
Abstract
Adipose tissue function changes with development. In the newborn, brown adipose tissue (BAT) is essential for ensuring effective adaptation to the extrauterine environment, and its growth during gestation is largely dependent on glucose supply from the mother to the fetus. The amount, location and type of adipose tissue deposited can also determine fetal glucose homeostasis. Adipose tissue first appears at around mid-gestation. Total adipose mass then increases through late gestation, when it comprises a mixture of white and brown adipocytes. BAT possesses a unique uncoupling protein, UCP1, which is responsible for the rapid generation of large amounts of heat at birth. Then, during postnatal life some, but not all, depots are replaced by white fat. This process can be utilised to investigate the physiological conversion of brown to white fat, and how it is re-programmed by nutritional changes in pre- and postnatal environments. A reduction in early BAT deposition may perpetuate through the life cycle, thereby suppressing energy expenditure and ultimately promoting obesity. Normal fat development profiles in the offspring are modified by changes in maternal diet at defined stages of pregnancy, ultimately leading to adverse long-term outcomes. For example, excess macrophage accumulation and the onset of insulin resistance occur in an adipose tissue depot-specific manner in offspring born to mothers fed a suboptimal diet from early to mid-gestation. In conclusion, the growth of the different fetal adipose tissue depots varies according to maternal diet and, if challenged in later life, this can contribute to insulin resistance and impaired glucose homeostasis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
In most species, adipose tissue makes up a comparatively small part of total fetal body weight and its size is largely dependent on the prevailing glucose environment [1]. Infants of diabetic mothers can be heavier [2] and may possess more adipose tissue, although the exact composition and distribution of this has yet to be detailed. In the fetus, adipose tissue usually remains inert and has a highly specialised role in ensuring that the newborn can effectively adapt to the cold challenge of the extrauterine environment [3]. This is achieved by the presence of a unique uncoupling protein, UCP1, which is only present in brown adipose tissue (BAT) and enables the rapid generation of heat [4]. BAT has a maximal rate of heat production of 300 watts/kg—much higher than the rate of 1 watt/kg in all other tissues [5]. It is very different from white adipose tissue, which represents an energy depot that can be mobilised when food availability is limited. Adipose tissue growth is greatest over the final few weeks of gestation [6], a not unexpected finding given the vital role it plays in enabling the newborn to effectively adapt to the thermal and nutritional challenges of life after birth. Recent findings that BAT is retained into adulthood [7, 8] and that its abundance is greatly diminished in obese individuals [9] has led to renewed interest in understanding its regulation, although to date little research has focused on the fetus. An increase in the amount of white fat and/or a reduction in the abundance of brown fat in early life would thus be one mechanism by which an individual would be at increased risk of later obesity, especially if this was accompanied by an increased number of white adipocyte cells [10].
The fetus grows and develops within a hypoxic and hypoglycaemic environment and has little need to thermoregulate, as its body temperature is primarily determined by that of its mother [5]. Adipose tissue deposition in utero could be restricted as a result of the relatively higher energy costs of lipid accretion compared with that of protein and carbohydrate [11]. Although in a majority of mammalian species fat mass is very low at birth, human fetuses are notable exceptions as they deposit large amounts of subcutaneous fat in late gestation [12]. This may have important implications in the face of delayed onset of lactation after delivery in women [13], during which time the infant can readily mobilise this extra fat. In most species, especially those with a short gestation period and offspring born with an immature hypothalamic–pituitary axis, minimal amounts of adipose tissue are deposited prior to birth and maturation of this tissue primarily occurs postnatally [14]. This could have important translational consequences, as the regulation of adipose tissue development differs before and after birth as oxygenation rises and nutrient supply is less constrained with the onset of suckling. Additionally, the composition of milk varies considerably between species [11].
Fetal adipose tissue development
The characteristics of adipose tissue in the fetus differ according to depot. In rodents, the primary BAT depot is interscapular, retains UCP1 throughout the life cycle and is located within a layer of white adipose tissue. In contrast, in both large mammals and humans, the major source of BAT in the fetus is around the central organs, a depot that is replaced by white fat after birth [15]. The main BAT depot in adult humans is in the supraclavicular region which weighs ∼40–50 g [7, 16, 17]. A comparable depot, in which UCP1 gene expression is substantially greater than all other adipose tissue locations, has now been identified in young sheep, where the sternal and clavicular areas meet the forelimb (Fig. 1). Its anatomical position immediately above the skeletal muscle at the front of the animal could be important developmentally, as it is an area in which fetal blood supply is preferentially maintained. As with humans, the proximity of this depot to major vessels supplying the brain is notable, and thus could be essential for ensuring the temperature of the brain is not compromised, especially after birth.

Anatomical location of brown and white adipose tissue depots in a 3-month-old sheep as defined by the relative abundance of UCP1 mRNA
Given the common ‘adipo-myocyte’ origins of brown adipocytes and skeletal myofibres [18], the close association between BAT and forelimb (but not hindlimb) skeletal muscle could provide new insights into the regulation of body composition in early life. Notably, BAT depots found around internal organs in the newborn (i.e. perirenal and pericardial) still retain detectable UCP1 in later life but at a much reduced level more characteristic of brown adipose in white (BRITE) tissue than a purely BAT depot [19]. As a consequence of the diverse range of BAT, BRITE and white adipose tissue depots that have been shown in mice to display distinct gene expression profiles, which in some cases are depot specific [20], it is clear we are now entering an exciting new phase of both adipose tissue and muscle developmental biology. The pronounced range of molecular markers now identified within nearly every type and depot of adipose tissue in young adult mice may reflect changes in early development [20]. This now needs examining across a range of species, together with the extent to which they are nutritionally, as well as thermally, sensitive. Moreover, in mice, genetic variability affects BRITE, but not BAT, development, suggesting very different regulation during development [21].
These recent advances in our understanding of adipose tissue biology explain why, to date, comparatively little progress has been made in our knowledge of how changes in the materno-fetal environment can affect fetal and offspring body composition and its relationship with glucose homeostasis. Interestingly, in view of the common origin of brown adipocytes and skeletal muscle [18], a close link between functional brown adipose tissue and muscle volume has recently been suggested in children and adolescents [22]. Furthermore, deletion of myostatin (a negative regulator of skeletal muscle mass) in mice prevents diet-induced obesity and not only promotes skeletal muscle growth, but also the appearance of BRITE cells [23]. These findings may be especially important given the recent discovery that, in adults, muscular activity can directly influence adipose tissue function [24]. This effect appears to be mediated by an increased rate of production of a previously unknown hormone, irisin, a membrane protein cleaved from FNDC5. Surprisingly, irisin promotes the expression of UCP1 in white, but not brown, adipose tissue. The magnitude of response differs across white fat depots in mice and is greatest within inguinal fat. It will now be necessary to investigate whether irisin is implicated in muscle development.

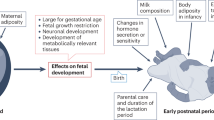
Fetal adipose tissue is made up of both brown and white adipocytes, the latter of which are laid down first. It is only as the endocrine responses essential for enabling effective adaptation to the extrauterine environment occur within the fetus that BAT characteristics become dominant (Fig. 2).This is the case in both ovine [25] and bovine [26] fetuses and is not unexpected, given that precocious maturation of fetal BAT would potentially place the fetus at risk from hyperthermia. The preceding deposition of white adipose tissue not only provides a protective layer of fat around internal organs, but ensures that sufficient lipid can be mobilised to provide the large amounts of energy required for non-shivering thermogenesis [27]. This could explain why white rather than BAT characteristics predominate [26]. A rise in UCP1 gene expression within the perirenal adipose tissue depot up to term is driven by a range of stimulatory endocrine factors, as summarised in the text box below. These factors are closely inter-related, with the pre-partum rise in cortisol perhaps having the greatest influence on adipose tissue development [28]. This process is clearly susceptible to changes in the maternal nutritional and endocrine environment, which can result in either an increase in the total amount of fat and/or a change in UCP1 abundance and therefore BAT, as summarised in Fig. 3.

Summary of the primary developmental stages for perirenal adipose tissue through fetal and postnatal life indicating the changes in its histological appearance (i.e. transition from multilocular cells that possess UCP1 in the fetus to large lipid-filled cells with no UCP1 at 30 days of age) in conjunction with the rapid rise and decline in UCP1 as observed in the sheep

Summary of the potential effects of either a compromised or stimulated maternal metabolic environment on placental function and fetal adipose tissue growth up to term as compared with controls. The effects of a change in maternal food intake relate to defined stages of gestation in pregnant sheep and are further compared with the effect of gestational diabetes in women

Glucocorticoids have different effects on BAT function depending on the conditions. For example, in vitro, corticosterone (or dexamethasone) inhibits UCP1 gene expression and translation [29], but in vivo, when combined with a rise in other endocrine factors, it has a pronounced stimulatory role [28]. The regulatory effects of glucocorticoids also vary between large and small mammals. Nutritional interventions in rodents are often considered to be ‘stressful’, so that many of their long-term outcomes are explained mechanistically by increased exposure of the fetus to corticosterone [30]. Large animal studies with a global reduction in food intake (but without causing micronutrient deficiencies [31]) have shown a suppression of maternal cortisol [32]. This adaptation is in order to reduce the maternal energy requirements and promote fat mobilisation. Moreover, as discussed in more detail below, the ability of the placenta to adapt to any nutritional intervention, and thus minimise changes in nutrient supply to the fetus, will be dependent on the timing of the maternal challenge [32].
Nutritional interventions during pregnancy and their impact on fetal adipose tissue
Compromised maternal metabolism
The extent to which a reduction in maternal dietary intake to suboptimal amounts has a direct effect on the placenta in terms of its total mass is dependent on the timing of the intervention. In sheep, targeted nutritional interventions coincident with the period of placental growth, i.e. 30–80 days’ gestation [33, 34], reduce placental growth over this period. Once the maternal dietary intake is restored, the normal decline in placental mass between mid- and late gestation is abolished and so placental weight is larger at term. Additionally, the normal glucose exchange capacity is enhanced, which could act to promote the fetal glucose supply [34, 35], and thus fetal adiposity [36]. These types of responses to a targeted change in maternal diet are clearly different from those seen under some clinical conditions of compromised placentation, such as preeclampsia, a primary characteristic of which is raised maternal plasma leptin [37]. In humans, this type of condition is usually accompanied by a degree of fetal hypoxia that could promote placental expression of LEP (encoding leptin). Overall, the range of responses is dependent on whether the onset of preeclampsia is early or late [38]. The duration and magnitude of placental compromise thus determines the extent to which fetal growth is restricted [39, 40]. Clearly, this is a very different challenge to that of manipulating macronutrient availability as studied in most animal models. In the human situation, it is also likely that the degree of postnatal catch-up growth [41] determines the magnitude of excess fat deposition later in life. To date, however, there are no studies that have closely followed up the effect of placental growth restriction on postnatal body composition, specifically, adipose tissue distribution and type.
It is not simply the amount of adipose tissue present in the fetus that determines glucose homeostasis. The relative proportion of brown and white adipose tissue, with their different characteristics, together with the magnitude of postnatal exposure to an obesogenic environment, are equally important. In sheep, when nutritionally programmed offspring are reared in a standard free-living environment where their activity is similar to that seen in the wild, little change in later fat mass occurs [42]. One advantage of using sheep for such studies is that the maternal diet can be manipulated at defined stages of fetal adipose tissue development. These investigations indicate that a reduction in the quantity of food consumed by the mother over the first half of gestation to suboptimal amounts results in greater fat deposition in the fetus at term [43]. Using this intervention, coincident with the first appearance of fetal adipose tissue, there was an accompanying reduction in the plasma concentrations of a range of maternal counter-metabolic hormones, including cortisol, leptin, thyroid hormones, insulin and IGF1, over the period of nutritional manipulation [43]. As a consequence, maternal glucose status was unaffected throughout pregnancy and fat deposition was enhanced, possibly as a result of increased sensitivity to IGF1. Long-term adverse outcomes were not observed until the offspring became obese and had reached young adulthood [44]. At this stage, pronounced evidence of adipose tissue dysfunction, primarily within the perirenal depot, in the form of macrophage accumulation, insulin resistance and ectopic lipid accumulation was apparent. The key components of fetal adipose tissue development that caused these effects were a combination of increased glucocorticoid action [45] (possibly mediated by reduced exposure to maternal cortisol) and enhanced white, as opposed to brown, adipocyte accumulation in utero [43].
When the mother consumes a suboptimal amount of food over the final month of gestation, birthweight and fat mass are reduced. This is highly likely to reflect a reduction in lipid stores rather than in brown adipocyte numbers [46]. Interestingly, the reduced amount of fat present in the newborn has a greater capacity to retain UCP1, which could be indicative of a protective mechanism against subsequent exposure to an obesogenic environment. Furthermore, although consumption of a suboptimal amount of food in late gestation suppresses glucocorticoid action in postnatal life [42], this adaptation is reversed in young adulthood when key inflammatory genes, together with LEP, UCP2 and the gene encoding the glucocorticoid receptor (NR3C1) are also upregulated, as summarised in Fig. 4. This includes enhanced expression of the genes for a range of inflammatory markers, including IL-6, toll-like receptor 4 and CD68 (a macrophage-specific molecule that also shows increased abundance over the first month of life) [47]. Consequently, targeted changes of the in utero nutritional environment could contribute to early perturbations in glucose/insulin signalling, as observed in the metabolic syndrome, even in the absence of any absolute changes in adipose tissue mass, at least in sheep [48]. One reason for this is that, in sheep, fat growth in is most rapid through lactation [15], when both maternal body weight [49] and parity [50] can have a much greater effect than changes in maternal diet. This could further explain why only very modest effects can be seen in the offspring during young adulthood compared with those seen in response to exposure to an obesogenic environment. However, any additional fat deposited is unlikely to have a major deleterious role, given the normal variations in adiposity with season, for example [51].

Summary of the main changes in gene expression within perirenal adipose tissue in young adult sheep following exposure to suboptimal maternal nutrition through late gestation with respect to (a) adipokine secretion (ADIPOQ), reactive oxygen species sensitivity (UCP2) and glucocorticoid action (NR3C1) and (b) inflammation and macrophage accumulation (TNF, IL6, CCL2, TLR4 and CD68). Offspring born to nutrient-restricted mothers are indicated by grey bars and those born to controls by white bars. *p < 0.05, **p < 0.01
Enhanced maternal metabolism
In humans, one common condition in which nutrient supply to the fetus will be increased is during gestational diabetes, when both the carbohydrate and lipid supply are raised [52]. At the same time, increased maternal body mass index can be accompanied by raised placental mass and infant birthweight [53, 54]. The extent to which these differences in maternal metabolism are accompanied by early changes in placental invasion, growth and function remain to be defined. One characterised maternal predictor of gestational diabetes is raised leptin at 16 weeks’ gestation [55], an adaptation that normally, but not always, persists up to term [56]. The increase in leptin is due in part to an enhanced synthetic capacity by the placenta [57] and by the visceral adipose tissue [58]. At term, placental expression of LEP and LEPR, which encodes the receptor, is raised in diabetic pregnancies [57]. This would be predicted to promote the proliferation and survival of trophoblast cells together with Janus kinase-signal transducers of transcription, mitogen-activated protein kinase and phosphoinositide 3-kinase pathways [59]. An adaptation of this type would be expected to amplify the secretion of inflammatory cytokines within both the maternal and fetal circulation [60]. Whether these placental changes directly impact on the adiposity or function of the fetus or the mother [61] remains to be investigated.
Stimulating maternal metabolism by either increasing the amount of food provided or chronic cold exposure promotes enhanced fetal adipose tissue development, enhancing the amount of perirenal BAT, at least in sheep [62]. UCP1 abundance is promoted through changes in sympathetic nervous activity and/or related endocrine systems, such as the prolactin receptor, which are, in turn, dependent on the amount of food consumed by the mother [62]. The positive effect of maternal cold exposure on fetal BAT deposition is time dependent [46]. Emerging research is focusing on the effects of excess maternal fat mass on fetal development. An important consideration in the development of such models is the type of intervention used to promote positive energy balance and, therefore, fat deposition in the mother. For example, initial studies have shown that allowing pregnant sheep to feed to appetite can increase birthweight. In this situation fat mass is actually reduced whilst BAT characteristics are enhanced [62]. This represents a positive fetal adaptation to an increase in a nutritionally balanced diet and would be predicted to prevent excess fat mass in the offspring. Currently, such studies in groups of offspring that are balanced experimentally for fetal number remain to be undertaken [63]. The number of fetuses is critical. Twins are smaller than singletons at birth, possess less adipose tissue and are more sensitive to the effects of changes in maternal diet. Once these constraints are removed immediately following birth, twins deposit more fat through into adulthood. To date, rodent models have focused on the effects of increased maternal intake of one or more macronutrient [64], reporting that this often has no effect on birthweight, but the differential response between fat depots has not been examined.
Fetal sex and its impact on fetal development and adiposity
In most species, adult females are usually smaller and possess more brown fat than males [8, 65]. In humans differences in body composition are apparent from late gestation when female fetuses possess more fat and less muscle [66]. Sex can impact on metabolic control right from conception, with female embryos having a slower rate of cell division and metabolism than males [67]. Furthermore, in vitro studies on bovine embryos indicate sex-related epigenetic differences, which would be predicted to extend beyond embryogenesis [68]. Energy balance regulation could feasibly be a system affected by such epigenetic modifications programmed in utero. This has been supported by recent work on adult mice showing that tyrosine phosphate epsilon, a phosphatase that downregulates both the insulin receptor and leptin signalling, may preferentially regulate body mass in female but not male mice [69]. This is proposed to occur via a resetting of hypothalamic leptin signalling. However, any differences only become apparent when animals are challenged with a high-fat diet or, in the case of female mice, ovariectomised. In rats, when mothers are fed a diet rich in fat, sugar and salt and the same diet is then given to the offspring, the resultant female offspring show increased adiposity with earlier onset of hyperglycaemia, hyperinsulinaemia and hyperlipidaemia [70]. The extent to which this type of adverse adaptation may affect fetal brown or white adipose tissue has yet to be examined. However, it is noteworthy that female rodents and humans possess more BAT [8, 65], whilst in newborn sheep plasma leptin is threefold higher in females than males [71]. In newborn humans, plasma leptin is ∼30% higher in females and this difference that decreases with age [72], a response that may be mediated in part through the type of feeding [73].
Epigenetic adaptations within adipose tissue
There is currently substantial interest in potential epigenetic adaptations within adipose tissue that could enhance an individual’s susceptibility to obesity. However, to date, apart from in the pancreas [74], there are no convincing data showing that changes in methylation status, especially in adipose tissue, in early life can have long-term consequences. This is exemplified by a recent study that used the widely adopted model of feeding a low-protein/high-carbohydrate diet to mice throughout pregnancy and lactation. The methylation status of the leptin receptor was shown to be reduced within the white adipose tissue of male offspring at 7 months of age that were born to nutritionally challenged mothers [75]. Despite this adaptation, the offspring were smaller, had less fat and showed reduced basal, but not postprandial, LEP expression and leptin secretion. The change in methylation status and the subsequent functional consequences in white fat were not described. Whether this is a response to the change in leptin dynamics, or a cause of them, was similarly not addressed. Importantly though, despite the ‘nutritionally programmed’ offspring eating more food per unit body weight than age-matched controls, they remained smaller [75]. This suggests that heat production may have been raised, and a more logical target tissue would therefore be brown, rather than white, fat. A focus on BAT could be important as it has been shown in mice that deletion of the histone H3 lysine 9-specific demethylase JmjC-domain-containing histone demethylase 2A (JHDM2A) results in an increase in body weight after 8 weeks of age [76]. This is associated with reduced BAT, making the animals more susceptible to cold-induced hypothermia and less able to oxidise fat, primarily because of a disruption of β-adrenergic stimulation [77]. Interestingly, the expression of this gene is barely detectable in ovine BAT, suggesting a species-specific role (M. Pope, H. Budge, M.E. Symonds, unpublished results). Given the highly variable differences in methylation status between individual animals of the same species and individual genes, and lack of apparent correlation with changes in gene expression with development [78], a comprehensive study of this process appears to be highly challenging.
In conclusion, adipose tissue growth and development is a complex process that appears to differ greatly between species. The current resurgence of interest in BAT and its potential role in energy balance throughout the life cycle means that future studies into fetal programming will need to be more comprehensive. This will require a greater emphasis on the tissue itself, further investigations into its relationship with white adipose tissue growth in depots where they are found together, plus more work on its role in glucose homeostasis.
Abbreviations
- BAT:
-
Brown adipose tissue
- BRITE:
-
Brown adipose in white
- UCP:
-
Uncoupling protein
References
Symonds ME, Lomax MA (1992) Maternal and environmental influences on thermoregulation in the neonate. Proc Nutr Soc 51:165–172
Schaefer-Graf UM, Pawliczak J, Passow D et al (2005) Birth weight and parental BMI predict overweight in children from mothers with gestational diabetes. Diabetes Care 28:1745–1750
Symonds ME, Mostyn A, Pearce S, Budge H, Stephenson T (2003) Endocrine and nutritional regulation of fetal adipose tissue development. J Endocrinol 179:293–299
Cannon B, Nedergaard J (2004) Brown adipose tissue: function and physiological significance. Physiol Rev 84:277–359
Power G (1989) Biology of temperature: the mammalian fetus. J Dev Physiol 12:295–304
Vernon RG (1986) The growth and metabolism of adipocytes. In: Buttery PJ, Haynes NB, Lindsay DB (eds) Control and manipulation of animal growth. Butterworths, London, pp 67–83
van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM et al (2009) Cold-activated brown adipose tissue in healthy men. N Engl J Med 360:1500–1508
Au-Yong IT, Thorn N, Ganatra R, Perkins AC, Symonds ME (2009) Brown adipose tissue and seasonal variation in humans. Diabetes 58:2583–2587
Vijgen GH, Bouvy ND, Teule GJ, Brans B, Schrauwen P, van Marken Lichtenbelt WD (2011) Brown adipose tissue in morbidly obese subjects. PLoS One 6:e17247
Spalding KL, Arner E, Westermark PO et al (2008) Dynamics of fat cell turnover in humans. Nature 453:783–787
Budge H, Symonds ME (2006) Fetal and neonatal nutrition—lipid and carbohydrate requirements and adaptations to altered supply at birth. In: Kurjak A, Chrervenak FA (eds) Textbook of perinatal medicine. Informa Healthcare, London, pp 1005–1014
Widdowson EM (1950) Chemical composition of newly born animals. Nature 116:626–628
Nommsen-Rivers LA, Heinig MJ, Cohen RJ, Dewey KG (2008) Newborn wet and soiled diaper counts and timing of onset of lactation as indicators of breastfeeding inadequacy. J Hum Lact 24:27–33
Cannon B, Connoley E, Obregon M-J, Nedergaard J (1988) Perinatal activation of brown adipose tissue. In: Kunzel W, Jesen A (eds) The endocrine control of the fetus. Springer, Berlin, pp 306–320
Clarke L, Buss DS, Juniper DS, Lomax MA, Symonds ME (1997) Adipose tissue development during early postnatal life in ewe-reared lambs. Exp Phys 82:1015–1027
Virtanen KA, Lidell ME, Orava J et al (2009) Functional brown adipose tissue in healthy adults. N Engl J Med 360:1518–1525
Cypess AM, Lehman S, Williams G et al (2009) Identification and importance of brown adipose tissue in adult humans. N Engl J Med 360:1509–1517
Seale P, Bjork B, Yang W et al (2008) PRDM16 controls a brown fat/skeletal muscle switch. Nature 454:961–967
Petrovic N, Walden TB, Shabalina IG, Timmons JA, Cannon B, Nedergaard J (2010) Chronic peroxisome proliferator-activated receptor γ (PPARγ) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J Biol Chem 285:7153–7164
Walden TB, Hansen IR, Timmons JA, Cannon B, Nedergaard J (2012) Recruited vs. nonrecruited molecular signatures of brown, “brite”, and white adipose tissues. Am J Physiol Endocrinol Metab 302:E19–E31
Xue B, Rim JS, Hogan JC, Coulter AA, Koza RA, Kozak LP (2007) Genetic variability affects the development of brown adipocytes in white fat but not in interscapular brown fat. J Lipid Res 48:41–51
Gilsanz V, Chung SA, Jackson H, Dorey FJ, Hu HH (2011) Functional brown adipose tissue is related to muscle volume in children and adolescents. J Pediatr 158:722–728
Zhang C, McFarlane C, Lokireddy S et al (2012) Inhibition of myostatin protects against diet-induced obesity by enhancing fatty acid oxidation and promoting a brown adipose phenotype in mice. Diabetologia 55:183–193
Boström P, Wu J, Jedrychowski MP et al (2012) A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481:463–468
Clarke L, Bryant MJ, Lomax MA, Symonds ME (1997) Maternal manipulation of brown adipose tissue and liver development in the ovine fetus during late gestation. Br J Nutr 77:871–883
Taga H, Chilliard Y, Meunier B et al (2011) Cellular and molecular large-scale features of fetal adipose tissue: is bovine perirenal adipose tissue brown? J Cell Physiol 227:1688–1700
Clarke L, Heasman L, Firth K, Symonds ME (1997) Influence of route of delivery and ambient temperature on thermoregulation in newborn lambs. Am J Physiol Regul Integr Comp Physiol 272:R1931–R1939
Mostyn A, Pearce S, Budge H et al (2003) Influence of cortisol on adipose tissue development in the fetal sheep during late gestation. J Endocrinol 176:23–30
Bakopanos E, Silva JE (2002) Opposing effects of glucocorticoids on β3-adrenergic receptor expression in HIB-1B brown adipocytes. Mol Cell Endocrinol 190:29–37
Budge H, Stephenson T, Symonds ME (2007) Maternal nutrient restriction is not equivalent to maternal biological stress. Curr Drug Targets 8:888–893
Dandrea J, Cooper S, Ramsay MM et al (2002) The effects of pregnancy and maternal nutrition on the maternal renin-angiotensin system in sheep. Exp Phys 87(3):353–359
Yiallourides M, Sebert SP, Wilson V et al (2009) The differential effects of the timing of maternal nutrient restriction in the ovine placenta on glucocorticoid sensitivity, uncoupling protein 2, peroxisome proliferator-activated receptor-gamma and cell proliferation. Reproduction 138:601–608
Ehrhardt RA, Bell AW (1995) Growth and metabolism of the ovine placenta during mid-gestation. Placenta 16:727–741
Heasman L, Clarke L, Firth K, Stephenson T, Symonds ME (1998) Influence of restricted maternal nutrition in early to mid gestation on placental and fetal development at term in sheep. Pediatr Res 44:546–551
Dandrea J, Wilson V, Gopalakrishnan G et al (2001) Maternal nutritional manipulation of placental growth and glucose transporter 1 (GLUT-1) abundance in sheep. Reproduction 122:793–800
Bispham J, Gardner DS, Gnanalingham MG, Stephenson T, Symonds ME, Budge H (2005) Maternal nutritional programming of fetal adipose tissue development: differential effects on messenger ribonucleic acid abundance for uncoupling proteins and peroxisome proliferator-activated and prolactin receptors. Endocrinology 146:3943–3949
Mise H, Sagawa N, Matsumoto T et al (1998) Augmented placental production of leptin in preeclampsia: possible involvement of placental hypoxia. J Clin Endocrinol Metab 83:3225–3229
Sitras V, Paulssen RH, Gronaas H et al (2009) Differential placental gene expression in severe preeclampsia. Placenta 30:424–433
Rolfo A, Many A, Racano A et al (2010) Abnormalities in oxygen sensing define early and late onset preeclampsia as distinct pathologies. PLoS One 5:e13288
van der Merwe JL, Hall DR, Wright C, Schubert P, Grove D (2010) Are early and late preeclampsia distinct subclasses of the disease—what does the placenta reveal? Hypertens Pregnancy 29:457–467
Ong KK, Loos RJ (2006) Rapid infancy weight gain and subsequent obesity: systematic reviews and hopeful suggestions. Acta Paediatr 95:904–908
Gnanalingham MG, Mostyn A, Symonds ME, Stephenson T (2005) Ontogeny and nutritional programming of adiposity in sheep: potential role of glucocorticoid action and uncoupling protein-2. Am J Physiol Regul Integr Comp Physiol 289:R1407–R1415
Bispham J, Gopalakrishnan GS, Dandrea J et al (2003) Maternal endocrine adaptation throughout pregnancy to nutritional manipulation: consequences for maternal plasma leptin and cortisol and the programming of fetal adipose tissue development. Endocrinology 144:3575–3585
Sharkey D, Gardner DS, Fainberg HP et al (2009) Maternal nutrient restriction during pregnancy differentially alters the unfolded protein response in adipose and renal tissue of obese juvenile offspring. FASEB J 23:1314–1324
Whorwood CB, Firth KM, Budge H, Symonds ME (2001) Maternal undernutrition during early to midgestation programs tissue-specific alterations in the expression of the glucocorticoid receptor, 11β-hydroxysteroid dehydrogenase isoforms, and type 1 angiotensin II receptor in neonatal sheep. Endocrinology 142:2854–2864
Pearce S, Budge H, Mostyn A, Symonds ME, Stephenson T (2005) Differential effects of maternal cold exposure and nutrient restriction on prolactin receptor and uncoupling protein 1 abundance in adipose. Adipocytes 1:57–64
Sharkey D, Symonds ME, Budge H (2009) Adipose tissue inflammation: developmental ontogeny and consequences of gestational nutrient restriction in offspring. Endocrinology 150:3913–3920
Sebert SP, Hyatt MA, Chan LL et al (2009) Maternal nutrient restriction between early and midgestation and its impact upon appetite regulation after juvenile obesity. Endocrinology 150:634–641
Symonds ME, Pearce S, Bispham J, Gardner DS, Stephenson T (2004) Timing of nutrient restriction and programming of fetal adipose tissue development. Proc Nutr Soc 63:397–403
Hyatt MA, Keisler DH, Budge H, Symonds ME (2010) Maternal parity and its effect on adipose tissue deposition and endocrine sensitivity in the postnatal sheep. J Endocrinol 204:173–179
Lincoln GA, Richardson M (1998) Photo-neuroendocrine control of seasonal cycles in body weight, pelage growth and reproduction: lessons from the HPD sheep model. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol 119:283–294
Larsson G, Spjuth J, Ranstam J, Vikbladh I, Saxtrup O, Astedt B (1986) Prognostic significance of birth of large infant for subsequent development of maternal non-insulin-dependent diabetes mellitus: a prospective study over 20–27 years. Diabetes Care 9:359–364
Jansson T, Powell TL (2007) Role of the placenta in fetal programming: underlying mechanisms and potential interventional approaches. Clin Sci (Lond) 113:1–13
Winder NR, Krishnaveni GV, Veena SR et al (2011) Mother’s lifetime nutrition and the size, shape and efficiency of the placenta. Placenta 32:806–810
Qiu C, Williams MA, Vadachkoria S, Frederick IO, Luthy DA (2004) Increased maternal plasma leptin in early pregnancy and risk of gestational diabetes mellitus. Obstet Gynecol 103:519–525
Briana DD, Malamitsi-Puchner A (2009) Reviews: adipocytokines in normal and complicated pregnancies. Reprod Sci 16:921–937
Uzelac PS, Li X, Lin J et al (2010) Dysregulation of leptin and testosterone production and their receptor expression in the human placenta with gestational diabetes mellitus. Placenta 31:581–588
Kleiblova P, Dostalova I, Bartlova M et al (2010) Expression of adipokines and estrogen receptors in adipose tissue and placenta of patients with gestational diabetes mellitus. Mol Cell Endocrinol 314:150–156
Maymo JL, Perez AP, Gambino Y, Calvo JC, Sanchez-Margalet V, Varone CL (2011) Review: Leptin gene expression in the placenta—regulation of a key hormone in trophoblast proliferation and survival. Placenta 32(Suppl 2):S146–S153
Miehle K, Stepan H, Fasshauer M (2012) Leptin, adiponectin, and other adipokines in gestational diabetes mellitus and preeclampsia. Clin Endocrinol (Oxf) 76:2–11
Lappas M, Yee K, Permezel M, Rice GE (2005) Release and regulation of leptin, resistin and adiponectin from human placenta, fetal membranes, and maternal adipose tissue and skeletal muscle from normal and gestational diabetes mellitus-complicated pregnancies. J Endocrinol 186:457–465
Budge H, Bispham J, Dandrea J et al (2000) Effect of maternal nutrition on brown adipose tissue and prolactin receptor status in the fetal lamb. Pediatr Res 47:781–786
Symonds ME, Budge H (2009) Nutritional models of the developmental programming of adult health and disease. Proc Nutr Soc 68:173–178
Armitage JA, Poston L, Taylor PD (2008) Developmental origins of obesity and the metabolic syndrome: the role of maternal obesity. Front Horm Res 36:73–84
Pearce S, Casteilla L, Gualillo O, Symonds ME, Stephenson T (2003) Differential effects of age and gender on the postnatal responsiveness of brown adipose tissue to prolactin administration in rats. Exp Phys 88(4):527–531
Hawkes CP, Hourihane JO, Kenny LC, Irvine AD, Kiely M, Murray DM (2011) Gender- and gestational age-specific body fat percentage at birth. Pediatrics 128(3):e645–e651
Mittwoch U (1993) Blastocysts prepare for the race to be male. Hum Reprod 8:1550–1555
Bermejo-Alvarez P, Rizos D, Rath D, Lonergan P, Gutierrez-Adan A (2008) Epigenetic differences between male and female bovine blastocysts produced in vitro. Physiol Genomics 32:264–272
Rousso-Noori L, Knobler H, Levy-Apter E et al (2011) Protein tyrosine phosphatase epsilon affects body weight by downregulating leptin signaling in a phosphorylation-dependent manner. Cell Metab 13:562–572
Bayol SA, Simbi BH, Bertrand JA, Stickland NC (2008) Offspring from mothers fed a ‘junk food’ diet in pregnancy and lactation exhibit exacerbated adiposity that is more pronounced in females. J Physiol 586:3219–3230
Mostyn A, Bispham J, Pearce S et al (2002) Differential effects of leptin on thermoregulation and uncoupling protein abundance in the neonatal lamb. FASEB J 16:1438–1440
Okereke NC, Uvena-Celebrezze J, Hutson-Presley L, Amini SB, Catalano PM (2002) The effect of gender and gestational diabetes mellitus on cord leptin concentration. Am J Obstet Gynecol 187:798–803
Petridou E, Mantzoros CS, Belechri M et al (2005) Neonatal leptin levels are strongly associated with female gender, birth length, IGF-I levels and formula feeding. Clin Endocrinol (Oxf) 62:366–371
Sandovici I, Smith NH, Nitert MD et al (2011) Maternal diet and aging alter the epigenetic control of a promoter-enhancer interaction at the Hnf4a gene in rat pancreatic islets. Proc Natl Acad Sci U S A 108:5449–5454
Jousse C, Parry L, Lambert-Langlais S et al (2011) Perinatal undernutrition affects the methylation and expression of the leptin gene in adults: implication for the understanding of metabolic syndrome. FASEB J 25:3271–3278
Inagaki T, Tachibana M, Magoori K et al (2009) Obesity and metabolic syndrome in histone demethylase JHDM2a-deficient mice. Genes Cells 14:991–1001
Tateishi K, Okada Y, Kallin EM, Zhang Y (2009) Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 458:757–761
Waterland RA, Kellermayer R, Rached MT et al (2009) Epigenomic profiling indicates a role for DNA methylation in early postnatal liver development. Hum Mol Genet 18:3026–3038
Casteilla L, Forest C, Robelin J, Ricquier D, Lombert A, Ailand G (1987) Characterisation of mitochondrial-uncoupling protein in bovine fetus and newborn calf. Am J Physiol 254:E627–E636
Bassett JM, Symonds ME (1998) β2-Agonist ritodrine, unlike natural catecholamines, activates thermogenesis prematurely in utero in fetal sheep. Am J Physiol Regul Integr Comp Physiol 275:R112–R119
Pearce S, Budge H, Mostyn A et al (2005) Prolactin, the prolactin receptor and uncoupling protein abundance and function in adipose tissue during development in young sheep. J Endocrinol 184:351–359
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
MES developed and wrote the manuscript; MP undertook the analysis summarised in Figs 1 and 2, drafted Figs 1, 2 and 3 and assisted in editing the article; DS undertook the analysis and produced Fig. 4 and assisted with editing the final article; HB contributed to the article’s development and to editing the article. All authors approved the final version for publication.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Symonds, M.E., Pope, M., Sharkey, D. et al. Adipose tissue and fetal programming. Diabetologia 55, 1597–1606 (2012). https://doi.org/10.1007/s00125-012-2505-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2505-5