigated in diabetic nephropathy [14, 15], little is known of the role of UII in the development of diabetes-associated atherosclerosis.
Given the potential pro-atherosclerotic effects of UII, we sought to examine the role of this peptide in diabetes-associated atherosclerosis by determining the effects of UII on endothelial cell function in a hyperglycaemic in vitro setting. We also determined whether chronic UT blockade with the non-peptidergic antagonist, SB-657510, could ameliorate the development of diabetes-associated atherosclerosis in the Apoe KO mouse [11, 16]. Finally, we assessed whether the UII system is specifically upregulated in human diabetes-associated atherosclerosis.
Methods
Endothelial cell treatment with UII and UT antagonism
Human aortic endothelial cells (HAECs) (Clonetics; Lonza Australia, Mt Waverly, VIC, Australia) were grown in 75 cm2 culture flasks (250,000 per flask with humidified incubation, 5% CO2 (vol./vol.), 37°C) in endothelial growth medium (EGM)-2 Bulletkit, supplemented with EGM and heparin (Lonza). Cells were transferred to six-well plates prior to UII/SB-657510 treatment. HAECs were used between passages two to six. Cells were grown in medium containing 25 mmol/l glucose (high-glucose conditions) or 5.6 mmol/l glucose (normal-glucose conditions; regular medium). Mannitol was used as an osmotic control (25 mmol/l). In all experiments, HAECs were used at 80 to 90% confluence. On the day of the experiment, cells were washed with fresh EGM-2, which was then replaced with EGM-2 containing 25 mmol/l glucose for 72 h. Cells were treated in the last 24 h with UII (10−8 to 10−11 mol/l) or, after 30 min pre-incubation with UII (10−8 mol/l), with the UT antagonist SB-657510 (10−8 to 10−10 mol/l) (GlaxoSmithKline, Philadelphia, PA, USA).
Endothelial monocyte adhesion assay
HUVECs were cultured and grown on fibrinogen (100 μg/ml) pre-coated coverslips in 24-well plates in normal- or high-glucose conditions (5 or 25 mmol/l). Once 70% confluent, HUVECs were treated for 24 h with UII (10−8 mol/l), SB-657510 (10−8 mol/l) or UII + SB-657510. At the end of the treatment, THP-1 cells (monocytic cell line) (1 × 106 cells/ml) were seeded on to the monolayer of HUVECs and incubated for 20 min (37°C). THP-1 cells pre-treated with phorbol 12-myristate 13-acetate (PMA) (1 μmol/l, 10 min, 37°C; Sigma-Aldrich, St Louis, MO, USA) were used as a positive control. Coverslips were transferred into wells containing 3% (wt/vol.) paraformaldehyde in PBS (30 min) before being washed and mounted onto slides. Adhered cells were counted using light microscopy.
Animals
Male Apoe KO mice (6 weeks old, backcrossed 20 times to a C57BL/6 background) (Animal Resource Centre, Canning Vale, WA, Australia) were housed at the Baker IDI Heart and Diabetes Research Institute and studied according to National Health and Medical Research Council (NHMRC) guidelines in line with international standards. Animals had unrestricted access to water and feed, and were maintained on a 12 h light/dark cycle on standard mouse chow (Barastoc; Ridley Agriproducts, St Arnaud, VIC, Australia).
Of the four groups of mice (n = 20/group), two groups were rendered diabetic with five daily injections of streptozotocin, 55 mg/kg (Sigma-Aldrich), in citrate buffer at 6 weeks of age. Diabetic and non-diabetic animals were either left untreated or treated with the UT antagonist SB-657510, 30 mg kg−1 day−1 [11], dissolved in 0.001% (vol./vol.) DMSO in drinking water. Systolic blood pressure was assessed by a non-invasive tail cuff system (IITC Life Science, Woodland Hills, CA, USA) in conscious mice after 19 weeks of study, as described previously [17].
After 20 weeks, animals were killed by i.p. injection of sodium pentobarbitone (100 mg/kg; Delvet, Seven Hills, NSW, Australia). Blood was collected and analysed by HPLC (BioRad, Richmond, CA, USA) for total glycated haemoglobin levels [18], and aortas were dissected out and cleaned (see electronic supplementary material [ESM], Detailed methods) for en face analysis, before being embedded for subsequent immunohistochemical studies. Plasma levels of total cholesterol and triacylglycerol were measured with a standard commercial enzymatic assay (LX20PRO; Beckman Coulter Diagnostics, Gladesville, NSW, Australia).
Aortic plaque area
En face analysis of the percentage of aortic intimal area covered by sudan IV stained atherosclerotic plaque was conducted as described previously [19].
Immunohistochemistry
Staining was conducted on paraffin-embedded sections as described previously [19] or as outlined in ESM (Detailed methods) using the primary antibodies listed in ESM Table 1.
Aorta photomicrographs were captured in a blinded manner using an Olympus BX-50 (Olympus Optical) and an RTV camera (Q-Imaging MicroPublisher 3.3, Surrey, BC, Canada) under identical light conditions, and the percentage of stained aortic area (excluding adventitia) was determined. Photomicrographs from whole cross-sections of human carotid endarterectomies were acquired on an FSX-100 box-type microscope (Olympus Optical, Tokyo, Japan) via stitching. Staining was digitally quantified (Image Pro-Plus version 6.0; Media Cybernetics, Silver Spring, MD, USA).
Quantitative RT-PCR
Total RNA from aorta was extracted with Trizol and analysed, and cDNA generated as described previously [20]. Samples were run using the Taqman system (ABI Prism 7500; Perkin-Elmer, Foster City, CA, USA); probes and primers were as listed in ESM Table 2. Gene expression was normalised to 18S and reported as ratios of gene expression in untreated control mice, which was given an arbitrary value of 1.
Protein extraction and western blot
Mouse aorta protein was extracted from the interphase and organic phase remaining after aortic RNA extraction and western blot performed (see ESM, Detailed methods). Membranes were visualised (BioRad gel-doc; Biolab Laboratories, Milan, Italy) and analysed using Quantity One software (version 4.6; BioRad).
Human carotid endarterectomy
Recruitment of individuals for this study was carried out at the Alfred Hospital, Melbourne, VIC, Australia. Ethics approval was obtained from the Alfred Human Research Ethics Committee (authorisation number 24/07). Informed consent was obtained from all participants. Diseased carotid artery tissue was removed during the carotid endarterectomy procedure. Participant data are listed in ESM Table 3. Carotid endarterectomy specimens were fixed in 4% (vol./vol.) formalin before being embedded in paraffin and decalcified with 10% (vol./vol.) nitric acid, prior to cutting and subsequent immunohistochemistry.
Statistical analysis
Data were analysed by one-way ANOVA with post-hoc comparisons of group means performed by Fisher’s least significant different method. Analyses were performed using SPSS version 17.0 (SPSS, Chicago, IL, USA). Unless otherwise specified, data are shown as mean ± SEM and a value of p < 0.05 was considered statistically significant.
Results
Effects of high glucose and UII on HAECs
A pilot study noted that 10−11 to 10−8 mol/l UII affected gene expression in HAECs, with 10−8 mol/l UII eliciting the greatest response (ESM Fig. 1a, b). This dose was therefore chosen for subsequent experiments. In additional experiments, 10−10 to 10−8 mol/l of the UT antagonist SB-657510 was found to modulate gene expression (ESM Fig. 1c, d). For simplification, only data from the 10−8 mol/l SB-657510 treatment group are shown (Fig. 1).

HAECs were grown in medium with normal glucose (NG; 5 mmol/l), high glucose (HG; 25 mmol/l) or mannitol (M; osmotic control), and treated or not treated (NT) with 10−8 mol/l UII or 10−8 mol/l UII + 10−8 mol/l SB-657510 (UII + SB). (a) Expression of Rela and (b) Ccl2 was increased in cells treated with UII in high-glucose medium, with SB-657510 treatment being associated with lower expression (n = 4–6). (c) Additionally HUVECs were grown in normal- or (d) high-glucose medium, and either not treated or stimulated for 24 h with UII (10−8 mol/l), SB-657510 (10−8 mol/l) or UII + SB-657510 before THP-1 cell adhesion was assessed. One group of THP-1 cells was pre-stimulated with PMA (positive control). n = 3–4. (a–d) *p < 0.05 vs NT and †p < 0.05 vs UII in cells grown in the same medium
In high-glucose medium, UII significantly increased RELA (encoding the p65 subunit of nuclear factor κB [NFκB]) and CCL2 (encoding macrophage chemoattractant protein 1 [MCP-1]) expression in HAECs. However, in normal-glucose conditions, UII had no significant effect on CCL2 expression. Treatment with UII and SB-657510 resulted in significantly reduced RELA and CCL2 expression (Fig. 1a, b).
NOS3 (encoding endothelial nitric oxide synthase [eNOS]) expression was significantly higher in UII-treated cells in normal- and in high-glucose medium (normal glucose 1.0 ± 0.1-fold induction, normal glucose + UII 10−8 mol/l 2.6 ± 0.3, p < 0.05; high glucose, no treatment [NT] 1.0 ± 0.1, high glucose + UII 10−8 mol/l 2.4 ± 0.2, p < 0.05). Furthermore, in cells treated with UII + SB-657510 (both 10−8 mol/l) NOS3 expression was significantly lower in normoglycaemic and hyperglycaemic settings (normal glucose UII + SB-657510 10−8 mol/l 0.8 ± 0.1, high glucose UII + SB-657510 10−8 mol/l 1.2 ± 0.1, p < 0.05 for both compared with UII alone).
Expression of UTS2R (encoding UT) was significantly upregulated in HAECs treated with 10−8 mol/l UII in normal-glucose medium (normal glucose, NT 1.0 ± 0.3-fold induction, normal glucose + UII 7.0 ± 2.0, p < 0.05, n = 5-6; high glucose, NT 2.2 ± 0.9, high glucose + UII 3.9 ± 1.3, NS, n = 4-5). Cells incubated with UII and SB-657510 (both 10−8 mol/l) had significantly lower UTS2R expression in normal- and high-glucose medium than that seen with UII alone (normal glucose UII + SB-657510 0.7 ± 0.4, n = 3; high glucose UII + SB-657510 0.7 ± 0.1, n = 5; p < 0.05 for both).
Expression of NOX1 and NOX4 was substantially greater in normal- and high-glucose medium when cells were treated with UII (NOX1: normal glucose, NT 1.0 ± 0.2, normal glucose + UII 3.5 ± 1.0, high glucose, NT 0.9 ± 0.2, high glucose + UII 4.9 ± 1.6, p < 0.0001; NOX4: normal glucose, NT 1.0 ± 0.1, normal glucose + UII 6.2 ± 1.1, high glucose, NT 0.4 ± 0.1, high glucose + UII 7.2 ± 0.6, p < 0001). This increase was not seen in cells treated with UII and SB-657510 (NOX1: normal glucose UII + SB-657510 10−8 mol/l 1.4 ± 0.2, high glucose UII + SB-657510 10−8 mol/l 1.1 ± 0.2, p < 0.001 for both; NOX4: normal glucose UII + SB-657510 10−8 mol/l 0.6 ± 0.1, p = 0.004, high glucose UII + SB-657510 10−8 mol/l 0.4 ± 0.0, p < 0.001).
Effects of high glucose and UII on endothelial monocyte adhesion (HUVECs)
UII treatment (10−8 mol/l) significantly increased cell adhesion under high-glucose medium conditions; however, this was not significantly different from values in normal-glucose medium (p = 0.078) (Fig. 1c). Pre-treatment with SB-657510 tended to reduce UII-mediated cell adhesion under normal-glucose conditions (p = 0.054) and significantly reduced adhesion in high-glucose conditions. In normal- and high-glucose conditions, SB-657510 (10−8 mol/l) alone did not influence THP-1 cell adhesion to HUVECs grown in normal- or high-glucose medium compared with untreated control cells (Fig. 1c, d). PMA, the positive control, significantly increased cell adhesion under normal- and high-glucose conditions (Fig. 1c, d).
In vivo studies: physiological and biochemical characteristics of Apoe KO mice
As expected, diabetic animals displayed polydipsia and polyuria; however, UT antagonism had no significant effect on these variables (data not shown).
Diabetic animals had lower body weight than non-diabetic control mice, and treatment with SB-657510 did not significantly alter body weight in diabetic animals compared with untreated diabetic animals. Non-diabetic controls treated with SB-657510 had a modest decrease in body weight (∼3 g) compared with untreated control animals (Table 1). Systolic blood pressure did not differ significantly between any groups (Table 1).
As expected, diabetic animals had significantly higher levels of total glycated haemoglobin and plasma glucose than non-diabetic animals (Table 1). Diabetic animals had significantly higher levels of total cholesterol, and LDL- and HDL-cholesterol than non-diabetic control animals. Treatment with SB-657510 did not alter any of these metabolic variables. Diabetic, but not control animals treated with SB-657510 had significantly lower levels of plasma triacylglycerol than untreated diabetic Apoe KO mice (Table 1).
Aortic plaque area in Apoe KO mice
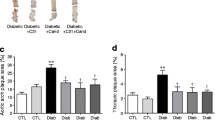
Diabetic animals had significantly more atherosclerosis than non-diabetic animals in terms of total plaque area as well as in individual segments of the aorta (Fig. 2). Diabetic animals treated with SB-657510 had significantly less total aortic plaque area, with significantly less atherosclerosis in the thoracic and abdominal aorta compared with untreated diabetic Apoe KO mice. However, differences did not reach statistical significance for plaque area in the aortic arch (p = 0.061) (Fig. 2). In non-diabetic control Apoe KO mice there were no significant changes in plaque area upon treatment (p = 0.6–0.9).

Examples of en face Apoe KO mouse aorta segments (a–d). Segments shown (left to right) are aortic arch, thoracic aorta and abdominal aorta from (a) Apoe control (C), (b) Apoe C + SB-657510-treated (SB), (c) Apoe diabetic (D) and (d) Apoe D + SB mice. (e) Digital analysis of the percentage of aortic intimal area covered by atherosclerosis in the total aorta, (f) aortic arch, (g) thoracic aorta and (h) abdominal aorta; n = 5–9; *p ≤ 0.05 vs Apoe C, †p ≤ 0.05 vs Apoe D and ‡p ≤ 0.05 vs Apoe D + SB-657510
UII system in Apoe KO mice
UII and UT
Semi-quantitative analysis of immunostaining for UII was significantly increased in aorta from diabetic Apoe KO mice compared with non-diabetic control animals (Fig. 3), with diabetic animals showing staining in the atherosclerotic plaque, the medial layer of the vascular wall and within the endothelium. Diabetic animals treated with SB-657510 had significantly less UII staining than untreated diabetic mice.

(a–d) Immunostaining for UII in aorta of Apoe KO mouse groups as follows: (a) non-diabetic control (Apoe C), (b) SB-657510-treated control (Apoe C + SB), (c) diabetic (Apoe D) and SB-657510-treated diabetic (Apoe D + SB). Scale bar 50 μm. (e) Digital quantification of staining showed significantly more UII staining in diabetic than in non-diabetic control animals, with SB-657510-treated animals showing significantly less staining than untreated diabetic animals; n = 4–8; *p < 0.05 vs Apoe C and †p < 0.05 vs Apoe D
Expression of Uts2r was significantly greater in diabetic than in non-diabetic control mouse aortas (diabetes 5.0 ± 1.0-fold induction vs control 1.0 ± 0.3, p = 0.004). SB-657510-treated groups showed no significant differences in Uts2r expression (diabetes + SB-657510 7.2 ± 1.2, control + SB-657510 1.4 ± 0.6) compared with their respective untreated groups (see preceding sentence).
Low levels of staining for the UT protein were found in mouse aorta, with levels in diabetic aorta not significantly elevated compared with non-diabetic aorta (diabetes 1.6 ± 0.3%; control 1.5 ± 0.5%). However, SB-657510-treated non-diabetic and diabetic groups had significantly less UT staining than their respective controls (diabetes + SB-657510 0.6 ± 0.1%, p = 0.011; control + SB-657510 0.6 ± 0.1% p = 0.024).
Signalling pathways
The downstream marker of UT activation, extracellular signal-regulated kinase (ERK) (also known as MAPK), was significantly increased in diabetic compared with non-diabetic aorta as reflected by measurement of its phosphorylated form (Fig. 4). This did not reach significance for total ERK (p = 0.062). Levels of phosphorylated ERK were significantly attenuated in the aorta of SB-657510-treated diabetic mice compared with untreated diabetic aorta; however, levels of total ERK were not significantly altered. The ratio of total ERK:phosphorylated ERK was not significantly different between untreated and treated diabetic animals (diabetes 0.15 ± 0.3, diabetes + SB-657510 0.15 ± 0.3).

(a) Western blot protein abundance of total ERK (t-ERK) (ERK 1 and 2 at 44 and 42 kDa respectively) and phospho-ERK (p-ERK) in aortas from diabetic (D) and non-diabetic control (C) Apoe KO mice that were treated or not with SB-657510 (+SB; 30 mg kg−1 day−1). (b) Quantification showing that phospho-ERK abundance was significantly greater in diabetic Apoe KO mice, with attenuation in SB-657510-treated diabetic animals. Quantification was relative to housekeeping protein (relative units); n = 2–3 per group; *p < 0.05 vs Apoe C, †p < 0.05 vs Apoe D and ‡p < 0.05 vs Apoe D + SB
In untreated diabetic animals, Nos3 expression in aorta was approximately 40% greater than that in diabetic animals treated with SB-657510 (diabetes 1.6 ± 0.4-fold induction, diabetes + SB-657510 0.6 ± 0.1, p < 0.05). However, non-diabetic control animals showed no significant difference compared with diabetic Apoe KO mice or with SB-657510 treatment in control animals (control 1.0 ± 0.3, control + SB-657510 0.8 ± 0.2).
Aortic cyclooxygenase-2 (COX2) immunostaining was elevated approximately twofold in diabetic Apoe KO mice compared with non-diabetic controls (p < 0.05) (Fig. 5a–e), with staining localised to the atheroma, endothelium and the media (Fig. 5c). Semi-quantitative analysis showed that diabetic animals treated with SB-657510 had significantly less COX2 staining in the aorta than untreated diabetic mice (Fig. 5e).

(a–e) Immunostaining for COX2 and (f–j) the oxidative stress marker nitrotyrosine in aorta of Apoe KO mice as follows: (a, f) non-diabetic control (Apoe C), (b, g) SB-657510-treated control (Apoe C + SB), (c, h) diabetic (Apoe D) and (d, i) SB-657510-treated diabetic (Apoe D + SB). Scale bars 50 μm. COX2 staining was found in atheroma, the endothelium (arrows) and media (arrow head). (e, j) Digital quantification of both stains showed significantly more staining in diabetic than in non-diabetic control animals, with SB-657510-treated diabetic animals showing significantly less staining than untreated diabetic animals; n = 4–8; *p < 0.05 vs Apoe C and †p < 0.05 vs Apoe D
Immunostaining for the marker of oxidative stress, nitrotyrosine, was significantly elevated in diabetic Apoe KO mice compared with non-diabetic controls (p < 0.05) (Fig. 5f–j). Diabetic animals treated with SB-657510 showed significantly less nitrotyrosine staining than untreated diabetic Apoe KO mice (Fig. 5j).
Expression of the downstream second messenger of UII, Rela, was also significantly decreased in aortas of diabetic animals treated with SB-657510 (diabetes 1.1 ± 0.3 vs diabetes + SB-657510 0.3 ± 0.1, p = 0.02).
Previously SB-657510 treatment has been shown to affect lipid uptake into the aorta of Apoe KO mice [11]. While gene expression of several lipid scavenger receptors was significantly elevated in the aorta of diabetic Apoe KO mice, treatment with SB-657510 did not result in any significant alterations in expression (ESM Table 4).
Effects of UT antagonism on macrophage markers in Apoe KO mice
Semi-quantitative analysis showed that immunostaining for MCP-1 was increased in aortas from diabetic animals, with staining being localised in the atherosclerotic plaque, vascular wall (medial layer) and endothelium. MCP-1 staining was significantly lower in diabetic animals treated with SB-657510 than in untreated diabetic animals (Fig. 6a–e).

(a–e) Immunostaining for MCP-1 and (f–j) the macrophage marker F4/80 in aorta of Apoe KO mice as follows: (a, f) non-diabetic control (Apoe C), (b, g) SB-657510-treated control (Apoe C + SB), (c, h) diabetic (Apoe D) and (d, i) SB-657510-treated diabetic (Apoe D + SB). Scale bars 50 μm. (e, j) Digital quantification of staining showed significantly more F4/80 and MCP-1 staining in diabetic than in non-diabetic control animals, with SB-657510-treated diabetic mice showing significantly less staining than untreated diabetic animals; n = 4–9; *p < 0.05 vs Apoe C and †p < 0.05 vs Apoe D
Semi-quantitative analysis showed that diabetic Apoe KO mice had significantly greater immunohistochemical staining for the macrophage marker F4/80 (Fig. 6f–j), with the majority of staining found within the atherosclerotic plaque, as well as in the adjacent vascular wall (Fig. 6h). The percentage area stained with F4/80 was significantly smaller in SB-657510-treated than in untreated diabetic Apoe KO mice (Fig. 6j).
Human carotid endarterectomy samples
Based on our findings in mouse studies, we assessed the abundance of UII and the UT, as well as MCP-1 in carotid endarterectomy specimens from non-diabetic and diabetic participants. Semi-quantitative analysis showed that UII staining was significantly elevated by at least fivefold in endarterectomies from diabetic individuals compared with non-diabetic specimens (Fig. 7c); however, UT staining was not significantly different between non-diabetic and diabetic participants (non-diabetic 2.3 ± 0.7%, diabetic 2.3 ± 0.8%). In addition, there was a sixfold increase in carotid MCP-1 staining in diabetic human vessels (Fig. 7), consistent with the results in mice for MCP-1 and UII staining. In human carotid endarterectomy sections, UII and MCP-1 staining was seen within the plaque (Fig. 7), with some staining also in the media.

(a, b) Human carotid endarterectomy sections showing immunohistochemical staining for UII and (d, e) MCP-1 in samples from non-diabetic control (a, d) and diabetic (b, e) individuals (arrow heads). Scale bars 500 μm. (c) Analysis of staining shows increases in UII and (f) MCP-1 in diabetic samples; n = 6–10; *p < 0.05 vs non-diabetic control
Discussion
UII is thought to play a role in atherosclerosis and may be particularly relevant in the diabetic setting, where plasma UII levels are elevated [21–23]. This is the first study to specifically investigate the role of UII and the UT in experimental and clinical diabetes-associated atherosclerosis. It reports for the first time that UII alters the response of endothelial cells studied in a high-glucose in vitro environment. Additionally, we have shown that antagonism of the UT with the highly specific, non-peptidergic antagonist, SB-657510, in diabetic Apoe KO mice decreased markers of macrophage infiltration and also significantly attenuated the development of atherosclerosis in the diabetic Apoe KO mouse, without significantly affecting key features of the diabetic milieu such as hyperglycaemia.
In vitro, diabetes resulted in an upregulation of inflammatory markers, including increased levels of the NFκB-dependent chemokine MCP-1 in HAECs. However, the effects were only observed in a high-glucose environment. Additionally, UII treatment resulted in a significant increase in monocyte adhesion to HUVECs, particularly in a high-glucose environment. This is in line with previous reports of UII increasing leucocyte adhesion in human coronary endothelial cells [24]. Furthermore, the UII-dependent effects observed in HAECS and HUVECs in a high-glucose environment were significantly attenuated by treatment with SB-657510. This suggests that UII may act differentially in the hyperglycaemic milieu, promoting pro-atherogenic responses. These positive in vitro findings therefore provide a strong rationale for performing complementary in vivo studies.
Previously, UII has been shown to act via cyclooxygenase [25], with the inducible form of cyclooxygenase, COX2, being elevated in carotid arteries of diabetic compared with non-diabetic patients [26]. COX2 is also a potential contributor to plaque vulnerability [27]. We found that COX2 immunostaining was increased in the aorta of diabetic Apoe KO mice and that diabetic animals treated with SB-657510 had significantly less COX2 staining. This suggests that UII may contribute to inflammation in diabetes-associated atherosclerosis partly via COX2.
It is well known that oxidative stress is a potential mediator of atherosclerosis, particularly in the diabetic context [28], and UII is known to act via NADPH oxidase (NOX)4 in smooth muscle cells [29, 30]. Indeed, in our in vitro studies, various NOX isoforms were activated, a phenomenon that appeared to be UII-dependent and attenuable by SB-657510. Importantly, we also assessed oxidative stress-mediated injury in vivo, using nitrotyrosine immunohistochemistry, and clearly demonstrated positive effects from UII blockade. This finding supports a central role for oxidative stress as a pathway for UII-mediated atherogenesis [31]. However, to date clinical treatments using antioxidants have not had major effects on the attenuation of cardiovascular disease or diabetic complications [32].
In the present study, UT antagonism attenuated the development of aortic atherosclerosis in diabetic Apoe KO mice (with the exception of the aortic arch). Previously, a 10 week treatment of high-fat-fed Apoe KO mice with SB-657510 was reported to result in a decrease of the atherosclerotic lesion area [11]. However, another study found that high-fat-fed Apoe KO mice lacking the UT had increased atherosclerosis [33]. Findings in UT/Apoe double KO mice should be interpreted with caution, as these animals have impaired lipid metabolism, including hyperlipidaemia and dysfunctional lipid regulation. In the present study, using a mouse model of type 1 diabetes, we found no evidence of this potentially confounding effect, as SB-657510 caused no significant changes in levels of total plasma cholesterol, LDL- or HDL-cholesterol. We did, however, detect a small decrease in triacylglycerol levels in SB-657510-treated diabetic (but not non-diabetic control) Apoe KO mice. This is consistent with a previous report, which indicated that another UT antagonist, palosuran, significantly decreased serum triacylglycerol in diabetic rats [14]. We also found that SB-657510 treatment did not significantly change the expression of various genes encoding lipid scavenger receptors and regulators in the aorta of Apoe KO mice, despite a previous report of altered acetyl-CoA acetyltransferase 1 (ACAT-1) levels in the aorta of high-fat-fed Apoe KO mice treated with SB-657510 [11]. Thus, the decrease in diabetes-associated atherosclerosis observed in our animal model is unlikely to be due to changes in lipid levels or aortic lipid regulators.
We report low levels of staining for the UT in non-diabetic mouse aorta and also in human carotid endarterectomy specimens. Thus, staining for the UT was not significantly elevated in diabetic compared with non-diabetic mouse aortas. This follows previous reports showing low levels of UT staining in rat vessels [34], and no change in UII binding between normal and atherosclerotic human coronary arteries, indicating that UT levels were unaltered [35]. However, given the pseudo-irreversible binding of UII to the UT [7] and the resultant, long-lasting effects of UII–UT activation seen in vivo [36], the levels of this receptor probably do not reflect the level of activation caused by UII.
Previous studies have reported that UII affects endothelial cell function via modulation of calcium signalling through a number of downstream pathways including protein kinase C and ERK activation [37]. Indeed, UII-treated HUVECs show a concentration-dependent increase in ERK phosphorylation [38]. In the present study, we have shown that ERK phosphorylation is increased in aortas from diabetic mice, with UT antagonist-treated diabetic animals having lower levels of the phosphorylated form of ERK. Similarly, we found that UT antagonism significantly attenuated expression of Rela, another downstream effector of UII [24], in the diabetic setting in vitro and in vivo. Thus UII is unlikely to contribute to the progression of diabetes-associated atherosclerosis via enhanced downstream signalling by molecules including ERK and NFκB.
UII and its receptor have both been linked to macrophage function, particularly the formation of foam cells [12]. This is consistent with our findings of increased CCL2 expression and monocyte adhesion in vitro, and the attenuation of these effects with UT antagonism in a high-glucose environment. Consistent with this, our own in vivo studies found evidence that UT antagonism reduced macrophage accumulation, as reflected by attenuated levels of various markers such as F4/80 and MCP-1 in the aorta of diabetic mice. Thus it is tempting to speculate that UT antagonism reduced diabetes-associated atherosclerosis in our animal model predominantly via a decrease in macrophage accumulation resulting from reduced abundance of the chemokine MCP-1.
Our results have clearly shown that MCP-1 and UII staining are elevated in the aorta of diabetic Apoe KO mice. We therefore conclude that examination of these proteins in human blood vessels is warranted. Previous studies have described increases in MCP-1 in diabetic endarterectomies [39]. However, to our knowledge, this is the first time that UII has been evaluated in vessels from diabetic patients and compared with atheroma from non-diabetic patients. Our findings strengthen the postulate that UII upregulation is a specific feature of diabetes-associated atherosclerosis and provide additional arguments for exploring UT antagonism in human diabetic participants with or at risk of macrovascular disease.
Due to the disappointing effects of the UT antagonist palosuran in diabetic nephropathy [15], enthusiasm for research into the therapeutic potential of targeting the UII system in diabetes has decreased. Palosuran, however, has a much lower affinity for the UT, and therefore alternative antagonists need to be considered. We had access to a highly specific UT antagonist, SB-657510, and were able to demonstrate an anti-atherosclerotic effect of this agent in the diabetic context, emphasising the potential role of the UII–UT axis as a target for protective therapies to reduce the burden of diabetic macrovascular complications.
Abbreviations
- COX2:
-
Cyclooxygenase-2
- EGM:
-
Endothelial growth medium
- eNOS:
-
Endothelial nitric oxide synthase
- ERK:
-
Extracellular signal-regulated kinase
- HAECs:
-
Human aortic endothelial cells
- KO:
-
Knockout
- MCP-1:
-
Macrophage chemoattractant protein 1
- NFκB:
-
Nuclear factor κB
- NHMRC:
-
National Health and Medical Research Council
- NOX:
-
NADPH oxidase
- NT:
-
No treatment
- PMA:
-
Phorbol 12-myristate 13-acetate
- UII:
-
Urotensin II
- UT:
-
UII receptor
References
Pearson D, Shively JE, Clark BR et al (1980) Urotensin II: a somatostatin-like peptide in the caudal neurosecretory system of fishes. Proc Natl Acad Sci USA 77:5021–5024
Ames RS, Sarau HM, Chambers JK et al (1999) Human urotensin-II is a potent vasoconstrictor and agonist for the orphan receptor GPR14. Nature 401:282–286
Liu Q, Pong SS, Zeng Z et al (1999) Identification of urotensin II as the endogenous ligand for the orphan G-protein-coupled receptor GPR14. Biochem Biophys Res Commun 266:174–178
Mori M, Sugo T, Abe M et al (1999) Urotensin II is the endogenous ligand of a G-protein-coupled orphan receptor, SENR (GPR14). Biochem Biophys Res Commun 265:123–129
Nothacker HP, Wang Z, McNeill AM et al (1999) Identification of the natural ligand of an orphan G-protein-coupled receptor involved in the regulation of vasoconstriction. Nat Cell Biol 1:383–385
Tal M, Ammar DA, Karpuj M, Krizhanovsky V, Naim M, Thompson DA (1995) A novel putative neuropeptide receptor expressed in neural tissue, including sensory epithelia. Biochem Biophys Res Commun 209:752–759
Douglas SA, Dhanak D, Johns DG (2004) From 'gills to pills': urotensin-II as a regulator of mammalian cardiorenal function. Trends Pharmacol Sci 25:76–85
Onan D, Hannan RD, Thomas WG (2004) Urotensin II: the old kid in town. Trends Endocrinol Metab 15:175–182
Maguire JJ, Kuc RE, Wiley KE, Kleinz MJ, Davenport AP (2004) Cellular distribution of immunoreactive urotensin-II in human tissues with evidence of increased expression in atherosclerosis and a greater constrictor response of small compared to large coronary arteries. Peptides 25:1767–1774
Bousette N, Patel L, Douglas SA, Ohlstein EH, Giaid A (2004) Increased expression of urotensin II and its cognate receptor GPR14 in atherosclerotic lesions of the human aorta. Atherosclerosis 176:117–123
Papadopoulos P, Bousette N, Al-Ramli W et al (2009) Targeted overexpression of the human urotensin receptor transgene in smooth muscle cells: Effect of UT antagonism in Apoe knockout mice fed with Western diet. Atherosclerosis 204:395–404
Watanabe T, Suguro T, Kanome T et al (2005) Human urotensin II accelerates foam cell formation in human monocyte-derived macrophages. Hypertension 46:738–744
Shiraishi Y, Watanabe T, Suguro T et al (2008) Chronic urotensin II infusion enhances macrophage foam cell formation and atherosclerosis in apolipoprotein E-knockout mice. J Hypertens 26:1955–1965
Clozel M, Hess P, Qiu C, Ding SS, Rey M (2006) The urotensin-II receptor antagonist palosuran improves pancreatic and renal function in diabetic rats. J Pharmacol Exp Ther 316:1115–1121
Vogt L, Chiurchiu C, Chadha-Boreham H et al (2010) Effect of the urotensin receptor antagonist palosuran in hypertensive patients with type 2 diabetic nephropathy. Hypertension 55:1206–1209
Douglas SA, Behm DJ, Aiyar NV et al (2005) Nonpeptidic urotensin-II receptor antagonists I: in vitro pharmacological characterization of SB-706375. Br J Pharmacol 145:620–635
Krege JH, Hodgin JB, Hagaman JR, Smithies O (1995) A noninvasive computerized tail-cuff system for measuring blood pressure in mice. Hypertension 25:1111–1115
Cefalu WT, Wang ZQ, Bell-Farrow A, Kiger FD, Izlar C (1994) Glycohemoglobin measured by automated affinity HPLC correlates with both short-term and long-term antecedent glycemia. Clin Chem 40:1317–1321
Watson AMD, Li J, Schumacher C et al (2010) The endothelin receptor antagonist avosentan ameliorates nephropathy and atherosclerosis in diabetic apolipoprotein E knockout mice. Diabetologia 53:192–203
Soro-Paavonen A, Watson AMD, Li J et al (2008) Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes 57:2461–2469
Totsune K, Takahashi K, Arihara Z, Sone M, Ito S, Murakami O (2003) Increased plasma urotensin II levels in patients with diabetes mellitus. Clin Sci (Lond) 104:1–5
Suguro T, Watanabe T, Kodate S et al (2008) Increased plasma urotensin-II levels are associated with diabetic retinopathy and carotid atherosclerosis in type 2 diabetes mellitus. Clin Sci (Lond) 115:327–334
Wang HX, Zeng XJ, Liu Y et al (2009) Elevated expression of urotensin II and its receptor in skeletal muscle of diabetic mouse. Regul Pept 154:89–90
Cirillo P, de Rosa S, Pacileo M et al (2008) Human urotensin II induces tissue factor and cellular adhesion molecules expression in human coronary endothelial cells: an emerging role for urotensin II in cardiovascular disease. J Thromb Haemost 6:726–736
Ishihata A, Ogaki T, Aita T, Katano Y (2005) Role of prostaglandins in urotensin II-induced vasodilatation in the coronary arteries of aged rats. Eur J Pharmacol 523:119–126
Szerafin T, Erdei N, Fulop T et al (2006) Increased cyclooxygenase-2 expression and prostaglandin-mediated dilation in coronary arterioles of patients with diabetes mellitus. Circ Res 99:e12–e17
Cipollone F, Prontera C, Pini B et al (2001) Overexpression of functionally coupled cyclooxygenase-2 and prostaglandin E synthase in symptomatic atherosclerotic plaques as a basis of prostaglandin E(2)-dependent plaque instability. Circulation 104:921–927
Lewis P, Stefanovic N, Pete J et al (2007) Lack of the antioxidant enzyme glutathione peroxidase-1 accelerates atherosclerosis in diabetic apolipoprotein E-deficient mice. Circulation 115:2178–2187
Diebold I, Petry A, Burger M, Hess J, Gorlach A (2011) NOX4 mediates activation of FoxO3a and matrix metalloproteinase-2 expression by urotensin-II. Mol Biol Cell 22:4424–4434
Djordjevic T, BelAiba RS, Bonello S, Pfeilschifter J, Hess J, Gorlach A (2005) Human urotensin II is a novel activator of NADPH oxidase in human pulmonary artery smooth muscle cells. Arterioscler Thromb Vasc Biol 25:519–525
Schaffer SW, Jong CJ, Mozaffari M (2012) Role of oxidative stress in diabetes-mediated vascular dysfunction: unifying hypothesis of diabetes revisited. Vasc Pharmacol 57:139–149
Golbidi S, Ebadi SA, Laher I (2011) Antioxidants in the treatment of diabetes. Curr Diabetes Rev 7:106–125
Bousette N, D’Orleans-Juste P, Kiss RS et al (2009) Urotensin II receptor knockout mice on an Apoe knockout background fed a high-fat diet exhibit an enhanced hyperlipidemic and atherosclerotic phenotype. Circ Res 105:686–695
Maguire JJ, Kuc RE, Kleinz MJ, Davenport AP (2008) Immunocytochemical localization of the urotensin-II receptor, UT, to rat and human tissues: relevance to function. Peptides 29:735–742
Katugampola SD, Kuc RE, Maguire JJ, Davenport AP (2002) G-protein-coupled receptors in human atherosclerosis: comparison of vasoconstrictors (endothelin and thromboxane) with recently de-orphanized (urotensin-II, apelin and ghrelin) receptors. Clin Sci (Lond) 103(Suppl 48):171S–175S
Watson AMD, Lambert GW, Smith KJ, May CN (2003) Urotensin II acts centrally to increase epinephrine and ACTH release and cause potent inotropic and chronotropic actions. Hypertension 42:373–379
Guidolin D, Albertin G, Oselladore B et al (2010) The pro-angiogenic activity of urotensin-II on human vascular endothelial cells involves ERK1/2 and PI3K signaling pathways. Regul Pept 162:26–32
Shi L, Ding W, Li D et al (2006) Proliferation and anti-apoptotic effects of human urotensin II on human endothelial cells. Atherosclerosis 188:260–264
Mas S, Martinez-Pinna R, Martin-Ventura JL et al (2010) Local non-esterified fatty acids correlate with inflammation in atheroma plaques of patients with type 2 diabetes. Diabetes 59:1292–1301
Acknowledgements
Thanks to J. Untario, E. Grixti and P. Chew for their technical assistance, to K. Gilbert, J. Mosele, D. Hoebee and A. McDougall for their assistance with the animals (all from Baker IDI Heart and Diabetes Institute, Australia), and to GlaxoSmithKline for supplying the UT antagonist SB-657510.
Funding
This work was supported by grants from Diabetes Australia Research Trust and the NHMRC of Australia (586678). A.M.D. Watson is supported by an NHMRC Australian Biomedical Fellowship (472698). M. Olukman was supported by the International Atherosclerosis Society. M.E. Cooper is an NHMRC Australian fellow and recipient of a JDRF Scholars Award. A. C. Calkin is supported by an Australian National Heart Foundation Fellowship (O-08M-3934). K.A.M. Jandeleit-Dahm and T. J. Allen are supported by NHMRC Senior Research Fellowships. This work is also supported in part by the Victorian Government’s Operational Infrastructure Support Program.
Duality of interest
D. J. Behm is an employee of GlaxoSmithKline and owns company stock. The other authors declare that they have no duality of interest associated with this manuscript.
Contribution statement
All authors contributed to the concept and design of the study, and to the acquisition, analysis and interpretation of data. All authors were also involved in the drafting or critical revision of the manuscript, and have approved the final version to be published.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 69 kb)
ESM Fig. 1
(PDF 25.1 kb)
ESM Table 1
(PDF 60 kb)
ESM Table 2
(PDF 11.9 kb)
ESM Table 3
(PDF 8 kb)
ESM Table 4
(PDF 9.69 kb)
Rights and permissions
About this article
Cite this article
Watson, A.M.D., Olukman, M., Koulis, C. et al. Urotensin II receptor antagonism confers vasoprotective effects in diabetes associated atherosclerosis: studies in humans and in a mouse model of diabetes. Diabetologia 56, 1155–1165 (2013). https://doi.org/10.1007/s00125-013-2837-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-2837-9