
Abstract
Aims/hypothesis
This study investigates the role of serine/threonine protein kinase 25 (STK25), a member of the sterile 20 (STE20) superfamily of kinases, in the regulation of skeletal muscle metabolism.
Methods
The effect of depleting STK25 in muscle cells was studied by reducing the mRNA and protein content of this target in the rat myoblast cell line L6 by small interfering (si)RNA. The changes in the mRNA and protein levels of several members of the fatty acid oxidative and glucose metabolic pathways were measured by quantitative real-time (qRT)-PCR and western blot. The rate of palmitate oxidation and glucose uptake was measured after transfection with siRNA for Stk25. Expression of STK25 was also evaluated in skeletal muscle biopsies from 41 white Europid men and women with normal and impaired glucose tolerance and type 2 diabetes using qRT-PCR.
Results
We demonstrate that partial depletion of STK25 increases the expression of uncoupling protein 3 (Ucp3), accompanied by increased lipid oxidation, in myoblasts. In addition, a reduced level of STK25 enhances the expression of Slc2a1 (also known as Glut1), Slc2a4 (also known as Glut4) and hexokinase 2, and correspondingly, improves insulin-stimulated glucose uptake in muscle cells. Consistent with these results, significantly higher STK25 levels were observed in the skeletal muscle of type 2 diabetic patients, compared with individuals with normal glucose tolerance.
Conclusions/interpretation
This is the first study indicating a possible role for STK25 in the regulation of glucose and lipid metabolism in L6 myoblasts. This kinase appears to be an interesting new mediator to be evaluated for therapeutic intervention in type 2 diabetes and related complications, as controlled increase in lipid oxidation and insulin-stimulated glucose uptake in skeletal muscle is favourable and can restore energy balance in metabolically compromised states.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The sterile 20 (STE20) serine/threonine protein kinase was first described in Saccharomyces cerevisiae as a mitogen-activated protein kinase kinase kinase kinase (MAP4K) involved in the mating pathway [1]. In mammals, more than 30 members of the STE20 superfamily of kinases have been described to date, characterised by a high degree of homology in the catalytic domain. They fall into two families, the p21-activated protein kinase (PAK) family with a C-terminal kinase domain, and the germinal centre kinase (GCK) family with an N-terminal kinase domain. These proteins are implicated in a wide range of biological responses, such as regulation of cell proliferation and differentiation, polarity, stress responses and cytoskeleton rearrangements [2].
STK25 (serine/threonine protein kinase 25), also referred to as YSK1 or SOK1, belongs to the GCKIII subfamily of STE20 proteins. STK25 has been shown to be slightly activated by reactive oxygen intermediates but not by growth factors or other environmental stresses, such as heat shock or osmolar stress [3]. Interestingly, STK25 does not activate any of the known MAPK pathways [3, 4] and thus this kinase might be involved in the regulation of a novel intracellular signalling pathway. Recently, STK25 was shown to be involved in two cellular processes: regulation of cell migration and modulation of cell death. STK25 was shown to associate with the Golgi matrix proteins Golgi matrix (GM)130 and cerebral cavernous malformation (CCM)3, and binding to GM130 was shown to activate the kinase by promoting autophosphorylation of STK25 [5]. STK25 pays a role in determining the proper localisation and morphology of the Golgi complex: inhibition of STK25 function by RNA interference perturbs perinuclear Golgi organisation, cell migration and invasion into type I collagen [5–7]. STK25 was shown to be normally localised on the Golgi complex, whereas in cells subjected to extreme stresses and caspases, it is cleaved and enters the nucleus, inducing cell death. In these conditions, downregulation of STK25 by RNA interference enhanced cell survival while overexpression of STK25 elicited apoptotic cell death [8]. The only known substrates for STK25 are myelin basic protein (MBP) [9], Golgi-associated 14-3-3ζ [5] and CCM3 [7]. To our knowledge, there are no studies suggesting a possible function for STK25 in metabolic regulation.
In this study, we demonstrate for the first time that partial depletion of STK25 by small interfering (si)RNA in a rodent skeletal muscle cell line leads to increased expression of Ucp3, accompanied by increased fatty acid oxidation. In addition, the expression of several key regulators of glucose metabolism was increased by reduced levels of STK25 in muscle cells, and insulin-stimulated glucose uptake was enhanced. In line with these findings, significantly higher STK25 mRNA levels were detected in skeletal muscle of patients with type 2 diabetes, which further supports the role of STK25 in the regulation of energy metabolism.
Methods
Cell culture
L6 myoblasts (Rattus norvegicus, American Type Culture Collection, Manassas, VA, USA) were maintained in DMEM (Lonza, Basel, Switzerland) including 4.5 g/l (25 mmol/l) glucose and l-glutamine, supplemented with 10% FBS (Invitrogen, San Diego, CA, USA).
RNA interference
L6 myoblasts were transfected with siRNA directed against Stk25 (s173550, s173551; Ambion, Austin, TX, USA) or scrambled siRNA (#AM4635; Ambion) as a control. Briefly, cells (125,000) were transfected with 30 nmol/l siRNA by using Lipofectamine RNAiMax (Invitrogen) according to the manufacturer’s instructions for reverse transfection using 12-well plates.
Quantitative real-time PCR
Relative quantification of mRNA was performed using the ABI Prism 7900HT Sequencing Detection System (Applied Biosystems, Foster City, CA, USA). See the electronic supplementary material [ESM] Methods for further details.
Western blot analysis
Western blot analysis was performed as previously described [10]. See the ESM Methods for further details on procedures and antibodies used.
Immunohistochemistry
L6 myoblasts were grown in Lab-Tek II Chamber glass slides (Nalgene Nunc International, Naperville, IL, USA). Cells were fixed for 20 min in 4% formaldehyde, permeabilised in 0.1% Triton X-100 for 7 min, blocked with 20% FBS for 30 min and probed with anti-STK25 antibody (anti-YSK1, sc-6865; Santa Cruz Biotechnology, Santa Cruz, CA, USA), diluted 1:50 in PBS containing 2% (wt/vol.) BSA, for 3 h at room temperature. The secondary antibody, conjugated with Alexa-594 (Molecular Probes, Eugene, OR, USA), diluted 1:100 in PBS containing 2% (wt/vol.) BSA, was added for 1 h. The cover slip was mounted using Duolink II Mounting Medium containing DAPI (Olink, Uppsala, Sweden). Microscopy analyses were performed at room temperature using the Leica SP5 confocal system, attached to a Leica DMI6000 microscope using a 63× oil immersion objective.
Transient transfections
L6 myoblasts cultured in 12-well plates were transfected with pFLAG-rStk25 using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Either 48 or 64 h post-transfection the cells were serum starved (DMEM with 1 g/l [5.5 mmol/l] glucose and 0.5% [wt/vol.] cell culture tested BSA [Sigma-Aldrich]) for 20 h or 3 h, respectively. Cells starved for 3 h were incubated with menadione (10 and 30 μmol/l, Sigma-Aldrich), H2O2 (0.2 and 0.5 mmol/l, Merck Chemicals, Darmstadt, Germany), thapsigargin (2 and 10 μmol/l, Sigma-Aldrich), human rTNF-α (50 ng/ml, Sigma-Aldrich), or mouse rIl6 (50 ng/ml, Sigma-Aldrich) for the indicated times, before all cells were lysed for western blot analysis.
Palmitate oxidation and glucose transport
Palmitate oxidation and glucose uptake were measured as previously described [11, 12]. See the ESM Methods for further details.
Quantitative RT-PCR analysis in muscle samples of human participants
Forty-one consecutively recruited white men (n = 23) and women (n = 18) were categorised into groups with normal glucose tolerance (NGT; n = 13, nine men, four women), impaired glucose tolerance (IGT; n = 14, eight men, six women), and type 2 diabetes (n = 14, six men, eight women) based on a 75 g oral glucose tolerance test. All the participants of the study gave their informed consent. The investigations were approved by the ethics committee of the University of Leipzig and were carried out in accordance with the Declaration of Helsinki. For participant characteristics and details on quantitative RT-PCR (qRT-PCR) analysis, see the ESM Methods.
Statistical analysis
Statistical significance between the groups was calculated with an unpaired Student’s t test, with a value of p < 0.05 considered statistically significant.
Results
Expression profiling of Stk25 in mouse, rat and human tissues
The relative expression of Stk25/STK25 mRNA in mouse, rat and human tissues was measured by qRT-PCR (Fig. 1a). Similarly to previous reports in which mRNA levels of Stk25 in rodents were assessed by northern blot [3, 4], Stk25 was identified in all tissues examined. In both mouse and rat tissues, the mRNA expression was highest in the small and large intestine and the brain, compared with heart, skeletal muscle, spleen, liver, lung, kidney and adipose tissues. STK25 mRNA was also quantified in the human metabolic tissues liver, fat and skeletal muscle, with expression being highest in skeletal muscle tissue (Fig. 1a). The western blot analysis in mouse tissues detected STK25 protein in all tissues examined, with the highest level in samples of brain and spleen (Fig. 1b). Only a weak correlation between Stk25 mRNA and protein expression was observed in different mouse tissues, indicating that post-transcriptional mechanisms are likely to be involved in controlling the abundance of STK25 protein.

Stk25 is broadly expressed in mouse, rat and human tissues. In myoblasts, STK25 protein is localised primarily in the cytoplasm. a Relative expression of Stk25/STK25 mRNA was assessed by qRT-PCR analysis in mouse (white bars), rat (black bars) and human (grey bars) tissues. The expression level of Stk25/STK25 in the liver of each species is set to 1. Mouse and rat data are means±SEM from three different animals. Human adipose tissue, liver and skeletal muscle data are means±SEM from three, three and 13 individuals, respectively. L, liver; Sp, spleen; Lu, lung; H, heart; LI, Large intestine; SI, small intestine; B, brain; WAT, white adipose tissue; K, kidney; SM, skeletal muscle. b Lysates of mouse tissues were analysed by western blot using anti-STK25 antibodies. Protein levels were analysed by densitometry and shown as bar histograms. The level of STK25 in the liver is set to 1. The data are shown as means±SEM of protein analysis in three or four different animals. A representative western blot is shown. c Relative expression level of Stk25 mRNA in mouse skeletal muscle tissue was assessed by qRT-PCR analysis with the soleus muscle (S) set to 1. Data are shown as means±SEM from three different animals. WQ, white quadriceps muscle; RQ, red quadriceps muscle; WG, white gastrocnemius muscle; RG, red gastrocnemius muscle. d Lysates of mouse muscle tissues were analysed by western blot using anti-STK25 antibodies. Protein levels were analysed by densitometry and shown as bar histograms. The level of STK25 in the soleus muscle (S) is set to 1. The data are shown as means±SEM of protein analysis in five different animals. A representative western blot is shown. e–g L6 myoblasts were processed for immunofluorescence, stained with anti-STK25 antibody (red, e), for DNA (DAPI, blue, f) or both (g), and analysed by confocal microscopy
In mice, the mRNA expression and protein production of Stk25/STK25 was analysed in red (type IIa) and white (type IIb) portions of the gastrocnemius and quadriceps muscles and compared with the soleus muscle, which is composed mostly of slow (type I) fibres. No significant differences were observed comparing these tissues for Stk25/STK25 levels (Fig. 1c,d).
STK25 protein in muscle cells is distributed throughout the cytoplasm
The cellular localisation of STK25 protein in myoblasts was studied by immunofluorescent staining of endogenous STK25 in the rat myoblast cell line L6, and analysed by confocal microscopy. In these cells, STK25 was uniformly distributed throughout the cytoplasm (Fig. 1e–g).
Partial depletion of STK25 in muscle cells increases expression of genes involved in lipid and glucose metabolism
The effect of depleting STK25 in muscle cells was studied by reducing the protein content of this target in the rat myoblast cell line L6 by approximately three- to fourfold via siRNA transfections (ESM Fig. 1). The mRNA expression of several members of the fatty acid oxidative and glucose metabolic pathways, as well as transcription factors involved in mitochondrial biogenesis, was quantified by qRT-PCR at four time points after transfection (Fig. 2).

Partial depletion of STK25 increases mRNA expression of genes involved in lipid and glucose metabolism in skeletal muscle cells. a–m. Expression of genes regulating lipid metabolism in skeletal muscle, Lpl (a), Cd36 (b), Cs (c), Cycs (d), mt-Co2 (e), Ucp2 (f), Ucp3 (g), Ppargc1a (h), Nrf1 (i), the oxidative stress marker Sod2 (j), as well as genes regulating glucose metabolism Slc2a1 (k), Slc2a4 (l) and Hk2 (m) was assessed in L6 myoblasts 48, 96, 144 and 192 h post-transfection with control siRNA (white bars) or anti-Stk25 siRNA (black bars) by qRT-PCR. The expression level of each gene in cells transfected with control siRNA at 48 h post-transfection is set to 1. Values are reported as means±SEM of transfected cells in three culture wells for each test condition. The data shown are representative of two independent transfection experiments with similar results. *p < 0.05; **p < 0.01 comparing cells transfected with control siRNA or anti-Stk25 siRNA at each time point
The entry of fatty acids into the muscle cells is regulated by selective transport of NEFAs across the membrane. Fatty acids are transported systemically as triacylglycerol and released by lipoprotein lipase (LPL), while the membrane protein cluster of differentiation 36 (CD36; also known as FAT) facilitates the major fraction of fatty acid uptake in muscle cells. The mRNA expression levels of Lpl and Cd36 were not affected by knockdown of Stk25 (Fig. 2a,b). The next rate-limiting step in lipid metabolism is the transport of the fatty acids into the mitochondria as acyl-CoA, which involves carnitine palmitoyltransferase 1 (CPT1). CPT1 is inhibited allosterically by malonyl-CoA, synthesised by acetyl-CoA carboxylase (ACC). The activity of ACC is regulated by reversible phosphorylation, where kinases such as AMP-activated protein kinase (AMPK) directly phosphorylate and inactivate ACC [13], thereby driving the entry of long-chain acyl-CoA into the mitochondria. No significant change in phosphorylation of ACC was detected after transfection with anti-Stk25 siRNA (ESM Fig. 2a,b), which is consistent with the idea that the uptake of acyl-CoA into the mitochondria is not affected. In the mitochondrial matrix, acyl-CoA is oxidised in a stepwise manner to form acetyl-CoA, which is converted to citrate by citrate synthase (CS), the rate-controlling enzyme of the tricarboxylic acid (TCA) cycle. The mRNA expression of Cs was not affected by partial knockdown of Stk25 (Fig. 2c). In the TCA cycle, the reducing equivalents are transferred to the electron transport chain to generate an electrochemical gradient across the mitochondrial inner membrane for oxidative phosphorylation. Quantification of the mRNA expression of cytochrome c (Cycs), a co-enzyme required for several enzymes in the electron transport chain for shuttling electrons, as well as cytochrome c oxidase subunit II (mt-Co2, also known as Cox2), a marker for respiratory Complex IV, indicated that these targets were not differentially regulated by partial depletion of STK25 (Fig. 2d,e). Uncoupling proteins 2 and 3 (UCP2 and UCP3) are mitochondrial inner membrane transporters, which are suggested to function in facilitation of fatty acid oxidation [14]. Stk25 knockdown markedly increased the expression of Ucp2 and Ucp3 compared with cells transfected with control siRNA (Fig. 2f,g). A statistically significant increase in Ucp2 mRNA was observed at the first time point analysed (48 h post-transfection), and was stable over the period of observation. A significant increase in the expression of Ucp3 was detected 144 h post-transfection and the increase was most pronounced (2.7-fold) at the last time point studied (192 h post-transfection). The transcriptional regulators peroxisome proliferator-activated receptor-gamma, coactivator 1, alpha (PPARGC1A, also known as PGC1-A) and nuclear respiratory factor 1 (NRF1) increase the expression of both nuclear and mitochondrial encoded genes involved in oxidative metabolism. No changes in the mRNA expression of these two targets were observed in cells transfected with anti-Stk25 siRNA (Fig. 2h,i). We next examined the expression of markers of oxidative stress. Superoxide dismutase 2 (SOD2) is a critical mitochondrial antioxidant defence against superoxide produced by respiration [15]. 4-HNE (4-hydroxy-2-nonenal) is one of the major highly reactive products of lipid peroxidation. 4-HNE reacts with mitochondrial proteins, and accumulation of 4-HNE adducts can be seen as a measure of oxidative damage in muscle tissue [16]. No difference in Sod2 mRNA expression or the level of 4-HNE adducts was observed in response to depletion of STK25 (Fig. 2j, ESM Fig. 2c,d).
Further, we quantified the mRNA levels for key targets regulating glucose metabolism in skeletal muscle. SLC2A1 (also known as GLUT1) and SLC2A4 (also known as GLUT4) are both key transporters for glucose uptake into skeletal muscle cells while hexokinase 2 (HK2) initiates the first, rate-limiting reaction of glycolysis and catalyses the formation of glucose 6-phosphate from glucose. The mRNA expression of Slc2a1, Slc2a4 and Hk2 in L6 myoblasts was significantly increased after partial depletion of STK25 (Fig. 2k–m). Increase in the expression levels of Ucp2, Ucp3, Slc2a1, Slc2a4 and Hk2 mRNA in response to reduction in Stk25 expression was confirmed using an independent anti-Stk25 siRNA (data not shown).
We confirmed the increased production of UCP3, SLC2A1, SLC2A4 and HK2 in cells transfected with anti-Stk25 siRNA at the protein level by western blot analysis (Fig. 3a–d). In accordance with mRNA data, protein levels of LPL, CD36, CS, cytochrome c (CYCS), cytochrome c oxidase subunit II (mt-CO2), NRF1 and SOD2 were not influenced by partial depletion of STK25 (ESM Fig. 3). Interestingly, the production of PPARGC1A protein was slightly increased by anti-Stk25 siRNA at the first two time points post-transfection (ESM Fig. 3f), even if no difference was observed at the mRNA level. PPARGC1A is known to induce Ucp2 and Ucp3 genes in muscle cells [17, 18], and thus this may be one mechanism for the increased expression of Ucp2 and Ucp3 mRNA observed. We could not detect any UCP2 protein at any of the time points studied.

Reduced level of STK25 increases protein levels of UCP3, SLC2A1, SLC2A4 and HK2, and enhances palmitate oxidation and insulin-stimulated glucose uptake in L6 myoblasts. a–d Cell lysates were analysed by western blot using antibodies specific for UCP3 (a), SLC2A1 (b), SLC2A4 (c) or HK2 (d) at indicated time points post-transfection of L6 myoblasts with control siRNA (white bars) or anti-Stk25 siRNA (black bars). Protein levels were analysed by densitometry and shown as bar histograms. The expression level of the corresponding protein in cells transfected with control siRNA at 48 h (SLC2A1, SLC2A4 and HK2) or 144 h (UCP3) post-transfection is set to 1. The data are shown as means±SEM of transfected cells in four culture wells for each test condition. *p < 0.05 comparing cells transfected with control siRNA or anti-Stk25 siRNA at each time point. A representative western blot is shown. e,f Palmitate oxidation (e) and 2-deoxyglucose uptake (f) were measured in L6 myoblasts 216 h or 144 h post-transfection, respectively, with control siRNA or anti-Stk25 siRNA. Values are reported as means±SEM of 8–16 determinations. The data point for measurement of fatty acid oxidation was selected based on the changes in the expression profile for Ucp3 (Figs 2g, 3a). The data point for measurement of glucose uptake was selected based on the changes in the expression of regulators of glucose metabolism (Figs 2k–m, 3b–d). A significant increase of approximately 20% in palmitate oxidation was observed even 192 h post-transfection with anti-Stk25 siRNA (data not shown). The data on glucose uptake shown are representative of two independent transfection experiments with similar results. **p < 0.01
As previously reported, a certain level of spontaneous differentiation occurs in L6 myoblasts even in the presence of 10% serum when cells are not subcultured and reach sub-confluence, which is accompanied by changes in the mRNA and protein expression of metabolic and myogenic targets [19–23]. In this study, significant downregulation of the expression of Cs, mt-Co2 and Nrf1 mRNA was seen in cells transfected both with control and with anti-Stk25 siRNA, comparing 48 and 192 h timepoints post-transfection, while Cd36, Ucp2, Ucp3, Ppargc1a, Sod2, Slc2a1 and Hk2 were upregulated over time. Similar alterations in expression profile over time took place even in mock transfected cells (transfection performed in an identical fashion, but with no siRNA added; data not shown), which supports the likelihood that these changes in mRNA expression are not related to the siRNA but rather reflect the partial differentiation process in cells occurring after transfection, when cells are not subcultured.
The total number of cells analysed at the four time points post-transfection did not differ, comparing the cells transfected with anti-Stk25 or control siRNA (data not shown), indicating that, under the experimental conditions applied, STK25 did not affect cell proliferation or survival.
Reduction in expression of Stk25 leads to increased β-oxidation and glucose uptake in muscle cells
The increased expression of Ucp3, as well as of regulators of glucose metabolism, would be expected to lead to enhancement of fat oxidation and glucose uptake in skeletal muscle cells. We therefore measured the rate of fatty acid (palmitate) oxidation and 2-deoxyglucose uptake in L6 myoblasts after transfection with siRNA for Stk25. In line with the increased expression of Ucp3, L6 myoblasts exhibit approximately 20% increase in palmitate oxidation after partial depletion of STK25, reaching the levels of oxidation achieved by the treatment with the reference substance phenformin (Fig. 3e). Phenformin is a biguanine, which stimulates fatty acid oxidation via activation of AMPK [24]. We could not detect any significant increase in glucose uptake in cells transfected with control siRNA following insulin treatment. This is surprising as significant stimulation with 100 nmol/l insulin has previously been reported in L6 myotubes [12]. In addition, no significant increase in glucose uptake was seen in basal conditions in cells transfected with anti-Stk25 siRNA, compared with cells transfected with control siRNA. However, compared with cells transfected by control siRNA, transfection with anti-Stk25 siRNA, combined with insulin treatment, resulted in a statistically significant increase in glucose uptake of approximately 50%, indicating that partial depletion of STK25 increases insulin-stimulated glucose uptake (Fig. 3f).
STK25 in skeletal muscle cells is activated by inflammatory and oxidative stress
To test the hypothesis that STK25 is involved in cellular stress responses, agonists for different classes of stimuli were evaluated for their ability to activate STK25 in L6 myoblasts. According to our data, STK25 undergoes autophosphorylation when immune complexes are incubated with γ-[32P]ATP for only a few (less than 5) min (data not shown). Since autophosphorylation leads to activation of this kinase [5], any differences in activity between control and stimulated cells, assessed by biochemical assay, will be masked by changes in the phosphorylation status of STK25 during the course of the incubation. Therefore, the activity of STK25 was measured based on the phosphorylation level of the threonine 174 residue, assessed in the lysate from L6 cells transiently transfected with pFLAG-rStk25. It has previously been demonstrated that phosphorylation of threonine 174 adequately reflects the activity level of STK25 [5].
Under the experimental conditions applied, treatment with proinflammatory cytokine TNF-α (50 ng/ml) but not with multifunctional myokine IL-6 (50 ng/ml), leads to a significant increase in phosphorylation of STK25 (Fig. 4a,b). In our study, menadione (10 μmol/l; a quinone, which is a potent intracellular generator of reactive oxygen intermediates [25]) and H2O2 (0.2 and 0.5 mmol/l) activated STK25 (Fig. 4c,d). No change in the phosphorylation of STK25 was observed in cells treated with thapsigargin (a strong inducer of mammalian endoplasmic reticulum stress through perturbation of calcium homeostasis [26]), or by serum starvation for 20 h (Fig. 4e,f).

Stimuli of inflammatory and oxidative stress increase the phosphorylation level of STK25 in L6 myoblasts. a–f L6 myoblasts were transfected with pFLAG-rStk25 and incubated with TNF-α (50 ng/ml, black bars) (a), IL-6 (50 ng/ml, black bars) (b), menadione (10 μmol/l, black bars and 30 μmol/l, grey bars) (c), H2O2 (0.2 mmol/l, black bars and 0.5 mmol/l, grey bars) (d), or thapsigargin (Tg, 2μmol/l, black bars and 10 μmol/l, grey bars) (e) at indicated time points, or serum starved for 20 h (black bars) (f). Cell lysates were analysed by western blot using antibodies specific for anti-phospo-Thr174-STK25 and STK25. No signal corresponding to the size of FLAG-tagged STK25 was observed in mock transfected cells. Protein levels were analysed by densitometry and the data are shown as the ratio of phospho-Thr174-STK25 to total STK25. The ratio of phospho-Thr174-STK25 to total STK25 without any stimuli at each time point is set to 1 (white bars). Values are reported as means±SEM of transfected cells in four to six culture wells for each test condition. A representative western blot is shown. *p < 0.05 comparing cells with stimuli added and basal condition at each time point
Expression of STK25 mRNA is significantly increased in skeletal muscle of type 2 diabetic patients
Expression of STK25 was quantified in skeletal muscle biopsies of individuals with type 2 diabetes, compared with the age- and BMI-matched individuals with either IGT or NGT (Table 1). Statistically significant higher STK25 mRNA levels were observed in skeletal muscle of type 2 diabetic patients, compared with participants with NGT. It is noteworthy that skeletal muscle STK25 mRNA expression in individuals with IGT was intermediate between the two extreme groups (Fig. 5a). There was no correlation between the BMI or WHR of the participants and STK25 mRNA expression levels (ESM Fig. 4), indicating that the increase of STK25 in the muscle of type 2 diabetic patients is not a consequence of obesity. In line with the suggested function of STK25 based on in vitro studies, expression of UCP3 and HK2 was decreased in the skeletal muscle of participants with IGT and/or type 2 diabetes compared with NGT controls (Fig. 5a).

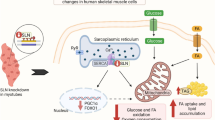
Expression of STK25 mRNA is significantly increased in the skeletal muscle of type 2 diabetic patients, indicating a potential therapeutic implication of this target in metabolic complications. a Expression of STK25, UCP3 and HK2 was quantified in skeletal muscle biopsies of individuals with type 2 diabetes (n = 14, grey bars), NGT (n = 13, white bars) and IGT (n = 14, black bars) by qRT-PCR. Values are reported as means±SEM. *p < 0.05, **p < 0.01. The p value comparing the UCP3 expression in patients with type 2 diabetes with participants with NGT was below 0.1. b Proposed model of the role of STK25 in the regulation of lipid and glucose metabolism in skeletal muscle: potential therapeutic implication in type 2 diabetes and related metabolic complications
Discussion
The importance of mitochondrial oxidative phosphorylation in the development of type 2 diabetes has recently been shown by gene expression profiling studies in the muscle tissues of diabetic and non-diabetic populations. Individuals with type 2 diabetes have reduced expression of genes of key proteins in oxidative metabolism and mitochondrial function in muscle [27]. Furthermore, individuals with insulin resistance but NGT display the same pattern of decreased expression of genes related to mitochondrial oxidative metabolism, suggesting that such characteristics are directly relevant to the development of type 2 diabetes [28]. One of the mitochondrial proteins showing decreased mRNA expression in diabetic individuals is UCP3 [29]. In addition, association studies of natural allele influences on obesity have shown that UCP2 and UCP3 are candidate obesity genes in humans [30].
UCPs are mitochondrial inner membrane transporters. While UCP1 is produced in brown adipose tissue, where it plays a pivotal role in the control of thermogenesis, UCP2 occurs in a large number of tissues, including skeletal muscle, whereas UCP3 is selectively produced in skeletal muscle, brown fat and to a lesser extent in the heart [31, 32]. UCP2 and UCP3 are suggested to regulate intramyocellular oxidative metabolism and whole-body energy expenditure. This is based on the finding that rodent skeletal muscle Ucp3 and Ucp2 mRNA levels are increased during fasting when muscle switches towards lipid use, and Ucp3 is increased by fatty acid loading [33]. Furthermore, overexpression of Ucp3 in rodent muscle cells increases the level of fatty acid oxidation [14]. In line with these results, overexpression of Ucp3 in skeletal muscle protected mice against fat-induced insulin resistance, reduced diet-induced obesity and improved glucose metabolism [34]. Interestingly, Ucp2 and Ucp3 mRNA expression in rat skeletal muscle was stimulated by insulin and contraction in vitro, an effect that could be mimicked by activation of AMPK by 5-amino-4-imidazolecarboxamideriboside (AICAR) [35].
In this study we demonstrate for the first time that reduction in the expression of STK25, a member of the Ste20 superfamily of kinases, leads to marked upregulation of Ucp2 and Ucp3 mRNA in L6 myoblasts. In line with mRNA data, increased levels of UCP3 protein were demonstrated in L6 cells transfected with anti-Stk25 siRNA. No UCP2 protein was detected in L6 cells, which questions the physiological relevance of this finding. The change in production level of UCP3 was accompanied by approximately 20% increase in palmitate oxidation. The statistically significant increase in lipid oxidation is in line with previous reports where moderate (2.2- to 2.5-fold) overproduction of UCP3 protein in L6 cells increased fatty acid oxidation by approximately 50% [14].
Further, we demonstrate that the levels of SLC2A4, the main insulin-sensitive glucose transporter in skeletal muscle, and HK2, the enzyme used to trap glucose in muscle, were increased in muscle cells in response to transfection with anti-Stk25 mRNA. The production of the second major glucose transporter in muscle, SLC2A1, was also upregulated in response to partial depletion of STK25. In line with these results, the insulin-stimulated glucose uptake was enhanced in response to reduced expression of Stk25. The impaired insulin effect in the skeletal muscle of type 2 diabetic patients has not been connected to a defect in the expression of the SLC2A1 or SLC2A4 genes. However, reduced basal, as well as insulin-stimulated, levels of HK2 mRNA have been reported [36]. In addition, exercise induces glucose uptake in skeletal muscle while increasing mRNA levels of SLC2A1, SLC2A4 and HK2 [37].
In myoblasts, STK25 protein was localised primarily to the cytoplasm, and therefore, the effect on the expression profile of its transcriptional targets is likely to be mediated via as yet unknown mediator(s), which transduce the signal to the nucleus.
Recent evidence suggests that inflammatory stress caused by TNF-α, as well as oxidative and endoplasmic reticulum stress, are fundamental steps in the progression of insulin resistance in skeletal muscle [38–42]. Previously, incubation with H2O2 has been shown to activate STK25 after immunoprecipitation from Ramos B cells or Madin–Darby canine kidney epithelial cells [9]. Interestingly, in this study the treatment of L6 myoblasts with the proinflammatory myokine, TNF-α, as well as menadione and H2O2, both causing oxidative stress, was shown to regulate the activity of STK25 at the level of phosphorylation.
Skeletal muscle accounts for a large proportion of body mass and is the major site of insulin-stimulated glucose disposal. Interestingly, transgenic mice expressing Ucp1 in muscle tissue at only 1–2% of the level in brown adipocytes were resistant to diet-induced obesity and did not develop insulin resistance [43]. Thus, the moderate changes in the expression of Ucp3, as well as genes regulating glucose metabolism by downregulation of Stk25 in skeletal muscle, are likely to influence the energy balance at the whole-body level. This was supported by the finding that STK25 mRNA was significantly increased in the skeletal muscle of patients with type 2 diabetes, compared with individuals with NGT. To further elucidate the function of STK25 in vivo, the phenotypic analysis of transgenic mice overexpressing Stk25 in metabolic tissues is ongoing. Our preliminary analysis of Stk25 transgenic mice suggests significantly impaired glucose use and insulin resistance, indicated by intraperitoneal glucose and insulin tolerance tests, respectively, after challenge with high fat diet (E. Cansby and M. Mahlapuu, unpublished data).
The siRNA transfection experiments described were performed in L6 myoblasts, which offer the advantage of being easy to transfect with high reproducibility and efficacy, compared with myotubes. Still, this is considered to be a limitation of the work as differentiated myotubes are more representative of skeletal muscle in vivo, compared with myoblasts. Moreover, the possibility that the partial depletion of STK25 modulated the differentiation process of L6 myoblasts, which contributed to the changes in gene and protein expression patterns observed, cannot be excluded.
Taken together, our data indicate that partial depletion of STK25, a member of the Ste20 superfamily of kinases, increases the expression of Ucp3, Slc2a1 (Glut1), Slc2a4 (Glut4) and Hk2 in the rodent myoblast L6 cell line, accompanied by enhanced fatty acid oxidation and insulin-stimulated glucose uptake. The study suggests that STK25 is an interesting new mediator to be evaluated for therapeutic intervention in type 2 diabetes and related complications, as controlled increase in skeletal muscle oxidative metabolism and glucose uptake would be important to improve the energy balance in metabolically compromised states (Fig. 5b).
Abbreviations
- 4-HNE:
-
4-Hydroxy-2-nonenal
- ACC:
-
Acetyl-CoA carboxylase
- AMPK:
-
AMP-activated protein kinase
- CS:
-
Citrate synthase
- GCK:
-
Germinal centre kinase
- HK2:
-
Hexokinase 2
- IGT:
-
Impaired glucose tolerance
- MAPK:
-
Mitogen-activated protein kinase
- NGT:
-
Normal glucose tolerance
- NRF1:
-
Nuclear respiratory factor 1
- PPARGC1A:
-
Peroxisome proliferator-activated receptor-gamma, coactivator 1 alpha
- qRT-PCR:
-
Quantitative real-time PCR
- SOD2:
-
Superoxide dismutase 2, mitochondrial
- STE20:
-
Sterile 20
- STK25:
-
Serine/threonine protein kinase 25
- UCP:
-
Uncoupling protein
References
Wu C, Whiteway M, Thomas DY, Leberer E (1995) Molecular characterization of Ste20p, a potential mitogen-activated protein or extracellular signal-regulated kinase kinase (MEK) kinase kinase from Saccharomyces cerevisiae. J Biol Chem 270:15984–15992
Pombo CM, Force T, Kyriakis J, Nogueira E, Fidalgo M, Zalvide J (2007) The GCK II and III subfamilies of the STE20 group kinases. Front Biosci 12:850–859
Pombo CM, Bonventre JV, Molnar A, Kyriakis J, Force T (1996) Activation of a human Ste20-like kinase by oxidant stress defines a novel stress response pathway. EMBO J 15:4537–4546
Osada S, Izawa M, Saito R et al (1997) YSK1, a novel mammalian protein kinase structurally related to Ste20 and SPS1, but is not involved in the known MAPK pathways. Oncogene 14:2047–2057
Preisinger C, Short B, De Corte V et al (2004) YSK1 is activated by the Golgi matrix protein GM130 and plays a role in cell migration through its substrate 14-3-3zeta. J Cell Biol 164:1009–1020
Fidalgo M, Fraile M, Pires A, Force T, Pombo C, Zalvide J (2010) CCM3/PDCD10 stabilizes GCKIII proteins to promote Golgi assembly and cell orientation. J Cell Sci 123:1274–1284
Voss K, Stahl S, Schleider E et al (2007) CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics 8:249–256
Nogueira E, Fidalgo M, Molnar A et al (2008) SOK1 translocates from the Golgi to the nucleus upon chemical anoxia and induces apoptotic cell death. J Biol Chem 283:16248–16258
Pombo CM, Tsujita T, Kyriakis JM, Bonventre JV, Force T (1997) Activation of the Ste20-like oxidant stress response kinase-1 during the initial stages of chemical anoxia-induced necrotic cell death. Requirement for dual inputs of oxidant stress and increased cytosolic [Ca2+]. J Biol Chem 272:29372–29379
Nerstedt A, Johansson A, Andersson CX, Cansby E, Smith U, Mahlapuu M (2010) AMP-activated protein kinase inhibits IL-6-stimulated inflammatory response in human liver cells by suppressing phosphorylation of signal transducer and activator of transcription 3 (STAT3). Diabetologia 53:2406–2416
Hansson PK, Asztely AK, Clapham JC, Schreyer SA (2004) Glucose and fatty acid metabolism in McA-RH7777 hepatoma cells vs. rat primary hepatocytes: responsiveness to nutrient availability. Biochim Biophys Acta 1684:54–62
Huppertz C, Fischer BM, Kim YB et al (2001) Uncoupling protein 3 (UCP3) stimulates glucose uptake in muscle cells through a phosphoinositide 3-kinase-dependent mechanism. J Biol Chem 276:12520–12529
Winder WW, Hardie DG (1996) Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am J Physiol 270:E299–E304
MacLellan JD, Gerrits MF, Gowing A, Smith PJ, Wheeler MB, Harper ME (2005) Physiological increases in uncoupling protein 3 augment fatty acid oxidation and decrease reactive oxygen species production without uncoupling respiration in muscle cells. Diabetes 54:2343–2350
Silva JP, Shabalina IG, Dufour E et al (2005) SOD2 overexpression: enhanced mitochondrial tolerance but absence of effect on UCP activity. EMBO J 24:4061–4070
Nabben M, Shabalina IG, Moonen-Kornips E et al (2011) Uncoupled respiration, ROS production, acute lipotoxicity and oxidative damage in isolated skeletal muscle mitochondria from UCP3-ablated mice. Biochim Biophys Acta 1807(9):1095–1105
Cha SH, Hu Z, Chohnan S, Lane MD (2005) Inhibition of hypothalamic fatty acid synthase triggers rapid activation of fatty acid oxidation in skeletal muscle. Proc Natl Acad Sci U S A 102:14557–14562
Wu Z, Puigserver P, Andersson U et al (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98:115–124
Yoshida T, Pan Y, Hanada H, Iwata Y, Shigekawa M (1998) Bidirectional signaling between sarcoglycans and the integrin adhesion system in cultured L6 myocytes. J Biol Chem 273:1583–1590
Garrels JI (1979) Changes in protein synthesis during myogenesis in a clonal cell line. Dev Biol 73:134–152
Shimokawa T, Kato M, Ezaki O, Hashimoto S (1998) Transcriptional regulation of muscle-specific genes during myoblast differentiation. Biochem Biophys Res Commun 246:287–292
Kim D, Jitrapakdee S, Thompson M (2007) Differential regulation of the promoter activity of the mouse UCP2 and UCP3 genes by MyoD and myogenin. J Biochem Mol Biol 40:921–927
Wright WE, Sassoon DA, Lin VK (1989) Myogenin, a factor regulating myogenesis, has a domain homologous to MyoD. Cell 56:607–617
Dimopoulos N, Watson M, Green C, Hundal HS (2007) The PPARdelta agonist, GW501516, promotes fatty acid oxidation but has no direct effect on glucose utilisation or insulin sensitivity in rat L6 skeletal muscle cells. FEBS Lett 581:4743–4748
Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ (1993) Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 75:241–251
Wu J, Ruas JL, Estall JL et al (2011) The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1alpha/ATF6alpha complex. Cell Metab 13:160–169
Mootha VK, Lindgren CM, Eriksson KF et al (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34:267–273
Patti ME, Butte AJ, Crunkhorn S et al (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A 100:8466–8471
Krook A, Digby J, O'Rahilly S, Zierath JR, Wallberg-Henriksson H (1998) Uncoupling protein 3 is reduced in skeletal muscle of NIDDM patients. Diabetes 47:1528–1531
Schonfeld-Warden NA, Warden CH (2001) Physiological effects of variants in human uncoupling proteins: UCP2 influences body-mass index. Biochem Soc Trans 29:777–784
Boss O, Samec S, Paoloni-Giacobino A et al (1997) Uncoupling protein-3: a new member of the mitochondrial carrier family with tissue-specific expression. FEBS Lett 408:39–42
Vidal-Puig A, Solanes G, Grujic D, Flier JS, Lowell BB (1997) UCP3: an uncoupling protein homologue expressed preferentially and abundantly in skeletal muscle and brown adipose tissue. Biochem Biophys Res Commun 235:79–82
Weigle DS, Selfridge LE, Schwartz MW et al (1998) Elevated free fatty acids induce uncoupling protein 3 expression in muscle: a potential explanation for the effect of fasting. Diabetes 47:298–302
Choi CS, Fillmore JJ, Kim JK et al (2007) Overexpression of uncoupling protein 3 in skeletal muscle protects against fat-induced insulin resistance. J Clin Invest 117:1995–2003
Pedersen SB, Lund S, Buhl ES, Richelsen B (2001) Insulin and contraction directly stimulate UCP2 and UCP3 mRNA expression in rat skeletal muscle in vitro. Biochem Biophys Res Commun 283:19–25
Vestergaard H, Bjorbaek C, Hansen T, Larsen FS, Granner DK, Pedersen O (1995) Impaired activity and gene expression of hexokinase II in muscle from non-insulin-dependent diabetes mellitus patients. J Clin Invest 96:2639–2645
Phillips SM, Han XX, Green HJ, Bonen A (1996) Increments in skeletal muscle GLUT-1 and GLUT-4 after endurance training in humans. Am J Physiol 270:E456–E462
Kohn LD, Wallace B, Schwartz F, McCall K (2005) Is type 2 diabetes an autoimmune-inflammatory disorder of the innate immune system? Endocrinology 146:4189–4191
Wellen KE, Hotamisligil GS (2005) Inflammation, stress, and diabetes. J Clin Invest 115:1111–1119
Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM (1995) Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 95:2409–2415
Saghizadeh M, Ong JM, Garvey WT, Henry RR, Kern PA (1996) The expression of TNF alpha by human muscle. Relationship to insulin resistance. J Clin Invest 97:1111–1116
Stump CS, Henriksen EJ, Wei Y, Sowers JR (2006) The metabolic syndrome: role of skeletal muscle metabolism. Ann Med 38:389–402
Li B, Nolte LA, Ju JS et al (2000) Skeletal muscle respiratory uncoupling prevents diet-induced obesity and insulin resistance in mice. Nat Med 6:1115–1120
Funding
This work was supported by grants from the Swedish Research Council, the European Foundation for the Study of Diabetes/Lilly research grant, the Novo Nordisk Foundation, the P. and A. Hedlunds Foundation, the L. Hierta Memorial Foundation and the M. Bergvall Foundation.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
AN, EC, AS and CXA performed the experiments, and analysed and interpreted the data. ML, MB and US interpreted the data and wrote parts of the manuscript. MM designed the study, interpreted the data and wrote the manuscript. All authors revised the manuscript and approved the final version.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Materials
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 126 kb)
ESM Fig. 1
(PDF 111 kb)
ESM Fig. 2
(PDF 180 kb)
ESM Fig. 3
(PDF 181 kb)
ESM Fig. 4
(PDF 76 kb)
Rights and permissions
About this article
Cite this article
Nerstedt, A., Cansby, E., Andersson, C.X. et al. Serine/threonine protein kinase 25 (STK25): a novel negative regulator of lipid and glucose metabolism in rodent and human skeletal muscle. Diabetologia 55, 1797–1807 (2012). https://doi.org/10.1007/s00125-012-2511-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2511-7