
Abstract
Aims/hypothesis
Chemokine (C-X-C motif) ligand 12 (CXCL12) (also known as stromal cell-derived factor-1 [SDF-1]-alpha) is a homeostatic chemokine with multiple roles in cell homing, tumour metastasis, angiogenesis and tissue regeneration after acute injuries. However, its role in chronic diseases remains poorly defined, e.g. in chronic glomerular diseases like diabetic glomerulosclerosis. We hypothesised that CXCL12 may have a functional role during the evolution of diabetic glomerulosclerosis, either by assisting glomerular repair or by supporting the maladaptive tissue remodelling in response to hyperglycaemia and glomerular hyperfiltration.
Methods
To define the functional role of CXCL12 in the progression of glomerular disease, we used the CXCL12-specific inhibitor NOX-A12, an l-enantiomeric RNA oligonucleotide (Spiegelmer). A mouse model of type 2 diabetes (db/db mice) was used. Male db/db mice, uni-nephrectomised at 6 weeks of age, received subcutaneous injections with a PEGylated form of NOX-A12, non-functional control Spiegelmer or vehicle on alternate days from 4 to 6 months of age.
Results
Immunostaining localised renal CXCL12 production to glomerular podocytes in db/db mice with early or advanced diabetic nephropathy. CXCL12 inhibition significantly reduced the degree of glomerulosclerosis, increased the number of podocytes, prevented the onset of albuminuria and maintained the peritubular vasculature without affecting blood glucose levels, body weight or glomerular macrophage infiltration.
Conclusions/interpretation
We conclude that podocytes produce CXCL12, which contributes to proteinuria and glomerulosclerosis in our mouse model of type 2 diabetes. This novel pathomechanism provides the first evidence that CXCL12 could be a therapeutic target in (diabetic) glomerulosclerosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetic nephropathy remains a leading cause of end-stage renal disease [1, 2]. Identifying additional pathomechanisms to improve the prevention and treatment of diabetic nephropathy is necessary as the current concept of angiotensin blockade does not always prevent disease progression [3]. In diabetic nephropathy the glomerular tuft undergoes a slow but progressive structural remodelling characterised by glomerular hypertrophy, diffuse and nodular accumulation of extracellular mesangial matrix, and podocyte damage [4]. The latter is thought to account for the progression of microalbuminuria in early stages to overt proteinuria and glomerulosclerosis in late stages of diabetic nephropathy [4]. Diabetic nephropathy onset and progression involve numerous pathomechanisms including the deposition of advanced glycation end-products, endothelial dysfunction and the increased local expression of growth factors and proinflammatory mediators [5]. Generally, chemokines belong to the latter group of factors because certain chemokines promote inflammation by recruiting and activating immune cells in diabetic nephropathy as in other types of kidney diseases [6–9]. For example, targeted deletion or therapeutic blockade of chemokine (C-C motif) ligand 2 (CCL2, also known as monocyte chemoattractant protein [MCP-1]) can prevent glomerulosclerosis by blocking macrophage recruitment to glomeruli in mouse models of type 1 or type 2 diabetes [10, 11].
Although structurally related, a subgroup of the chemokine superfamily, known as homeostatic chemokines, displays functions that are independent of tissue inflammation. Homeostatic chemokines are rather constitutively expressed as they contribute to the physiological homing and migration of immune cells in the bone marrow or lymphoid organs [12]. For example, chemokine (C-X-C motif) ligand 12 (CXCL12, also known as stromal cell-derived factor [SDF]-1) was first identified as a lymphocyte homing chemokine [13]. CXCL12 mediates its biological effects via the chemokine (C-X-C motif) receptors (CXCRs), CXCR4/fusin and CXCR7 [14]. The expression of CXCL12 (and CXCR4) is controlled by hypoxia-regulated factor-α, a transcription factor that orchestrates intrarenal expression in tissues when exposed to hypoxia or oxidative stress [15]. In fact, some biological effects of CXCL12 support tissue reoxygenation and regeneration. For example, CXCL12 supports vascular integrity similarly to vascular endothelial growth factor (VEGF) [16], induces angiogenesis [17], recruits bone marrow-derived progenitor cells [18–20] or promotes cell survival [21]. In this regard CXCL12 was shown to be a crucial mediator of repair in a number of different disease models, e.g. of pancreatic beta cell loss in type 1 diabetes [21], endovascular injury [19], vascular occlusion [18, 22] and ischaemic acute renal failure [20]. However, in chronic disease states injury response mechanisms are often maladaptive and instead promote disease progression to organ failure. For example, CXCL12-mediated fibrocyte recruitment aggravates bleomycin-induced pulmonary fibrosis [23, 24] or CXCL12-driven neoangiogenesis drives diabetic eye disease [17], haemangioblastoma and renal cancer [25]. Furthermore, CXCL12 attracts circulating tumour cells as a tissue-based mechanism for cancer metastasis [26]. These studies suggest that CXCL12 creates microenvironments that maintain niches for homing of other cells including progenitor cell recruitment during repair. However, this function may also drive tissue- and disease-specific maladaptive pathomechanisms.
The role of CXCL12 in glomerular disease is still to be explored. Nephrotoxic serum nephritis specifically induces glomerular CXCL12 expression [27]. In addition, transgenic overexpression of Cxcr4 (by knocking out Von-Hippel-Lindau gene) induced podocyte proliferation and glomerular crescent formation in mice [27]. In lupus-like immune complex glomerulonephritis of NZB/NZW mice, glomerular CXCL12 expression mainly originated from podocytes [28]. Blocking CXCL12 prevented glomerulonephritis, which was attributed to less autoantibody production and T cell recruitment to glomeruli [28]. So far nothing is known about CXCL12 in diabetic nephropathy. Diabetic nephropathy is devoid of podocyte proliferation or autoimmunity; hence, the aforementioned studies hardly predict the predominant functional role of CXCL12 in diabetic nephropathy. We speculated that progressive remodelling of the glomerular structure to glomerulosclerosis, a morphological variant of wound healing, might involve CXCL12 signalling. Based on the available data from other disease states, it appeared unclear whether CXCL12 predominantly protects from diabetic nephropathy (e.g. by maintaining tissue integrity and supporting regeneration) or whether it contributes to progression of diabetic nephropathy (e.g. by enhancing glomerulosclerosis). Our data support the latter by identifying a novel pathomechanism of glomerulosclerosis and CXCL2 as a potential therapeutic target in diabetic nephropathy.
Methods
CXCL12 antagonistic Spiegelmer NOX-A12
The nucleotide sequence of the Spiegelmer NOX-A12 (5′-GCGUGGUGUGAUC UAGAUGUAUUGGCUGAUCCUAGUCAGGUACGC-3′) was obtained from in vitro selection experiments that were essentially carried out as described [11]. NOX-A12 binds to CXCL12 with a subnanomolar affinity. Inhibition of CXCL12 was determined using a chemotaxis assay with Jurkat cells employing 0.3 nmol/l CXCL12, 3 h cell migration and increasing concentrations of NOX-A12. NOX-A12 inhibits murine CXCL12 with a half-maximal inhibitory concentration (IC50) of 300 pmol/l (data not shown). For in vivo application, NOX-A12 and a non-functional control Spiegelmer composed of the reverse NOX-A12 sequence were 5′-terminally modified with 40-kDa branched polyethylene glycol (PEGylated).
Animal studies
Male diabetic C57BLKS db/db mice or non-diabetic C57BL/6 mice aged 5 weeks were obtained from Taconic (Ry, Denmark) and housed in filter top cages with a 12 h dark/light cycle. All animals had unlimited access to food and water throughout the study. At the age of 6 weeks uni-nephrectomy (removal of one kidney) or sham surgery was performed through a 1 cm flank incision as previously described in db/db mice [29]. In mice undergoing sham surgery the kidney was left in situ. At the age of 4 months, db/db mice with one kidney that had documented blood glucose levels > 11 mmol/l and creatinine/albumin ratios > 3 (ratio in age-matched wild-type mice 0.1) were divided into four groups (n = 10–12). Thereafter on every alternate days for 8 weeks, each of these groups received either nil (no injections) or subcutaneous injections of 50 mg/kg formulated NOX-A12, control Spiegelmer in 5% (wt/vol.) glucose or 5% (wt/vol.) glucose alone as a vehicle control. The treatment dose and injection intervals were identical to those in our previous db/db mouse studies with similar formulations of Spiegelmers targeting other chemokines [11]. Treatment was continued for 8 weeks, after which tissues were removed for histopathological evaluation 3 to 4 h after the last Spiegelmer injection. Blood and urine samples were obtained at monthly intervals for estimation of urinary albumin (ELISA; Bethyl Labs, Montgomery, TX, USA) as well as serum and urinary creatinine (Jaffé reaction; DiaSys Diagnostic Systems, Holzheim, Germany). Blood glucose levels were monitored using a glucometer (Accu check; Roche, Mannheim, Germany). Plasma levels of CXCL12 were determined at the end of treatment period by ELISA (R&D System, Wiesbaden, Germany). All animal experiments were approved by the local government authorities.
Histopathological evaluation
For histopathological evaluation, kidneys were removed from each mouse and fixed in 10% (vol./vol.) formalin in PBS, and embedded in paraffin. Sections of 2 μm were stained with periodic acid–Schiff’s reagent. Glomerular sclerotic lesions were assessed using a semi-quantitative score by a blinded observer as follows: 0 = no lesion, 1 = <25% sclerotic, 2 = 25–49% sclerotic, 3 = 50–74% sclerotic and 4 = 75–100% sclerotic, respectively. We analysed 15 glomeruli per section. The indices for interstitial volume and tubular dilatation were determined by superimposing a 100-point grid on ten non-overlapping cortical fields and expressed as percentage of cells as described previously [11]. Immunostainings were performed as described [30] using the following primary antibodies: CXCL12 (1:100; R&D), MAC2 (macrophages, 1:5,000; Cederlane, Burlington, ON, Canada), Wilms tumour (WT)-1 (podocytes, 1:200; Santa Cruz, Santa Cruz, CA, USA), Ki-67 (proliferating cells, 1:50; Dianova, Hamburg, Germany) and mouse endothelial cell antigen 32 (MECA32) (interstitial endothelial cells, 1:5; School of Medicine, Stanford University, CA, USA). MAC2 or WT-1-positive glomerular cells were counted in 15 glomeruli and MAC2-positive interstitial cells were counted in 15 non-overlapping high-power fields (×400) by a blinded observer. Absolute numbers of vessels or interstitial macrophages were counted in 15 non-overlapping high-power fields (×200) of MECA32 stained sections. Ki67-positive nuclei were counted in 20 glomeruli or in 20 non-overlapping high-power fields (×200).
RNA preparation and real-time quantitative (TaqMan) RT-PCR
Total RNA was isolated from kidneys using an RNA extraction kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. After quantification, RNA quality was assessed using agarose gels. From isolated RNA, cDNA was prepared using reverse transcriptase (Superscript II; Invitrogen, Carlsbad, CA, USA). Real-time-PCR was performed using SYBRGreen PCR master mix and was analysed with a Light Cycler 480 (Roche). All gene expression values were normalised using 18S RNA as a housekeeping gene. All primers (Electronic supplementary material [ESM], Table 1) used for amplification were from Metabion (Martinsried, Germany).
Statistical analysis
Data are presented as mean ± SEM. Comparison of groups was performed using ANOVA and the post-hoc Bonferroni’s correction was used for multiple comparisons. A value of p < 0.05 was considered to indicate statistical significance.
Results
CXCL12 is produced by podocytes in db/db mice
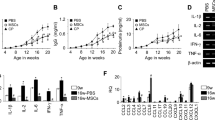
We wondered whether Cxcl12 is expressed in kidneys of db/db mice and first analysed Cxcl12 mRNA expression throughout the progression of kidney disease in male db/db mice. Cxcl12 mRNA was detectable in renal cortex preparations from 6-week-old db/db mice (Fig. 1b). The renal Cxcl12 mRNA levels did not significantly differ from those of sham-operated or uni-nephrectomised 6-month-old db/db mice or age-matched wild-type mice (Fig. 1b). By contrast, renal cortex Ccl2 mRNA expression levels significantly increased with the onset of type 2 diabetes and uni-nephrectomy in db/db mice (Fig. 1a), indicating that the homeostatic chemokine CXCL12 may have a different functional role to that of the proinflammatory chemokine CCL2 in kidney disease of db/db mice. CXCL12 immunostaining was positive in collecting ducts at the renal papilla, the uroepithelium along the renal pelvis and in endothelial and smooth muscle cells of intrarenal vessels (Fig. 2a). In the renal cortex, CXCL12 staining was also detected in glomeruli of 6-week-old db/db mice. At higher magnification, the glomerular staining was seen to originate from the cells on the outside of the glomerular capillaries, i.e. the visceral epithelial cells or podocytes (Fig. 2b, c). Co-staining with fluochrome-labelled antibodies against CXCL12 and WT-1 confirmed podocyte-specific CXCL12 production in glomeruli and ruled out CXCL12 production by mesangial cells or parietal epithelial cells (Fig. 3a–f). In 6-month-old uni-nephrectomised db/db mice with type 2 diabetes symptoms, the identical CXCL12 staining pattern and staining intensity were seen as compared with 6-week-old sham-operated db/db mice (Fig. 2c). Thus, podocytes are the major source of CXCL12 production in glomeruli of db/db mice.

Ccl2 (a) and Cxcl12 (b) mRNA expression in kidneys. Quantitative real-time RT-PCR analysis was performed on total cDNA derived from renal cortex of 6-week-old (grey bars) or 6-month-old (black bars) sham-operated (2K) db/db mice, 6-month-old db/db mice with one kidney (1K, hatched bars) or 6-month-old sham-operated (2K), non-diabetic, wild-type mice (white bars). cDNA was amplified using primers specific to murine Ccl2 and Cxcl12 mRNA for 40 PCR cycles and were corrected for the respective 18S rRNA expression. The data shown are means ± SEM. *p < 0.05 versus 6-week-old sham-operated (grey bars)

CXCL12 expression in kidneys of db/db mice. Paraffin-embedded renal sections from kidneys of 6-week-old sham-operated (2K) db/db and 6-month-old sham-operated C57BL/6 mice, and from 6-month-old db/db mice with one kidney (1K) were stained for CXCL12 and are shown at three different magnifications. a Renal medulla, papilla and pelvis (magnification ×10); (b) renal cortex (magnification ×100); and (c) single glomeruli (magnification ×400). Arrows indicate positive staining signals in visceral glomerular epithelia, i.e. podocytes

CXCL12 and WT-1 co-staining in kidney sections. Fluorescence microscopy of renal sections of 6-month-old db/db mice using a phycoerythrin (PE)-labelled antibody for WT-1 identified podocytes by red nuclear staining and a FITC-labelled anti-CXCL12 antibody identified CXCL12 expression by green cytoplasmatic staining (magnification ×400) (a). At higher magnification (×1,000) DAPI stained nuclei blue (b), of which only few represent WT-1-positive podocytes (red) (c, d). Co-staining for CXCL12 (green) (e) demonstrates that CXCL12 is present in the cytoplasm of podocytes with WT-1-positive nuclei (f)
CXCL12 blockade reduces diffuse glomerulosclerosis in db/db mice
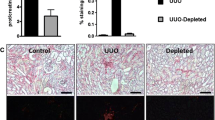
To test whether CXCL12 blockade affects glomerular pathology in db/db mice, we initiated subcutaneous injections with anti-CXCL12 or control Spiegelmer (50 mg/kg, every alternate day) at an age of 4 months in uni-nephrectomised db/db mice. Injections were continued for 8 weeks. Urine samples were collected every month and at the end of the treatment schedule tissues were harvested to assess the renal pathology. To assure Spiegelmer exposure and its biological activity, we determined plasma CXCL12 levels at the end of the study. Plasma CXCL12 levels remained undetectable in sham-operated db/db mice and in vehicle- or control Spiegelmer-treated db/db mice with one kidney. Anti-CXCL12 Spiegelmer injections increased the plasma levels of CXCL12 in db/db mice with one kidney (Fig. 4). This finding is consistent with our previous observations with other Spiegelmers and indicates that the CXCL12 antagonist retains CXCL12 in the circulation [11, 31]. Body weight and blood glucose levels did not differ among the injected db/db mice groups with one kidney (ESM Table 2). Renal histomorphology in 6-month-old sham-operated db/db mice showed moderate glomerulosclerosis as compared with age-matched wild-type mice; this progressed to diffuse glomerulosclerosis upon early uni-nephrectomy of db/db mice (Fig. 5). CXCL12 inhibition reduced the extent of glomerulosclerosis in uni-nephrectomised db/db mice to the level of age-matched sham-operated db/db mice (Fig. 5d). The control Spiegelmer had no effect. Thus, CXCL12 blockade started at 4 months of age reduces diffuse glomerulosclerosis in uni-nephrectomised db/db mice at 6 months of age.

Serum CXCL12 levels in Spiegelmer-treated db/db mice. Serum CXCL12 levels were determined in 6-month-old sham-operated (2K) and uni-nephrectomised (1K) db/db mice by ELISA. Data are means ± SEM. *p < 0.05 for difference between anti-CXCL12 Spiegelmer versus control Spiegelmer group

Renal pathology in 6-month-old sham-operated (2K) db/db mice or db/db mice with one kidney (1K). a Renal sections from mice of the different groups were stained with periodic acid–Schiff’s reagent. Stains show representative glomeruli from each group (magnification ×400). b Ki-67 immunostaining representative of mice from each group (original magnification ×400). c MECA32 immunostaining representative of mice from each group (original magnification ×100). d Periodic acid–Schiff’s stains were scored for extent of glomerulosclerosis as described in the Methods. From each mouse 15 glomeruli from each renal section were graded by that score. Values are percentage of each score as mean ± SEM from all mice in each group. White bar, score 0; light grey bar, score 1; medium grey bar, score 2; dark grey bar, score 3; black bar, score 4. Note that uni-nephrectomy was associated with a shift towards higher scores of glomerulosclerosis and that control Spiegelmer had no effect on glomerulosclerosis compared with vehicle-treated db/db mice with one kidney. *p < 0.05 for difference between anti-CXCL12 Spiegelmer vs control Spiegelmer-treated db/db mice with one kidney (1K)
CXCL12 blockade prevents proteinuria in db/db mice
In diabetic nephropathy glomerular pathology is usually associated with increasing levels of proteinuria indicative of progressive structural damage at the glomerular filtration barrier [32]. Thus, the beneficial effect of CXCL12 blockade on glomerular structure should be associated with less proteinuria. We quantified proteinuria in db/db mice by calculating albumin/creatinine ratios from urine samples taken at the second week of Spiegelmer injections and at the end of the study. Anti-CXCL12, but not the control Spiegelmer or vehicle prevented the progressive increase of urinary albumin/creatinine ratio in db/db mice (Fig. 6a). Proteinuria usually results from podocyte dysfunction or loss; hence, we determined the number of glomerular podocytes by counting WT-1-positive cells in 15 glomerular cross-sections per animal. CXCL12 blockade was associated with a significantly higher number of WT-1-positive podocytes (Fig. 6b, c) and some parietal epithelial cells in db/db mice with one kidney. Thus, CXCL12 inhibition improves the glomerular pathology and proteinuria associated with higher numbers of podocytes in db/db mice.

Proteinuria progression in db/db mice. a Proteinuria was determined after 2 and 8 weeks of Spiegelmer injections in vehicle-treated (black boxes), control Spiegelmer-treated (white boxes) and anti-CXCL12 Spiegelmer-treated (white triangles) uni-nephrectomised db/db mice. b The number of podocytes was quantified by counting WT-1-positive nuclei in glomeruli of db/db mice with one kidney at 6 months of age. Data represent means ± SEM. *p < 0.05 for difference between anti-CXCL12 Spiegelmer versus control Spiegelmer-treated animals. c Representative images of WT-1 stains, at original magnification of ×400
CXCL12 blockade does not affect infiltrating macrophages or proliferating cells in glomeruli of db/db mice with one kidney
Chemokine-mediated glomerular pathology in db/db mice can be mediated by macrophage recruitment [10, 11]. We therefore evaluated the number of glomerular macrophages by immunostaining for MAC2. MAC2-positive cells were virtually absent in glomeruli of sham-operated wild-type mice, but were increased in vehicle-treated db/db mice with one kidney (Fig. 7a). Neither control nor anti-Spiegelmer injections affected the number of MAC2-positive cells in glomeruli of db/db mice with one kidney (Fig. 7a). In addition, the numbers of Ki-67-positive proliferating cells were not affected in this compartment (Figs. 5b and 7c). Thus, blockade prevents glomerular pathology and dysfunction in db/db mice independently of infiltrating glomerular macrophages or glomerular cell proliferation.

Renal morphometry in 6-month-old mice. Renal sections from mice of all groups were stained for MAC2 (a), Ki-57 (c, d) and MECA32 (b). Values are shown as numbers of positive cells in 15 glomeruli (a, c) or as cells per ten high-power fields (hpf) (b, d) in sham-operated (2K) and uni-nephrectomised (1K) db/db mice at 6 months of age. e Morphometric analysis of renal sections was performed as described in methods. Values (a–e) represent means ± SEM. *p < 0.05 for difference between control Spiegelmer-treated and anti-CXCL12 Spiegelmer animals
CXCL12 blockade and tubulointerstitial pathology in db/db mice with one kidney
Advanced diabetic nephropathy is also associated with progressive tubulointerstitial injury. We quantified tubular dilatation, tubular cell damage and interstitial volume as markers of tubulointerstitial damage by blinded morphometry; we also quantified the numbers of Ki-67 proliferative tubular cells and MECA32-positive peritubular capillary cross-sections as markers of interstitial pathology in db/db mice at 6 months of age. In db/db mice with one kidney significant changes were observed in the numbers of Ki-67 proliferative tubular cells and MECA32-positive peritubular capillary cross-sections, but not in tubular dilatation, tubular cell damage and interstitial volume (Fig. 7b, e). As such, the histomorphological abnormalities of the tubulointerstitial compartment were rather mild in db/db mice with one kidney. However, CXCL12 blockade significantly increased the numbers of peritubular cross-sections and improved the tubular cell damage score, but not to the level of sham-operated wild-type mice (Figs. 5 and 7b, e). Control Spiegelmer was ineffective in this regard. CXCL12 blockade, however, did not significantly affect any of the other histomorphological variables of tubulointerstitial damage (Fig. 7e) such as the overall low number of interstital MAC2-positive macrophages as well as serum creatinine levels in 6-month-old db/db mice with one kidney (not shown). Thus, CXCL12 blockade significantly improves tubular damage and the peritubular vasculature density in db/db mice.
Discussion
The known functions of CXCL12 do not allow reliable prediction of its potential role in chronic glomerulopathies. In the present study we show that adult db/db mice constitutively produce CXCL12 in podocytes and that transient blockade of CXCL12 prevents the progression of glomerulosclerosis and proteinuria in animals with symptoms of type 2 diabetes. These data confirm the functional importance of podocytes in the progression of (diabetic) glomerular disease [33] and for the first time demonstrate a pathogenic role of podocyte-derived CXCL12 in (diabetic) glomerulosclerosis.
Our finding that adult C57BL/6 and db/db mouse kidneys produce CXCL12 selectively in podocytes and not in any of the other glomerular cell types is consistent with a previous report that described glomerular CXCL12 production in autoimmune-nephritic NZB/NZW F1 mice [28]. In contrast, podocytes did not stain positive for CXCL12 in another study using female SCID mice [34]. Podocyte CXCL12 staining was also absent in human renal biopsies from children with various inflammatory disease entities [35]. Glomerular CXCL12 staining was reported to localise to mesangial cells in healthy human kidneys [36]. However, in that study the histomorphological illustrations are more consistent with CXCL12 expression in podocytes than with that in mesangial cells. Furthermore, CXCL12 staining in adult human kidney shows prominent CXCL12 expression in podocytes only (S. Segerer, unpublished data). In db/db mice podocyte-specific expression of CXCL12 corresponds to the glomerular expression pattern of VEGF, another hypoxia-inducible factor-1-regulated proangiogenic factor with similar functional roles in angiogenesis and hypoxia control [37]. Podocyte-derived CXCL12 and VEGF both regulate glomerular capillary formation during renal development [38], further suggesting a role for CXCL12 in regulating glomerular structure by glomerular cell–cell interactions. Accordingly, outside the kidney, CXCL12 produced by distinct cells creates appropriate microenvironments for other cell types. For example, in the bone marrow, stromal or endothelial cell-derived CXCL12 creates a niche for haemopoetic stem cells [39–41]. The CXCL12-dependent mechanism creates the necessary microenvironment for tumour metastasis engraftment [42, 43], the homing of memory T cells to lymph nodes [44] or the specific recruitment of bone marrow cells that orchestrate angiogenesis [45]. We found that the progression of kidney disease in db/db mice did not increase renal Cxcl12 mRNA expression levels. Renal CXCL12 immunostaining, although not a reliable quantitative assessment of protein levels, also did not increase from young to older db/db mice. These expression data suggest that glomerular CXCL12 is needed to maintain the glomerular structure in db/db mice and that blocking CXCL12 might have detrimental effects. However, it was recently shown that adding an anti-CXCL12-specific antibody to low-ciclosporin treatment significantly reduced renal allograft glomerulosclerosis, compared with ciclosporin monotherapy in a Fischer 344 to Lewis rat chronic renal allograft nephropathy model [46]. Unfortunately, glomerular CXCL12 immunostaining was not reported in this study, but the authors referred to the anti-CXCL12-mediated improvement of allograft glomerulosclerosis to reduce arteriolar intimal thickening. Balabanian et al. documented podocyte CXCL12 expression in autoimmune-nephritic NZB/NZW F1 mice and also reported less glomerular disease and proteinuria upon CXCL12 blockade with a neutralising antibody [28]. This was mainly attributed to the effects of CXCL12 blockade on systemic autoimmunity and immune complex disease, which does not allow a safe conclusion on the local role of CXCL12 expression in glomerular pathology in this model. In our study, the model of type 2 diabetic nephropathy was independent of systemic allo- or autoimmunity and is thought to represent tissue remodelling as a consequence of hyperglycaemia and glomerular hyperfiltration [47]. Hence, the preventive effect of CXCL12 blockade on glomerulosclerosis in db/db mice should largely relate to the effects of blocking CXCL12 at the glomerular level. This would argue for a novel pathogenic role of podocyte-derived CXCL12 in (diabetic) glomerulosclerosis. This, again, would be consistent with the role of VEGF in rodent diabetic nephropathy, as VEGF is also produced by podocytes and acts locally on the glomerular vasculature, while VEGF blockade reduces glomerular pathology and proteinuria in experimental diabetic nephropathy [48]. Our study does not define the underlying maladaptive function of CXCL12, but we did find that CXCL12 blockade was associated with higher podocyte numbers. This could either relate to reduced podocyte death or increased podocyte regeneration. Hence, CXCL12 may either regulate podocyte cell cycle or podocyte regeneration from parietal epithelial cells [49, 50]. Confirmation of this would require a podocyte-specific knockout of CXCL12 in diabetic mice and, given the role of podocyte CXCL12 on renal development, this knockout would need to be conditional. Remarkably, CXCL12 blockade did not affect renal macrophage numbers. Examination of macrophage markers indicated a change in phenotype with increased expression of both M1 and M2 markers (data not shown), making it difficult to interpret whether this altered macrophage function affected disease progression.
In summary, here we show that adult db/db mice constitutively produce CXCL12 in podocytes and that transient blockade of CXCL12 prevents the progression of glomerulosclerosis and proteinuria in this mouse model of type 2 diabetes. These data demonstrate for the first time that CXCL12 plays pathogenic role in (diabetic) glomerulosclerosis, and we postulate that glomerular podocytes are an important source of this chemokine.
Abbreviations
- CCL2:
-
Chemokine (C-C motif) ligand 2
- CXCL12:
-
Chemokine (C-X-C motif) ligand 12
- CXCR:
-
Chemokine (C-X-C motif) receptor
- MECA32:
-
Mouse endothelial cell antigen 32
- VEGF:
-
Vascular endothelial growth factor
- WT:
-
Wilms tumour
References
Zimmet P, Alberti KG, Shaw J (2001) Global and societal implications of the diabetes epidemic. Nature 414:782–787
Ritz E, Rychlik I, Locatelli F, Halimi S (1999) End-stage renal failure in type 2 diabetes: a medical catastrophe of worldwide dimensions. Am J Kidney Dis 34:795–808
Svensson M, Sundkvist G, Arnqvist HJ et al (2003) Signs of nephropathy may occur early in young adults with diabetes despite modern diabetes management: results from the nationwide population-based Diabetes Incidence Study in Sweden (DISS). Diabetes Care 26:2903–2909
Qian Y, Feldman E, Pennathur S, Kretzler M, Brosius FC 3rd (2008) From fibrosis to sclerosis: mechanisms of glomerulosclerosis in diabetic nephropathy. Diabetes 57:1439–1445
Yamagishi S, Fukami K, Ueda S, Okuda S (2007) Molecular mechanisms of diabetic nephropathy and its therapeutic intervention. Curr Drug Targets 8:952–959
Ruster C, Wolf G (2008) The role of chemokines and chemokine receptors in diabetic nephropathy. Front Biosci 13:944–955
Galkina E, Ley K (2006) Leukocyte recruitment and vascular injury in diabetic nephropathy. J Am Soc Nephrol 17:368–377
Tesch GH (2008) MCP-1/CCL2: a new diagnostic marker and therapeutic target for progressive renal injury in diabetic nephropathy. Am J Physiol Renal Physiol 294:F697–F701
Anders HJ, Vielhauer V, Schlondorff D (2003) Chemokines and chemokine receptors are involved in the resolution or progression of renal disease. Kidney Int 63:401–415
Chow FY, Nikolic-Paterson DJ, Ma FY, Ozols E, Rollins BJ, Tesch GH (2007) Monocyte chemoattractant protein-1-induced tissue inflammation is critical for the development of renal injury but not type 2 diabetes in obese db/db mice. Diabetologia 50:471–480
Ninichuk V, Clauss S, Kulkarni O et al (2008) Late onset of Ccl2 blockade with the Spiegelmer mNOX-E36–3’PEG prevents glomerulosclerosis and improves glomerular filtration rate in db/db mice. Am J Pathol 172:628–637
Moser B, Wolf M, Walz A, Loetscher P (2004) Chemokines: multiple levels of leukocyte migration control. Trends Immunol 25:75–84
Bleul CC, Farzan M, Choe H et al (1996) The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 382:829–833
Burns JM, Summers BC, Wang Y et al (2006) A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med 203:2201–2213
Palm F (2006) Intrarenal oxygen in diabetes and a possible link to diabetic nephropathy. Clin Exp Pharmacol Physiol 33:997–1001
Freeburg PB, Abrahamson DR (2003) Hypoxia-inducible factors and kidney vascular development. J Am Soc Nephrol 14:2723–2730
Butler JM, Guthrie SM, Koc M et al (2005) SDF-1 is both necessary and sufficient to promote proliferative retinopathy. J Clin Invest 115:86–93
Ceradini DJ, Kulkarni AR, Callaghan MJ et al (2004) Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med 10:858–864
Schober A, Knarren S, Lietz M, Lin EA, Weber C (2003) Crucial role of stromal cell-derived factor-1alpha in neointima formation after vascular injury in apolipoprotein E-deficient mice. Circulation 108:2491–2497
Togel F, Isaac J, Hu Z, Weiss K, Westenfelder C (2005) Renal SDF-1 signals mobilization and homing of CXCR4-positive cells to the kidney after ischemic injury. Kidney Int 67:1772–1784
Yano T, Liu Z, Donovan J, Thomas MK, Habener JF (2007) Stromal cell derived factor-1 (SDF-1)/CXCL12 attenuates diabetes in mice and promotes pancreatic beta-cell survival by activation of the prosurvival kinase Akt. Diabetes 56:2946–2957
Jin DK, Shido K, Kopp HG et al (2006) Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med 12:557–567
Phillips RJ, Burdick MD, Hong K et al (2004) Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest 114:438–446
Xu J, Mora A, Shim H, Stecenko A, Brigham KL, Rojas M (2007) Role of the SDF-1/CXCR4 axis in the pathogenesis of lung injury and fibrosis. Am J Respir Cell Mol Biol 37:291–299
Zagzag D, Krishnamachary B, Yee H et al (2005) Stromal cell-derived factor-1alpha and CXCR4 expression in hemangioblastoma and clear cell-renal cell carcinoma: von Hippel-Lindau loss-of-function induces expression of a ligand and its receptor. Cancer Res 65:6178–6188
Burger JA, Kipps TJ (2006) CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood 107:1761–1767
Ding M, Cui S, Li C et al (2006) Loss of the tumor suppressor Vhlh leads to upregulation of Cxcr4 and rapidly progressive glomerulonephritis in mice. Nat Med 12:1081–1087
Balabanian K, Couderc J, Bouchet-Delbos L et al (2003) Role of the chemokine stromal cell-derived factor 1 in autoantibody production and nephritis in murine lupus. J Immunol 170:3392–3400
Ninichuk V, Khandoga AG, Segerer S et al (2007) The role of interstitial macrophages in nephropathy of type 2 diabetic db/db mice. Am J Pathol 170:1267–1276
Patole PS, Schubert S, Hildinger K et al (2005) Toll-like receptor-4: renal cells and bone marrow cells signal for neutrophil recruitment during pyelonephritis. Kidney Int 68:2582–2587
Kulkarni O, Pawar RD, Purschke W et al (2007) Spiegelmer inhibition of CCL2/MCP-1 ameliorates lupus nephritis in MRL-(Fas)lpr mice. J Am Soc Nephrol 18:2350–2358
Adler AI, Stevens RJ, Manley SE, Bilous RW, Cull CA, Holman RR (2003) Development and progression of nephropathy in type 2 diabetes: the United Kingdom Prospective Diabetes Study (UKPDS 64). Kidney Int 63:225–232
Li JJ, Kwak SJ, Jung DS et al (2007) Podocyte biology in diabetic nephropathy. Kidney Int Suppl:S36–S42
Mazzinghi B, Ronconi E, Lazzeri E et al (2008) Essential but differential role for CXCR4 and CXCR7 in the therapeutic homing of human renal progenitor cells. J Exp Med 205:479–490
Lotan D, Sheinberg N, Kopolovic J, Dekel B (2008) Expression of SDF-1/CXCR4 in injured human kidneys. Pediatr Nephrol 23:71–77
Hoffmann U, Banas B, Kruger B et al (2006) SDF-1 expression is elevated in chronic human renal allograft rejection. Clin Transplant 20:712–718
Hitchon C, Wong K, Ma G, Reed J, Lyttle D, El-Gabalawy H (2002) Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis Rheum 46:2587–2597
Eremina V, Quaggin SE (2004) The role of VEGF-A in glomerular development and function. Curr Opin Nephrol Hypertens 13:9–15
Peled A, Petit I, Kollet O et al (1999) Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science 283:845–848
Avecilla ST, Hattori K, Heissig B et al (2004) Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat Med 10:64–71
Sugiyama T, Kohara H, Noda M, Nagasawa T (2006) Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 25:977–988
Sipkins DA, Wei X, Wu JW et al (2005) In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 435:969–973
Bartolome RA, Ferreiro S, Miquilena-Colina ME et al (2009) The chemokine receptor CXCR4 and the metalloproteinase MT1-MMP are mutually required during melanoma metastasis to lungs. Am J Pathol 174:602–612
Scimone ML, Felbinger TW, Mazo IB, Stein JV, von Andrian UH, Weninger W (2004) CXCL12 mediates CCR7-independent homing of central memory cells, but not naive T cells, in peripheral lymph nodes. J Exp Med 199:1113–1120
Grunewald M, Avraham I, Dor Y et al (2006) VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell 124:175–189
Gao C, Huan J (2008) SDF-1 plays a key role in chronic allograft nephropathy in rats. Transplant Proc 40:1674–1678
Breyer MD, Bottinger E, Brosius FC 3rd et al (2005) Mouse models of diabetic nephropathy. J Am Soc Nephrol 16:27–45
Wolf G, Chen S, Ziyadeh FN (2005) From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes 54:1626–1634
Appel D, Kershaw DB, Smeets B et al (2009) Recruitment of podocytes from glomerular parietal epithelial cells. J Am Soc Nephrol 20:333–343
Ronconi E, Sagrinati C, Angelotti ML et al (2009) Regeneration of glomerular podocytes by human renal progenitors. J Am Soc Nephrol 20:322–332
Note added in proof
The role of CXCL12 and CXCR4 in renal development has now been demonstrated by Takabatake et al. in the following article: Takabatake Y, Sugiyama T, Kohara H et al. (2009). The CXCL12 (SDF-1)/CXCR4 axis is essential for the development of renal vasculature. J Am Soc Nephrol 20:1714–1723.
Acknowledgements
Parts of this project were prepared as a doctoral thesis at the Faculty of Medicine, University of Munich by S. G. Sayyed. The expert technical support of D. Draganovici, J. Mandelbaum and E. Radomska, University of Munich, is gratefully acknowledged. We thank S. Vonhoff and the chemistry group at NOXXON Pharma, Berlin, for providing the oligonucleotides. The study was partially funded by NOXXON Pharma. H.-J. Anders was funded by the EU Integrated Project INNOCHEM (FP6-518167) and the Else Kröner-Fresenius Stiftung. H. Hägele was funded by the Faculty of Medicine, University of Munich (FöFoLe thesis programme). S. Segerer was supported by the Hartmann Müller Stiftung.
Duality of interest
D. Eulberg and S. Klussmann are employees of NOXXON Pharma. All other authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
S. G. Sayyed and H. Hägele contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
Primer sequences used for RT-PCR (PDF 8.87 kb)
ESM Table 2
Body weight and blood glucose levels in db/db mice at 0 to 8 weeks of treatment (PDF 13.6 kb)
Rights and permissions
About this article
Cite this article
Sayyed, S.G., Hägele, H., Kulkarni, O.P. et al. Podocytes produce homeostatic chemokine stromal cell-derived factor-1/CXCL12, which contributes to glomerulosclerosis, podocyte loss and albuminuria in a mouse model of type 2 diabetes. Diabetologia 52, 2445–2454 (2009). https://doi.org/10.1007/s00125-009-1493-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-009-1493-6