
Abstract
Aims/hypothesis
We examined in skeletal muscle (1) whether fatty acid transport protein (FATP) 1 channels long-chain fatty acid (LCFA) to specific metabolic fates in rats; and (2) whether FATP1-mediated increases in LCFA uptake exacerbate the development of diet-induced insulin resistance in mice. We also examined whether FATP1 is altered in insulin-resistant obese Zucker rats.
Methods
LCFA uptake, oxidation and triacylglycerol esterification rates were measured in control and Fatp1-transfected soleus muscles to determine FATP1-mediated lipid handling. The effects of FATP1 on insulin sensitivity and triacylglycerol accumulation were determined in high-fat diet-fed wild-type mice and in muscle-specific Fatp1 (also known as Slc27a1) overexpressing transgenic mice driven by the muscle creatine kinase (Mck [also known as Ckm]) promoter. We also examined the relationship between FATP1 and both fatty acid transport and metabolism in insulin-resistant obese Zucker rats.
Results
Transient Fatp1 overexpression in soleus muscle increased (p < 0.05) palmitate transport (24%) and oxidation (35%), without altering triacylglycerol esterification or the intrinsic rate of palmitate oxidation in isolated mitochondria. In Mck/Fatp1 animals, Fatp1 mRNA and 15-(p-iodophenyl)-3-R,S-methylpentadecanoic acid uptake in skeletal muscle were upregulated (75%). However, insulin sensitivity and intramuscular triacylglycerol content did not differ between wild-type and Mck/Fatp1 mice following a 16 week high-fat diet. In insulin-resistant obese Zucker rats, LCFA transport and triacylglycerol accumulation were increased (85% and 24%, respectively), but this was not attributable to Fatp1 expression, as neither total cellular nor sarcolemmal FATP1 content were altered.
Conclusions/interpretation
Overexpression of Fatp1 in skeletal muscle increased the rate of LCFA transport and channelled these lipids to oxidation, not to intramuscular lipid accumulation. Therefore, skeletal muscle FATP1 overabundance does not predispose animals to diet-induced insulin resistance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Skeletal muscle insulin resistance is a cardinal feature of type 2 diabetes. In recent years, the accumulation of intramuscular bioactive lipids has been shown to impair skeletal muscle insulin sensitivity, as reviewed by others [1]. Suggestions [2, 3] that mitochondrial dysfunction is a determinant of intramuscular lipid accumulation and insulin resistance have been questioned [4–8]. In contrast, evidence for the excess influx of fatty acids [6, 9–11] and for an inability to completely oxidise them [12, 13] provides an alternative explanation for intramuscular lipid accumulation in insulin-resistant muscle.
The uptake of fatty acids into muscle is now known to occur via a highly regulated, protein-mediated mechanism. A number of insulin-sensitive fatty acid transporters have been identified in skeletal muscle [14], including fatty acid translocase (FAT/CD36), plasma membrane-associated fatty acid binding protein (FABPpm), and fatty acid transport proteins (FATP) 1 and 4, as identified in a review [15]. The functional impact of the co-production of these fatty acid transporters in skeletal muscle is unclear. However, it has been speculated that: (1) different transporters have different fatty acid specificities; (2) some transporters may collaborate to transfer fatty acids across the plasma membrane; and (3) selected transporters may channel fatty acids preferentially into different metabolic fates (e.g. oxidation or esterification). Several studies have associated skeletal muscle sarcolemmal FAT/CD36 with insulin resistance in animals and in humans [6, 9–11].
FATP1 has also been implicated in the development of insulin resistance, as Fatp1 (also known as Slc27a1) knockout (KO) mice are resistant to diet-induced insulin resistance [16, 17]. However the mechanism for this protection is uncertain, as skeletal muscle fatty acid uptake was not reduced [16, 17]. In contrast, fatty acid influx into white adipose tissue was markedly reduced, whereas, paradoxically, fatty acid influx in heart and liver was increased [16]. Genetically altering lipid metabolism at the whole body level via ablation or overexpression of selected genes can influence metabolism in two different ways, either by directly altering energy supply and storage, and/or by altering signalling pathways that affect gene expression and protein content in an endocrine and exocrine manner, as highlighted by others (review) [18, 19]. Therefore, interpretation of such experiments may be complex, as ablation of a gene in one tissue can affect metabolism in other tissues [20–23]. Since ablation of Fatp1 seems to reorganise lipid metabolism in various tissues [16], it is unclear whether the protection against diet-induced insulin resistance is attributable to ablation of Fatp1 in muscle or to adaptive alterations in lipid metabolism in other tissues. Although studies in cell lines indicate that FATP1 channels fatty acids to storage [24], its role in vivo may differ; for example, in the heart, FATP1 stimulates fatty acid oxidation [25]. A similar effect in skeletal muscle would suggest that FATP1 does not contribute to insulin resistance, as has been suggested [16, 17].
To better understand the role of muscle FATP1 in intramuscular lipid metabolism and insulin sensitivity, we examined the effects of muscle-specific FATP1 overabundance using two different approaches. First, we transiently transfected Fatp1 cDNA into a single muscle in vivo to study the functional role of this transporter in fatty acid metabolism, unencumbered by possible changes in lipid metabolism in other tissues or in the circulating substrate and endocrine milieu, as the contralateral muscle serves as a control. Second, we generated a muscle-specific Fatp1 overexpressing transgenic mouse under the control of the Mck (also known as Ckm) promoter and examined whole-body and skeletal muscle insulin sensitivity following a 16 week high-fat diet. Finally, we examined FATP1, long-chain fatty acid (LCFA) transport and oxidation in obese Zucker rats, a well-known model of skeletal muscle insulin resistance and intramuscular lipid accumulation. We hypothesised that muscle-specific Fatp1 overexpression would predispose animals to high-fat diet-induced insulin resistance. However, our results indicate that, in muscle, FATP1 channels fatty acids preferentially into oxidation. Thus contrary to our hypothesis, FATP1 overabundance does not exacerbate diet-induced insulin resistance, as intramuscular triacylglycerol do not accumulate. Moreover, insulin resistance and enhanced intramuscular accumulation in obese Zucker rats are not attributable to FATP1, as muscle total cellular and sarcolemmal contents were not altered.
Methods
Animals
We used 10-week-old female Sprague–Dawley rats (University of Guelph bred) for the soleus muscle Fatp1 electrotransfection experiments (n = 5). Mck/Fatp1 transgenic FVB/N mice were developed, as described below, from our own breeding colony (n = 5 − 11). The soleus muscles of 10-week-old lean female and obese Zucker (fa/fa) rats (Charles River, Baie d’Urfé, QC, Canada) were excised and used to study fatty acid metabolism (n = 5). All animals were housed in a climate- and temperature-controlled room (12 h light–dark cycle), and given free access to rat chow and water prior to the start of experiments. The institutional Animal Care and Use Committees approved all procedures at either NIH (Bethesda, MD, USA) or the University of Guelph (Guelph, ON, Canada).
Transient electrotransfection of Fatp1
Electrotransfection experiments were performed as previously described [26–30]. The original Fatp1 cDNA clone was a gift from J. Schaffer (Washington University School of Medicine, St. Louis, MO, USA). The initial Fatp1 cDNA clones were used as templates in subsequent PCR amplifications designed to insert 5′ Kozak sequences and 3′ Myc epitope tags to the full-length open reading frame of Fatp1. The Fatp1 expression construct was then cloned into the EcoR1 site of pcDNA3.1 (Invitrogen, Carlsbad, CA, USA) and clones were sequenced to verify correct insert orientation, identity of the insert, and presence of the Kozak and epitope sequences. Animals were then anaesthetised with isoflurane and the soleus muscle was electrotransfected with: (1) pcDNA3.1 plasmid containing Fatp1 cDNA (250 μg in 50% [vol./vol.] saline); or (2) empty pcDNA3.1 plasmid (250 μg pcDNA in 50% [vol./vol.] saline). Rats were provided with an analgesic (Temgesic) and allowed to recover for 2 weeks.
Giant vesicles and fatty acid transport
Giant vesicles were prepared from control and Fatp1 transfected solei, and rates of palmitate uptake were determined in giant vesicles as previously described [6, 9, 10, 31].
Whole-muscle palmitate oxidation and esterification rates
Palmitate esterification and oxidation were determined using an incubated soleus muscle preparation as reported previously [6, 32].
Citrate synthase
Muscle samples were homogenised in a 100 mmol/l potassium phosphate buffer [33] and citrate synthase activity was assayed spectrophotometrically (412 nm; 37°C) as previously reported [34].
Isolation of mitochondria and mitochondrial palmitate oxidation
Differential centrifugation was used to obtain subsarcolemmal (SS) mitochondria and intermyofibrillar (IMF) mitochondria [4, 6]. In brief, SS (supernatant fraction) and IMF (pellet) mitochondria were separated by centrifugation at 800 g (10 min). The pellet was treated with a protease (Sigma-Aldrich, St Louis, MO, USA), and then SS and IMF mitochondria were subsequently isolated by centrifuging twice at 10,000 g (10 min). Palmitate oxidation was measured in a sealed system as previously described [4, 6].
Western blotting
Whole-muscle homogenates were prepared as previously described [27, 35]. Samples were separated by electrophoresis on 10% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membranes. We used the same antibodies against FABPpm and FAT/CD36 as those used previously [6, 9, 27]. Commercially available antibodies were used to detect FATP1 and FATP4 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), cytochrome c oxidase complex IV (Invitrogen, Burlington, ON, Canada), and complexes I, II, III and IV of the electron transport chain (Mitosciences, Eugene, OR, USA). Blots were quantified using chemiluminescence and an imaging system (ChemiGenius-2; SynGene, Cambridge, UK).
Generation of Mck/Fatp1 transgenic animals
The full-length of mouse Fatp1 cDNA (accession number U15976) cloned in pcDNA 3.1 plasmid (p2211) was obtained from H. Lodish (Whitehead Institute for Biomedical Research, Cambridge, MA, USA). XhoI and NotI restriction sites on 5′ and 3′ end of Fatp1 were introduced by PCR using the following primers: 5′ primer sequence 5′CCGCTCGAGGCCACCATGCGGGCTCCTGGAGCAGGA3′; and 3′ primer sequence 5′AAGGAAAAAAGCGGCCGCTCAGAGTGAGAAGTCGCCTGC3′. The linear PCR products (1,974 bp) were used for further cloning.
The vector containing Mck promoter/enhancer region and poly A sequence [p2208, pBS SK(−)] was a gift from C. R. Kahn (Joslin Diabetes Center, Boston, MA, USA). Plasmid p2208 was extensively modified before subcloning of Fatp1. One XhoI at the 5′ end of Mck promoter sequence was first removed. A modified plasmid (p2215) was digested with SacI (at 5′ end) and XhoI (at 3′ end) to obtain a 9.5 kb sequence containing the full length of Mck promoter (6.5 kb). The poly A sequence (257 bp) was prepared by digesting p2215 with Not I (at 5′ end) and Sac I (at 3′ end). The final clones (p2250, 11.6 kb) were created by ligating three pieces of linear DNA together in the following order: (1) 9.5 kb sequence containing Mck promoter sequence; (2) Fatp1 PCR products; and (3) 257 bp poly A sequence. The sequence identity of coding and non-coding sequences of Fatp1 in p2250 was confirmed by DNA sequencing analysis. The final construct, a linear 8.5 kb sequence, was generated by digesting p2250 with Asp718 and SacI, and the construct was diluted in 10 mmol/l Tris pH 7.4 and 0.1 mmol/l EDTA solution (5 ng/μl) for micro-injection (electronic supplementary material [ESM] Fig. 1). Southern and northern blotting was used to confirm specificity of Fatp1 transgenic animals. Briefly, DNA and RNA were extracted from rapidly frozen tissue in liquid nitrogen as previously described [4, 35]. Integrity of isolated samples was checked by separating them on 1% agarose gel prior to analysis.
15-(p-Iodophenyl)-3-R,S-methylpentadecanoic acid uptake
In vivo 15-(p-iodophenyl)-3-R,S-methylpentadecanoic acid (BMIPP) uptake was measured as described previously [36], with minor modifications. 125I-labelled BMIPP was obtained from F. F. Knapp, Jr (Oak Ridge National Laboratory, Oak Ridge, TN, USA). A dose of 0.37 mBq was injected into the tail vein of mouse and mice were killed within 30 min after the injection. Tissue uptake of BMIPP was expressed as per cent of injected dose per gram of tissue.
Whole-body metabolic phenotype following a high-fat diet
Mice (7 weeks old, age- and-sex matched) were placed on a high-fat diet (58% energy from fat; Research Diets, New Brunswick, NJ, USA) for an additional 16 weeks. After an overnight fast, basal glucose was measured with a glucometer (Glucometer Elite; Bayer, New York, NY, USA), and serum triacylglycerol and circulating fatty acids determined with commercially available kits (Sigma-Aldrich, St Louis, MO, USA and Roche Diagnostics, Penzberg, Germany, respectively). An intraperitoneal glucose tolerance test (IPGTT), an insulin tolerance test and a triacylglycerol clearance test were performed as previously described [21].
Assessment of skeletal muscle insulin action by euglycaemic–hyperinsulinaemic clamp
After an overnight fast, we performed a hyperinsulinaemic–euglycaemic clamp over a 2-h period as previously described [17, 37]. A bolus of 2-deoxy-d-[1-14C]glucose was administered at the 75 min mark of the clamp period to determine tissue-specific glucose uptake as previously reported [37].
Triacylglycerol concentrations
Intracellular triacylglycerol concentrations were determined biochemically as previously described [38]. Lipids were extracted in a standard Folch solution and hydrolysis with base, triacylglycerol was measured radiometrically using a glycerol kinase assay [39].
Statistics
All data are presented as the mean ± SEM. Unpaired t tests and paired t tests were used where appropriate, with statistical significance accepted at p < 0.05.
Results
Transient overexpression of Fatp1 and whole skeletal muscle metabolism
To examine the effects of muscle-specific Fatp1 overexpression without affecting other tissues, we transfected Fatp1 cDNA into one soleus musle, while the contralateral soleus muscle was sham-transfected with an empty vector. At 2 weeks after transfection, FATP1 total protein levels were increased ∼70% (Fig. 1a, c), with a ∼30% increase in plasma membrane FATP1 protein (Fig. 1b, c). No compensatory changes occurred in FATP4, FAT/CD36 or FABPpm protein contents (Fig. 1a, b). The increase in sarcolemmal FATP1 resulted in proportionate increases (p < 0.05) in the rate of palmitate transport (24% higher) into giant sarcolemmal vesicles, and in rates of palmitate oxidation (35% higher), while rates of triacylglycerol synthesis were not altered in Fatp1-transfected muscles (Fig. 1c).

The effects of transient Fatp1 overexpression on soleus muscle lipid metabolism. The soleus muscle of female Sprague–Dawley rats were transfected with 250 μg Fatp1 cDNA; the contralateral muscle was transfected with an equal volume of an empty vector and served as a control. a Representative blots of selected fatty acid transporters in whole-muscle homogenate and b on sarcolemmal membranes at 2 weeks after transfecting Fatp1 into soleus muscle. Con., control; Tran., transfected. c Skeletal muscle lipid metabolism following Fatp1 transfection. Data are expressed relative to control (100%) as mean ± SEM. However, the absolute values for these experiments were: palmitate transport 5.0 ± 0.5 vs 6.2 ± 0.4 pmol mg−1 (15 s)−1, wild-type and Fatp1-transfected, respectively; palmitate oxidation 20.2 ± 1.3 vs 27.1 ± 0.9 nmol (mg wet weight)−1 (40 min)−1, wild-type and Fatp1-transfected, respectively; triacylglycerol (TAG) esterification 161.3 ± 6.7 vs 141.3 ± 6.9 nmol (mg wet weight)−1 (h)−1 wild-type and Fatp1-transfected, respectively. n = 5 for all experiments; however for palmitate transport, giant vesicles from eight to nine animals were pooled for each of the five independent experiments; *p < 0.05 for difference from control. White bars, control muscle; black bars, transfected muscle. d Representative blots of selective electron transport chain complexes
A recent report on L6E9 myotubes suggests that FATP1 is also present on mitochondrial membranes, where it regulates the rate of mitochondrial LCFA oxidation [40]. To determine whether the increase in palmitate oxidation observed at the whole-muscle level was a result of intracellular lipid trafficking or due to changes in mitochondrial FATP1 protein, we measured palmitate oxidation and FATP1 protein content in isolated SS and IMF mitochondria in control and Fatp1-transfected muscles. In this tissue, Fatp1 transfection did not increase FATP1 protein on mitochondrial membranes (ESM Fig. 2) and did not alter rates of palmitate oxidation in SS (control 34 ± 3 vs Fatp1-transfected 33 ± 3 nmol mg−1 h−1) or IMF (control 63 ± 7 vs Fatp1-transfected 64 ± 6 nmol mg−1 h−1) mitochondria. In addition, transfection did not increase total cellular content of electron transport chain proteins (Fig. 1d) or citrate synthase activity (control 43 ± 4 vs Fatp1-transfected 39 ± 3 μmol [g wet weight]−1 min−1), indicating an increase in mitochondrial content was not responsible for the increase in palmitate oxidation observed following transient Fatp1 transfection. Together, these results suggest that the increased rates of whole-muscle palmitate oxidation result from increased sarcolemmal transport and fatty acid delivery to mitochondria.
Generation of Fatp1 transgenic animals
Results from Fatp1-transfected soleus muscle suggest that FATP1 is unlikely to contribute to intramuscular triacylglycerol accumulation. To examine this further, Mck/Fatp1 transgenic mice were generated. Southern blot analysis of tail DNA using a transgene-specific probe was used to identify transgenic animals (Fig. 2a). Northern blot analysis with a transgene-specific probe demonstrated that transgene mRNA was present in skeletal and cardiac muscles (Fig. 2b), and absent in other tissues (Fig. 2b). Northern blot analysis using an Fatp1 probe designed to span exons 5, 6 and 7 of endogenous and transgene-derived Fatp1 mRNA, confirmed pronounced overexpression of Fatp1 in skeletal and cardiac muscles (Fig. 2c).

Generation of Mck/Fatp1 transgenic animals. a Southern blot analysis of tail DNA using a transgene (TG)-specific probe to exon 4 confirmed successful generation of transgenic animals. WT, wild-type. b Northern blot analysis using a transgene-specific probe confirmed the specificity of Fatp1 overexpression. S. intestine, small intestine. c Compared with wild type (WT) animals, northern blot analysis with a probe that detects endogenous and transgene-derived Fatp1 mRNA confirmed the pronounced overexpression of Fatp1 in skeletal muscle (Quad, quadriceps; Gastroc, gastrocnemius) and heart of Mck/Fatp1 animals
Metabolic phenotype of Mck/Fatp1 mice on a chow diet
To evaluate the phenotype of Mck/Fatp1 mice, we examined BMIPP uptake by muscle and other tissues. BMIPP uptake confirmed a functional role for skeletal muscle FATP1 in sarcolemmal LCFA transport and the specificity of Mck/Fatp1 overexpression, as uptake of BMIPP into the muscle (75% higher) and heart (87% higher) was increased (p < 0.05) in Mck/Fatp1 animals, while BMIPP uptake into fat pads, liver, kidney and lung were not altered (Fig. 3). Despite this, there were no differences in weight gain, blood glucose, serum triacylglycerol, serum NEFA concentrations and organ weights (heart, liver, spleen, kidney, skeletal muscle and perigonadal fat pad) between wild-type and Mck/Fatp1 mice fed a chow diet (data not shown).

Tissue-specific uptake of 125I-labelled BMIPP in vivo in mice overexpressing Fatp1 in skeletal muscle. Data are expressed relative to wild-type (100%) as means±SEM. However, the absolute values, expressed as % of injected dose/g tissue, are as follows: quadriceps (Quad) 2.9 ± 0.3 vs 5.1 ± 0.3 for wild-type and Mck/Fatp1, respectively; heart 16.7 ± 2.3 vs 31.3 ± 2.6 for wild-type and Mck/Fatp1, respectively; fat 8.2 ± 1.2 vs 6.0 ± 0.9 for wild-type and Mck/Fatp1, respectively; liver 18.2 ± 1.6 vs 17.2 ± 1.9 for wild-type and Mck/Fatp1, respectively; kidney 9.0 ± 0.8 vs 8.2 ± 0.3 for wild-type and Mck/Fatp1, respectively; lung 8.9 ± 0.5 vs 8.0 ± 0.2 for wild-type and Mck/Fatp1, respectively. *p < 0.05 for difference from wild-type; n = 5 for all experiments. White bars, wild-type animals; black bars, Mck/Fatp1 animals
Metabolic phenotype of Mck/Fatp1 mice on a high-fat diet
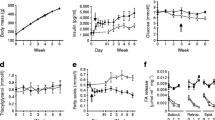
As previous reports have shown that whole-body Fatp1 ablation protects against high-fat diet-induced insulin resistance [16, 17], we examined whether overexpression of Fatp1, specifically in skeletal muscle, predisposes to or exacerbates the effects of a high-fat diet. Body weights in wild-type and Mck/Fatp1 animals did not differ at the start or finish of the 16-week feeding intervention (ESM Table 1). As expected, a high-fat diet increased blood glucose (Fig. 4a) and NEFA concentrations in wild-type and Mck/Fatp1 mice; however, blood glucose, serum insulin, triacylglycerol and NEFA concentrations did not differ in wild-type and Mck/Fatp1 mice before or after the high-fat diet (Fig. 4a, b, ESM Table 1).

Effect of high-fat feeding on blood glucose (a) and NEFA (b) concentrations in wild-type mice (white bars) and mice overexpressing Fatp1 (Mck/Fatp1) (black bars). Chow-fed values (Pre high-fat) are from the same animals prior to commencing high-fat feeding. Blood was sampled in non-fasting condition. Data are expressed as means±SEM; n = 5 for all experiments; *p < 0.05 for difference from chow-fed
Lipid metabolism of Mck/Fatp1 animals following a high-fat diet
Since the Mck/Fatp1 mice exhibited elevated rates of skeletal muscle LCFA uptake, we examined tissue accumulation of triacylglycerol in response to a high-fat diet. Fasting serum triacylglycerol concentration (ESM Table 1), triacylglycerol clearance rates (Fig. 5a) and triacylglycerol content in skeletal and heart muscles (Fig. 5b) were not different between wild-type and Mck/Fatp1 animals following a high-fat diet.

Comparison of triacylglycerol (TAG) clearance (a) and tissue-specific triacylglycerol content (b) in wild-type (solid line and white bars) mice and in mice overexpressing Fatp1 (Mck/Fatp1) (dashed line and black bars) following a 16-week high-fat diet. Data are expressed as means±SEM; n = 5 for all experiments; *p < 0.05 for difference from wild-type
Insulin sensitivity of Mck/Fatp1 mice following a high-fat diet
We next examined insulin sensitivity in the Mck/Fatp1 animals during the high-fat diet. Whole-body glucose tolerance (Fig. 6a, b) and insulin tolerance (Fig. 6c) did not differ between wild-type and Mck/Fatp1 animals on a 16-week high-fat diet. In addition, the ability of insulin to suppress endogenous hepatic glucose production (Table 1) and glucose infusion rates (Table 1) determined during a 2 h euglycaemic–hyperinsulinaemic clamp in conscious animals did not differ between wild-type and Mck/Fatp1. During the clamp there was a trend towards increased accumulation of the non-metabolisable glucose analogue 2-deoxy-d-[1-14C]glucose in the muscle (p = 0.056) of Mck/Fatp1 mice (Table 1), whereas insulin-stimulated glucose uptake was not different in white or brown adipose tissue (Table 1).

Comparison of glucose (a) and insulin (c) tolerance in wild-type (continuous lines) mice and in mice overexpressing Fatp1 (Mck/Fatp1) (dashed lines) following a 16-week high-fat diet. b AUC during the IPGTT. Animals were fasted for 6 h in the light cycle prior to commencing experiments. Data are expressed as mean ± SEM; n = 5 for all experiments; †p = 0.14
FATP1 and intracellular lipid handling in obese Zucker rats
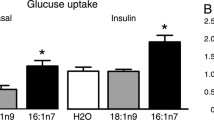
The foregoing experiments suggest that increasing skeletal muscle levels of FATP1 result in increased rates of fatty acid transport into muscle, but do not cause peripheral insulin resistance because of a concomitant increase in LCFA oxidation. To further examine this, we measured the plasma membrane content of FATP1, LCFA transport rates and intracellular lipid handling in obese Zucker rats, a model of insulin resistance. Accordingly, plasma membrane palmitate transport was increased (by 85%; p < 0.05), without compensatory changes in fatty acid oxidation; instead triacylglycerol esterification was increased by about 25% (p < 0.05; Fig. 7). However, these changes were not attributable to FATP1, as total muscle homogenate and sarcolemmal FATP1 protein levels were not altered (Fig. 7).

Skeletal muscle lipid metabolism in obese Zucker rats. Representative blots of whole muscle homogenates (Hom) and sarcolemmal (PM) FATP1 in lean (L, white bars) and obese (O, black bars) animals. Data are expressed relative to lean animals (100%) as means±SEM. However, the absolute numbers for these experiments are as follows: palmitate transport 4.7 ± 0.8 vs 9.4 ± 1.8 pmol (mg protein)−1 (15 s)−1, lean and obese, respectively; palmitate oxidation 68.0 ± 6.6 vs 68.2 ± 5.4 nmol (mg wet weight)−1 (40 min)−1, lean and obese, respectively; triacylglycerol (TAG) esterification 157.8 ± 6.9 vs 196.2 ± 7.0 nmol (mg wet weight)−1 (h)−1, lean and obese, respectively. n = 5 for all experiments; however, for palmitate transport, giant vesicles from eight to nine animals were pooled for each of the five independent experiments; *p < 0.05 for difference from lean
Discussion
Novel insights
This study showed that the skeletal muscle fatty acid transporter FATP1 channels fatty acids to oxidation and not intramuscular lipid accumulation. As a result, increasing muscle FATP1 does not predispose animals to diet-induced insulin resistance.
FATP-mediated intramuscular lipid trafficking
There are several skeletal muscle fatty acid transporters, creating a potentially redundant system, as previously reviewed [15]. Thus, it has been speculated that different fatty acid transporters traffic lipids to different metabolic fates. The current data indicate that FATP1 increases LCFA transport and channels exogenous lipids into oxidation, rather than into lipid storage (Fig. 1c). This is supported by a number of observations in our Fatp1-transfected soleus muscle, showing that: (1) muscle palmitate transport and (2) oxidation were increased independently of (3) changes in mitochondrial content and the intrinsic rates of palmitate oxidation by mitochondria, or (4) of rates of triacylglycerol esterification and/or content. Cardiac specific overabundance of FATP1 also supports our results, as this increased LCFA uptake and oxidation, not triacylglycerol accumulation [25]. In liver [41] and adipose [42] tissue, acyl-CoA synthetase long-chain family member 1 (ACSL1)/FATP1 has also been suggested to direct fatty acids towards oxidation. In contrast, experiments in 293 cells [24] and in human myotubes [43] suggested that FATP1 channels exogenous LCFAs into triacylglycerol storage. These disparities may reflect different experimental systems, as the regulation of fatty acid metabolism could be quite different in cultured cells, due to their low oxidative capacity and low rate of fatty acid metabolism compared with mature mammalian muscle. Taken together, our data suggest that within rodent skeletal muscle, FATP1 targets LCFAs to oxidation. However, the mechanism of action by which ACSL1/FATP1 directs fatty acids to oxidation remains unknown. The catalytic activity of ACSL1 has been suggested to activate AMP-activated protein kinase due to a change in cell energy status, specifically a rise in the AMP/ATP ratio [44], which may also partially account for the apparent FATP1-mediated targeting of fatty acids towards oxidation in the current study.
Mitochondrial FATP1
Although it has been observed that FATP1 resides on mitochondrial membranes in myotubes, regulating LCFA oxidation in an unknown manner [40], this is not supported by findings of the present study in mature mammalian muscle. In mitochondria isolated from Fatp1-transfected muscle, palmitate oxidation was not altered and no upregulation of mitochondrial Fatp1 was observed. To our knowledge, Fatp1 does not contain a mitochondrial targeting sequence, and we did not include a targeting sequence in our vector. Other investigators also omitted a mitochondrial targeting sequence when upregulating mitochondrial Fatp1 in L6E9 cells [40]. While the lack of a targeting sequence in the current study may partially explain negative findings at the mitochondria level, we have previously used identical methods to overexpress Fabppm (also known as Got2), which also does not contain a mitochondrial targeting sequence, resulting in increased FABPpm levels on sarcolemmal and mitochondrial membranes [26]. Hence, the lack of a detectable increase in FATP1 on muscle mitochondria is unlikely to be due to methodological issues. In addition, we show that several markers of mitochondrial content (subunits of the electron transport chain) were not increased in response to transient transfection with Fatp1. Thus, our present results indicate that changes in whole-muscle LCFA oxidation following Fatp1 transfection result from changes in sarcolemmal transport and an increased delivery of LCFAs to mitochondria, as opposed to an enhanced intrinsic rate of fatty acid oxidation by mitochondria due to an increase in mitochondrial FATP1. This further emphasises that plasma membrane FATP1 does indeed channel exogenous lipids to mitochondria for oxidation.
Skeletal muscle FATP1 in the development of diet-induced insulin resistance
Skeletal muscle insulin resistance has been associated with intramuscular lipid accumulation, possibly as a result of bioactive lipids (e.g. ceramide and diacylglycerol) interfering with insulin signalling and sensitivity, as indicated by a review [1]. It has been suggested that upregulation of fatty acid transport into muscle could contribute to skeletal muscle lipid oversupply and increases in intramuscular lipids [6, 9, 10]. In this respect, FATPs could represent a key mechanism in the aetiology of insulin resistance. However, muscle FATP1, unlike other fatty acid transporters, is not correlated with indices of carbohydrate or lipid metabolism [27, 35] and the transport effectiveness of FATP1 is low [27, 45]. These observations suggest that alterations in this transporter may have limited effects on lipid metabolism in vivo. In addition, channelling fatty acids to mitochondrial oxidation, as demonstrated in the present study, further reduces the likelihood that FATP1 contributes to the development of insulin resistance in skeletal muscle. A rise in fatty acid oxidation has been shown to protect against diet-induced insulin resistance [46]. It could therefore be speculated from the current data that overabundance of FATP1 and the subsequent targeting of fatty acids towards oxidation would protect against diet-induced insulin resistance. In support of this notion, following a high-fat diet in mice overexpressing Fatp1, there was a trend for the following to be improved: glucose uptake during an IPGTT test (p = 0.14), glucose infusion rates during a euglycaemic–hyperinsulinaemic clamp (p = 0.15) and skeletal muscle insulin-stimulated glucose uptake during the clamp procedures (p = 0.056). We have only measured an increase in fatty acid oxidation in Fatp1-transfected muscle, similar to its apparent role in the liver [42] and adipose tissue [41]. A limitation of the current study is that we did not determine rates of fatty acid oxidation in Mck/Fatp1 mice. However, we assume that fatty acid oxidation was also likely to have been increased in muscles of these animals. Therefore we conclude that overexpression of Fatp1 in muscle did not exacerbate the development of diet-induced insulin resistance, as fatty acid oxidation was stimulated, while intramuscular lipids did not accumulate.
The conclusion that FATP1 has a minor role, if any, in the development of insulin resistance is in marked contrast to previous experiments in Fatp1-KO mice, which indicated that ablating Fatp1 protects against diet-induced insulin resistance, yet does not alter fatty acid uptake or selected bioactive lipids (ceramide, diacylglycerol) [17]. However, Fatp1 is highly expressed in white adipose tissue, and Fatp1 ablation appears to preferentially affect the metabolic function of this tissue [16]. Therefore, in view of the apparent significance of FATP1 in adipose tissue [16], but not in muscle [27], it is unclear whether the metabolic phenotype observed following high-fat feeding in Fatp1-KO animals was a direct result of alterations in adipose tissue metabolism, rather than in skeletal muscle [16]. While it remains possible that ablating Fatp1 affords some protection and/or provides a potential therapeutic target for the treatment of insulin resistance, FATP1 in muscle does not appear to contribute to the underlying aetiology of this pathology. This is further emphasised in insulin-resistant obese Zucker rats, in which LCFA transport was increased and intramuscular triacylglycerol accumulated. While increases in plasma membrane FAT/CD36 content appear to contribute to this insulin-resistant phenotype [6, 10], neither FATP1 protein nor its sarcolemmal content is altered in muscle of obese Zucker rats, as seen here and elsewhere [6].
Summary
The present study questions the role of skeletal muscle FATP1 in diet-induced insulin resistance. In muscle-specific Fatp1-overexpressing animals, rates of fatty acid transport into muscle were increased, but these animals were not predisposed to insulin resistance, an effect we attribute to targeting the additional lipids to oxidation and not to storage. This FATP1-associated channelling of fatty acids to oxidation probably prevented additional lipotoxicity despite increased plasma membrane transport.
Abbreviations
- ACSL1:
-
Acyl-CoA synthetase long-chain family member 1
- BMIPP:
-
15-(p-Iodophenyl)-3-R,S-methylpentadecanoic acid
- FABPpm:
-
Plasma membrane-associated fatty acid binding protein
- FAT/CD36:
-
Fatty acid translocase
- FATP:
-
Fatty acid transport protein
- IMF:
-
Intermyofibrillar
- IPGTT:
-
Intraperitoneal glucose tolerance test
- KO:
-
Knockout
- LCFA:
-
Long-chain fatty acid
- Mck/Fatp1 :
-
Muscle-specific Fatp1 overexpressing
- Subsarcolemmal:
-
transgenic mice
- SS:
-
Subsarcolemmal
References
Holland WL, Knotts TA, Chavez JA, Wang LP, Hoehn KL, Summers SA (2007) Lipid mediators of insulin resistance. Nutr Rev 65:S39–S46
Kelley DE, He J, Menshikova EV, Ritov VB (2002) Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51:2944–2950
Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, de Kelley (2005) Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 54:8–14
Holloway GP, Gurd BJ, Snook LA, Lally J, Bonen A (2010) Compensatory increases in nuclear PGC1alpha protein are primarily associated with subsarcolemmal mitochondrial adaptations in ZDF rats. Diabetes 59:819–828
Holloway GP, Thrush AB, Heigenhauser GJ et al (2007) Skeletal muscle mitochondrial FAT/CD36 content and palmitate oxidation are not decreased in obese women. Am J Physiol Endocrinol Metab 292:E1782–E1789
Holloway GP, Benton C, Mullen KL et al (2009) In obese rat muscle transport of palmitate is increased and is channeled to triacylglycerol storage despite an increase in mitochondrial palmitate oxidation. Am J Physiol Endocrinol Metab 296:E738–E747
Boushel R, Gnaiger E, Schjerling P, Skovbro M, Kraunsoe R, Dela F (2007) Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia 50:790–796
Mogensen M, Sahlin K, Fernstrom M et al (2007) Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 56:1592–1599
Bonen A, Parolin ML, Steinberg GR et al (2004) Triacylglycerol accumulation in human obesity and type 2 diabetes is associated with increased rates of skeletal muscle fatty acid transport and increased sarcolemmal FAT/CD36. FASEB J 18:1144–1146
Luiken JJ, Arumugam Y, Dyck DJ et al (2001) Increased rates of fatty acid uptake and plasmalemmal fatty acid transporters in obese Zucker rats. J Biol Chem 276:40567–40573
Aguer C, Mercier J, Yong Wai Man C et al (2010) Intramyocellular lipid accumulation is associated with permanent relocation ex vivo and in vitro of fatty acid translocase (FAT)/CD36 in obese patients. Diabetologia 53:1151–1163
Koves TR, Ussher JR, Noland RC et al (2008) Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab 7:45–56
Consitt LA, Bell JA, Koves TR et al (2010) PGC-1α overexpression increases lipid oxidation in myocytes from extremely obese individuals. Diabetes 59:1407–1415
Jain S, Schwenk RW, Glatz JFC, Luiken JJFP, Bonen A (2009) Additive effects of insulin and muscle contraction on fatty acid transport and fatty acid transporters, FAT/CD36, FATPpm, FATP1, 4 and 6. FEBS Lett 583:2294–2300
Glatz JFC, Luiken JJFP, Bonen A (2010) Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiol Rev 90:367–417
Wu Q, Ortegon AM, Tsang B, Doege H, Feingold KR, Stahl A (2006) FATP1 is an insulin-sensitive fatty acid transporter involved in diet-induced obesity. Mol Cell Biol 26:3455–3467
Kim JK, Gimeno RE, Higashimori T et al (2004) Inactivation of fatty acid transport protein 1 prevents fat-induced insulin resistance in skeletal muscle. J Clin Investig 113:756–763
Mutch DM, Fauconnot L, Grigorov M, Fay LB (2006) Putting the ‘ome’ in lipid metabolism. Biotechnol Annu Rev 12:67–84
German JB, Gillies LA, Smilowitz JT, Zivkovic AM, Watkins SM (2007) Lipidomics and lipid profiling in metabolomics. Curr Opin Lipidol 18:66–71
Mutch DM, Crespy V, Clough J et al (2006) Hepatic cytochrome P-450 reductase-null mice show reduced transcriptional response to quercetin and reveal physiological homeostasis between jejunum and liver. Am J Physiol Gastrointest Liver Physiol 291:G63–G72
Mutch DM, Klocke B, Morrison P et al (2007) The disruption of hepatic cytochrome p450 reductase alters mouse lipid metabolism. J Proteome Res 6:3976–3984
Bruning JC, Michael MD, Winnay JN et al (1998) A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell 2:559–569
Kuriyama H, Liang G, Engelking LJ, Horton JD, Goldstein JL, Brown MS (2005) Compensatory increase in fatty acid synthesis in adipose tissue of mice with conditional deficiency of SCAP in liver. Cell Metab 1:41–51
Hatch GM, Smith AJ, Xu FY, Hall AM, Bernlohr DA (2002) FATP1 channels exogenous FA into 1,2,3-triacyl-sn-glycerol and down-regulates sphingomyelin and cholesterol metabolism in growing 293 cells. J Lipid Res 43:1380–1389
Chiu HC, Kovacs A, Blanton RM et al (2005) Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ Res 96:225–233
Holloway GP, Lally J, Nickerson JG et al (2007) Fatty acid binding protein facilitates sarcolemmal fatty acid transport but not mitochondrial oxidation in rat and human skeletal muscle. J Physiol 582:393–405
Nickerson JG, Alkhateeb H, Benton CR et al (2009) Greater transport efficiencies of the membrane fatty acid transporters FAT/CD36 and FATP4 than FABPPM and FATP, and differential effects on fatty acid esterification and oxidation in rat skeletal muscle. J Biol Chem 284:16522–16530
Clarke DC, Miskovic D, Han XX et al (2004) Overexpression of membrane-associated fatty acid binding protein (FABPpm) in vivo increases fatty acid sarcolemmal transport and metabolism. Physiol Genomics 17:31–37
Kramer HF, Witczak CA, Taylor EB, Fujii N, Hirshman MF, Goodyear LJ (2006) AS160 regulates insulin- and contraction-stimulated glucose uptake in mouse skeletal muscle. J Biol Chem 281:31478–31485
Schertzer JD, Plant DR, Lynch GS (2006) Optimizing plasmid-based gene transfer for investigating skeletal muscle structure and function. Mol Ther 13:795–803
Bonen A, Luiken JJ, Liu S et al (1998) Palmitate transport and fatty acid transporters in red and white muscles. Am J Physiol 275:E471–E478
Dyck DJ, Peters SJ, Glatz J et al (1997) Functional differences in lipid metabolism in resting skeletal muscle of various fiber types. Am J Physiol 272:E340–E351
Srere PA (1969) Citrate synthase. In: Lowenstein JM (ed) Methods in enzymology. Academic, New York
Bergmeyer HU (1974) Methods of enzymatic analysis. Verlag Chemie Weinheim, New York
Benton CR, Nickerson JG, Lally J et al (2008) Modest PGC-1alpha overexpression in muscle in vivo is sufficient to increase insulin sensitivity and palmitate oxidation in SS, not IMF, mitochondria. J Biol Chem 283:4228–4240
Coburn CT, Knapp FF Jr, Febbraio M, Beets AL, Silverstein RL, Abumrad NA (2000) Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J Biol Chem 275:32523–32529
Chen M, Haluzik M, Wolf NJ et al (2004) Increased insulin sensitivity in paternal Gnas knockout mice is associated with increased lipid clearance. Endocrinology 145:4094–4102
Chou CJ, Haluzik M, Gregory C et al (2002) WY14,643, a peroxisome proliferator-activated receptor alpha (PPARalpha) agonist, improves hepatic and muscle steatosis and reverses insulin resistance in lipoatrophic A-ZIP/F-1 mice. J Biol Chem 277:24484–24489
Gavrilova O, Leon LR, Marcus-Samuels B et al (1999) Torpor in mice is induced by both leptin-dependent and -independent mechanisms. Proc Natl Acad Sci USA 96:14623–14628
Sebastian D, Guitart M, Garcia-Martinez C et al (2009) Novel role of FATP1 in mitochondrial fatty acid oxidation in skeletal muscle cells. J Lipid Res 50:1789–1799
Ellis JM, Li LO, Wu PC et al (2010) Adipose acyl-CoA synthetase-1 directs fatty acids toward beta-oxidation and is required for cold thermogenesis. Cell Metab 12:53–64
Li LO, Ellis JM, Paich HA et al (2009) Liver-specific loss of long chain acyl-CoA synthetase-1 decreases triacylglycerol synthesis and beta-oxidation and alters phospholipid fatty acid composition. J Biol Chem 284:27816–27826
Garcia-Martinez C, Marotta M, Moore-Carrasco R et al (2005) Impact on fatty acid metabolism and differential localization of FATP1 and FAT/CD36 proteins delivered in cultured human muscle cells. Am J Physiol Cell Physiol 288:C1264–C1272
Liu Q, Gauthier MS, Sun L, Ruderman N, Lodish H (2010) Activation of AMP-activated protein kinase signaling pathway by adiponectin and insulin in mouse adipocytes: requirement of acyl-CoA synthetases FATP1 and Acsl1 and association with an elevation in AMP/ATP ratio. FASEB J 24:4229–4239
DiRusso CC, Li H, Darwis D, Watkins PA, Berger J, Black PN (2005) Comparative biochemical studies of the murine fatty acid transport proteins (FATP) expressed in yeast. J Biol Chem 280:16829–16837
Bruce CR, Brolin C, Turner N et al (2007) Overexpression of carnitine palmitoyltransferase I in skeletal muscle in vivo increases fatty acid oxidation and reduces triacylglycerol esterification. Am J Physiol Endocrinol Metab 292:E1231–E1237
Acknowledgements
This work was funded by the Canadian Institutes of Health Research (to A. Bonen) and the Natural Sciences and Engineering Research Council of Canada (to A. Bonen and G. P. Holloway). Mck/Fatp1 transgenic mouse studies were funded by the intramural programme of the National Institute of Diabetes, Digestive and Kidney Diseases (to M. L. Reitman, C. J. Chous, O. Gavrilova and M. Haluzik). A. Bonen is the Canada Research Chair in Metabolism and Health.
Duality of interest
The authors declare there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
G. P. Holloway and C. J. Chou contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
Cloning strategy for Mck. The plasmid p2211 contains a full-length of mouse Fatp1 cDNA cloned in pcDNA3.1. XhoI and NotI restriction sites on the 5′ and 3′ ends of Fatp1 were introduced by PCR. The final products (1,941 bp) were prepared for further cloning. The vector p2208 contains Mck promoter/enhancer region and poly A sequence cloned in pBS SK(−). One XhoI at the 5′ end of Mck promoter sequence was first removed (p2215). Then, p2215 was digested with SacI (at 5′ end) and XhoI (at 3′ end) to obtain a 9.5 kb sequence containing the full length of Mck promotor (6.5 kb). The poly A sequence (257 bp) was prepared by digesting p2215 with NotI (at 5′ end) and SacI (at 3′ end). The final clones (p2250, 11.6 kb) were created by ligating the 9.5 kb sequence containing Mck promoter sequence, the Fatp1 PCR products and 257 bp poly A sequence together. A linear 8.5 kb sequence was generated by digesting p2250 with Asp718 and SacI, and the construct was prepared for micro-injection (PDF 50 kb)
ESM Fig. 2
The soleus muscle of female Sprague–Dawley rats were transfected with 250 μg of Fatp1 cDNA; the contralateral muscle was transfected with an equal volume of an empty vector and served as a control. a Representative blots of FATP1 and the associated Myc-tag. Con., control; HOM, homogenate; Trans., transfected. b Palmitate oxidation in isolated SS and IMF mitochondria following transient transfection. White bars, control muscles; black bars, transfected muscles. c Representative blots showing successful overabundance of FABPpm protein, as well the associated Flag-tag, in isolated SS and IMF mitochondria, suggesting technical limitations are unlikely to be responsible for our lack of FATP1 overabundacne in mitochondria. n = 5 for all experiments; *p < 0.05 for difference from SS (PDF 224 kb)
ESM Table 1
Metabolic characteristics of mice following 16-week high-fat diet (PDF 63 kb)
Rights and permissions
About this article
Cite this article
Holloway, G.P., Chou, C.J., Lally, J. et al. Increasing skeletal muscle fatty acid transport protein 1 (FATP1) targets fatty acids to oxidation and does not predispose mice to diet-induced insulin resistance. Diabetologia 54, 1457–1467 (2011). https://doi.org/10.1007/s00125-011-2114-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-011-2114-8