
Abstract
Aims/hypothesis
Ablation of gastric inhibitory polypeptide (GIP) receptor action is reported to protect against obesity and associated metabolic abnormalities. The aim of this study was to use prediabetic ob/ob mice to examine whether 60 days of chemical GIP receptor ablation with (Pro3)GIP is able to counter the development of genetic obesity-related diabetes.
Materials and methods
Young (5–7 weeks) ob/ob mice received once daily i.p. injections of either saline vehicle or (Pro3)GIP (25 nmol kg−1 day−1) over a 60 day period. Food intake, body weight and circulating glucose and insulin were measured at frequent intervals. At 60 days, glucose tolerance, response to native GIP, postprandial responses, insulin sensitivity, HbA1c, circulating hormones and plasma lipids were assessed.
Results
Body weight and food intake in (Pro3)GIP-treated mice did not differ from ob/ob controls. GIP receptor blockade significantly improved non-fasting glucose (p < 0.001), HbA1c (p < 0.05), glucose tolerance (p < 0.001), meal tolerance (p < 0.001) and insulin sensitivity (p < 0.05). Remarkably, (Pro3)GIP treatment prevented the age-related development of diabetes, as none of these parameters differed significantly between treated ob/ob mice and normal age-matched lean controls. Circulating levels of glucagon, corticosterone, adiponectin and total cholesterol were unchanged by (Pro3)GIP, while levels of triacylglycerol, LDL-cholesterol and resistin were decreased (p < 0.05) compared with those in control ob/ob mice. Plasma and pancreatic insulin concentrations were generally lower after (Pro3)GIP treatment than in control ob/ob mice (p < 0.01), but plasma insulin levels remained substantially raised (p < 0.001) compared with those observed in lean controls.
Conclusions/interpretation
These data indicate that sustained GIP receptor antagonism provides an effective means of preventing the development of many of the metabolic abnormalities of obesity-driven diabetes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The most widely accepted role of gastric inhibitory polypeptide (GIP), also known as glucose-dependent insulinotropic polypeptide, is potentiation of glucose-induced insulin secretion from pancreatic beta cells [1]. GIP is secreted from intestinal K cells in response to feeding and, together with glucagon-like peptide 1 (GLP-1), accounts for the full incretin effect [2]. GIP and GLP-1 exert these effects through binding to specific beta cell receptors, activating adenylate cyclase and other signal recognition pathways, leading to insulin exocytosis [3]. GIP and GLP-1 have also been reported to stimulate beta cell neogenesis, differentiation and proliferation whilst affording protection from cytokine attack and apoptosis [4–6]. These various attributes, plus reports of GLP-1-stimulated decreases of gastric emptying, glucagon secretion and appetite [7], have focused much attention on the potential of incretin hormones for the treatment of type 2 diabetes [8, 9].
Both GLP-1 and GIP are rapidly degraded in the circulation by the ubiquitous enzyme dipeptidyl-peptidase IV (DPPIV) [10], but problems of short half-life and pharmacokinetics have been overcome by the development of peptide mimetics possessing N-terminal modifications that make them resistant to DPPIV but able to activate incretin hormone receptors [8, 9, 11]. One such GLP-1 mimetic, exendin-4, has been approved for clinical use in the USA under the tradename Byetta (Amylin Pharmaceuticals, San Diego, CA), and a second palmitoylated derivative of GLP-1, liraglutide, is in late clinical development [6]. Similar exploitation of GIP is at the late preclinical stage, complicated to some extent by the debate that GIP but not GLP-1 action is specifically impaired in type 2 diabetes [12, 13]. This insensitivity appears most pronounced in response to continuous infusion rather than bolus injection of native GIP [14], possibly implicating receptor desensitisation. However, it is now accepted that there is no specific molecular defect of the GIP receptor in established diabetes, first-degree relatives or gestational diabetes [15–17]. Recognition that GLP-1 action is also impaired, to a lesser extent than GIP action, in type 2 diabetes [18], suggests that the diminished insulin response is linked to generalised beta cell secretory dysfunction, presumably stemming from beta cell glucose insensitivity [19].
Although further work is required to determine whether DPPIV-resistant GIP molecules can overcome beta cell insensitivity and eventually make it to the clinic, possibly in combination with GLP-1 mimetics to activate both arms of the enteroinsular axis, other less well-recognised actions of GIP may afford other unexpected therapeutic opportunities [11]. The GIP receptor is expressed in various extrapancreatic sites, including adipose tissue, bone, intestine, heart, stomach and brain [20]. In relation to this, GIP has been shown to inhibit gastric acid secretion [21], attenuate glucagon-stimulated glucose production [22], decrease hepatic insulin extraction [23] stimulate glucose uptake in muscle [24] and increase both fatty acid synthesis and lipoprotein lipase activity in adipocytes [25, 26]. The particularly potent stimulation of GIP secretion after high-fat feeding [27] suggests a role for GIP in fat metabolism [20]. Consistent with this, plasma GIP concentrations have been reported to be elevated in obesity-related type 2 diabetes [28] and animals with obesity-related diabetes [29]. Experimental evidence in mice also indicates that inhibition of GIP signalling by genetic knockout of the GIP receptor prevents diet-induced obesity through the preferential use of fat as an energy substrate [30]. Furthermore, it has been shown that short-term administration of the specific and stable GIP receptor antagonist (Pro3)GIP [31] can reverse many of the established metabolic abnormalities in adult ob/ob mice [32].
In the present study we have employed young prediabetic ob/ob mice to determine whether prolonged 60 day chemical GIP receptor ablation using daily injections of (Pro3)GIP is able to counter the development and progression of obesity-related diabetes in this commonly employed animal model. The results obtained indicate that sustained GIP receptor antagonism provides an effective means of preventing the development of many of the metabolic abnormalities of obesity-driven forms of diabetes.
Materials and methods
Animals Equal numbers of young male and female obese diabetic (ob/ob) mice derived from the colony maintained at Aston University (Birmingham, UK) [33] were used at 5–7 weeks of age. Animals were age-matched, divided into groups and housed individually in an air-conditioned room at 22 ± 2°C with a 12 h light:12 h dark cycle (08:00–20:00 h). Age-matched, normal, lean control mice from the same colony were used for comparative purposes as appropriate. The effects of repeated daily injection of (Pro3)GIP in these lean control mice has previously been shown to result in mild impairment of glucose homeostasis [34]. Drinking water and a standard rodent maintenance diet (Trouw Nutrition, Cheshire, UK) were freely available. All animal experiments were carried out in accordance with the UK Animals (Scientific Procedures) Act 1986.
Synthesis, purification and characterisation of (Pro3)GIP
(Pro3)GIP was sequentially synthesised on an Applied Biosystems (Foster City, CA) automated peptide synthesiser (Model 432 A), as reported previously [31]. (Pro3)GIP was purified by reversed-phase HPLC on a Waters Millenium 2010 chromatography system (Software version 2.1.5) and subsequently characterised using electrospray ionisation mass spectrometry (ESI-MS), as described elsewhere [31].
Experimental protocols for in vivo studies
Over a 60 day period, the ob/ob mice received once daily i.p. injections (17:00 h) of either saline vehicle (0.9% [w/v] NaCl) or (Pro3)GIP (25 nmol/kg body weight). This dose of (Pro3)GIP was chosen on the basis of earlier studies showing blockade of glucoregulatory effects of exogenous and endogenous GIP in ob/ob mice [35, 36]. The once daily dosing regimen has also proved effective in longer-term studies [32, 34]. Food intake and body weight were recorded daily, whereas plasma glucose and insulin concentrations were monitored at intervals of 3–7 days. Blood for HbA1c and plasma for measurement of cholesterol, triacylglycerol, glucagon, corticosterone and circulating adipokines were taken on day 60. Intraperitoneal glucose tolerance (18 mmol/kg body weight), metabolic response to native GIP (25 nmol/kg body weight) and insulin sensitivity (50 U/kg body weight) tests were performed at the end of the study period. Mice fasted for 18 h were used to examine the metabolic response to 15 min feeding on day 60. All acute experiments commenced at 10:00 h. In a separate series, pancreatic tissues were excised at the end of the 60 day treatment for measurement of insulin content following extraction with 5 ml/g of ice-cold acid ethanol (750 ml ethanol, 235 ml water, 15 ml concentrated HCl). Blood samples taken from the cut tip of the tail vein of conscious mice at the times indicated in the figures were immediately centrifuged using a Beckman microcentrifuge (Beckman Instruments, Galway, Ireland) for 30 s at 13,000×g. The resulting plasma was then aliquoted into fresh Eppendorf tubes and stored at −20°C prior to analysis. Where indicated, identical experiments were performed using age-matched, normal, lean control mice. Non-fasting plasma glucose levels in the ob/ob mice were 10.9 ± 0.8 vs 7.2 ± 0.5 mmol/l in the lean controls at the beginning of the experiment (p < 0.01).
Biochemical analysis
Plasma glucose was assayed by an automated glucose oxidase procedure [31] using a Beckman Glucose Analyzer II (Beckman Instruments). Plasma and pancreatic insulin were assayed by a modified dextran-coated charcoal radioimmunoassay [37]. Plasma triacylglycerol and cholesterol levels were measured using a Hitachi Automatic Analyser 912 (Boehringer Mannheim, Mannheim, Germany). HbA1c was determined using a kit purchased from Chirus (Watford, UK). Glucagon, adiponectin and resistin were measured using radioimmunoassay kits from Linco Research (St Charles, MI, USA). Corticosterone was measured similarly using a kit from MP Biomedicals (Heidelberg, Germany). All analyses were carried out according to the manufacturers’ instructions.
Statistical analyses
Results are expressed as means±SEM. Data were compared using ANOVA, followed by a Student–Newman–Keuls post-hoc test. AUC analyses were calculated using the trapezoidal rule with baseline subtraction [31]. LDL-cholesterol was calculated using the Friedewald Equation [38]. Groups of data were considered to be significantly different if the p value was less than 0.05.
Results
Effects of (Pro3)GIP on food intake, body weight, non-fasting plasma glucose and insulin and HbA1c concentrations
The administration of (Pro3)GIP for 60 days had no effect on the food intake of ob/ob mice, and a pattern towards decreased food consumption was observed as the age-related hyperphagia declined (Fig. 1a). The food intake of ob/ob mice (Fig. 1) was consistently greater than that of lean controls (5.4 ± 0.3 g mouse−1 day−1) (p < 0.001). At the start of the study the body weight of the ob/ob mice was 42.3 ± 2.5 g compared with 36.8 ± 1.2 g for lean controls (p = 0.067). Although there was a general trend for reduced body weight gain in ob/ob mice treated with (Pro3)GIP, the difference between the two ob/ob groups, corresponding to 8.4% of the body weight of saline controls, failed to reach significance (p = 0.072) over the study period (Fig. 1b). The corresponding weight gain of lean controls over this period was minimal (2.5 ± 0.1 g). The plasma glucose concentrations of (Pro3)GIP-treated ob/ob mice remained stable throughout the study and were similar to those of lean controls at 60 days (Fig. 2a,c). In comparison, untreated ob/ob mice exhibited hyperglycaemia, and glucose concentrations were significantly raised in these mice compared with (Pro3)GIP-treated mice, from day 14 onwards. Consistent with this pattern, HbA1c was only significantly elevated (p < 0.05) in the untreated ob/ob group at 60 days (Fig 2d). Plasma insulin had a tendency to be lower with (Pro3)GIP treatment, and on day 44 was significantly decreased (p < 0.05) compared with the levels observed control ob/ob mice (Fig. 2b). However, both groups exhibited quite marked hyperinsulinaemia (p < 0.001) compared with lean mice (Fig. 2e).

Effects of daily (Pro3)GIP administration on food intake (a) and body weight (b) of ob/ob mice. Parameters were measured for 5 days prior to and for 60 days during (indicated by black bar) treatment with saline (squares) or (Pro3)GIP (25 nmol kg−1 day−1; triangles). Food intake and body weight gain of normal lean control mice were 5.4 ± 0.3 and 2.5 ± 0.1 g, respectively. Values are means±SEM for groups of 7–8 mice

a, b Effects of daily (Pro3)GIP administration on non-fasting glucose profile (a) and insulin profile (b) of ob/ob mice. Parameters were measured for 5 days prior to and for 60 days during treatment with saline (squares) or (Pro3)GIP (25 nmol kg−1 day−1; triangles). c–e Final 60 day measures of glucose (c), HbA1c (d) and insulin (e) in (Pro3)GIP-treated ob/ob mice (black bars), saline-treated ob/ob mice (white bars) and untreated lean control mice (hatched bars). HbA1c concentrations were assessed on day 60. Values are means±SEM for groups of 7–8 mice. *p < 0.05, **p < 0.01, ***p < 0.001 vs saline-treated ob/ob mice; ††† p < 0.001 vs (Pro3)GIP-treated ob/ob mice
Effects of (Pro3)GIP on intraperitoneal glucose tolerance and response to native GIP
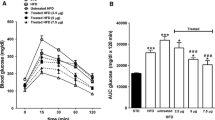
Daily administration of (Pro3)GIP for 60 days resulted in significantly reduced (p < 0.001) plasma glucose concentrations at 0, 15, 30 and 60 min as compared with untreated ob/ob mice following i.p. glucose (Fig. 3a). This was corroborated by a significantly (p < 0.001) decreased 0–60 min AUC value (Fig. 3c). Indeed, the i.p. glucose tolerance of (Pro3)GIP-treated ob/ob mice at 60 days was similar to that of normal lean mice. As shown in Fig. 3b, corresponding plasma insulin concentrations were also significantly reduced (p < 0.05) in the (Pro3)GIP-treated group compared with the untreated ob/ob group, as was the 0–60 min AUC (Fig. 3d). However, insulin levels in the two groups were considerably greater (p < 0.001) than those observed in normal lean mice. Interestingly, a similar pattern was observed when ob/ob mice treated for 60 days were administered glucose together with native GIP (25 nmol/kg body weight), consistent with the antagonism of the action of GIP by extended (Pro3)GIP administration (data not shown).

Effects of daily (Pro3)GIP administration on glucose tolerance (a) and plasma insulin (b) response to glucose in (Pro3)GIP-treated ob/ob mice (triangles), saline-treated ob/ob mice (squares) and untreated lean control mice (diamonds). Tests were conducted after daily treatment for 60 days. Glucose (18 mmol/kg body weight) was administered at the time indicated by the arrow. Plasma glucose (c) and insulin AUC values (d) for 0–60 min post-injection are also shown for the (Pro3)GIP-treated ob/ob mice (black bars), saline-treated ob/ob mice (white bars) and untreated lean control mice (hatched bars). Values are means±SEM for groups of eight mice. *p < 0.05, **p < 0.01, ***p < 0.001 vs saline-treated ob/ob mice; ††† p < 0.001 vs (Pro3)GIP-treated ob/ob mice
Effects of (Pro3)GIP on metabolic response to feeding, insulin sensitivity and pancreatic insulin content
The plasma glucose response to 15 min feeding was significantly lowered at 15, 30, 60 and 105 min (p < 0.05 to p < 0.001) in ob/ob mice treated for 60 days with (Pro3)GIP compared with control ob/ob mice. This translated into a significantly (p < 0.001) decreased overall glycaemic excursion, despite similar food intakes (0.4–0.6 g mouse−1 15 min−1). Plasma insulin levels were not significantly different between the two groups of ob/ob mice. However, the glycaemic and insulinotropic responses of both groups were significantly different from those of normal lean mice (data not shown).
As shown in Fig. 4a, insulin sensitivity was significantly (p < 0.05) improved in ob/ob mice in terms of AUC measures and post-injection values following treatment with (Pro3)GIP for 60 days. The hypoglycaemic action of this dose of insulin was similar in (Pro3)GIP-treated ob/ob mice and normal lean controls (Fig. 4a). Compared with untreated ob/ob mice, (Pro3)GIP-treated ob/ob mice had a significantly decreased (p < 0.01) pancreatic insulin content, with values similar to normal lean mice at 60 days (Fig. 4b). Pancreatic weight was similar in all three groups.

a Effects of daily (Pro3)GIP administration on plasma glucose in (Pro3)GIP-treated ob/ob mice (25 nmol kg−1 day−1, triangles), saline-treated ob/ob mice (squares) and untreated lean control mice (diamonds). Insulin (50 U/kg body weight) was administered by intraperitoneal injection at the time indicated by the arrow. b Plasma glucose AUC values. c, d Pancreatic weight (c) and insulin content (d) in (Pro3)GIP-treated ob/ob mice (black bars), saline-treated ob/ob mice (white bars) and untreated lean control mice (hatched bars). Tests were conducted after daily treatment for 60 days. Values are means±SEM for groups of 7–8 mice. *p < 0.05, **p < 0.01 vs saline-treated ob/ob mice; † p < 0.05 vs (Pro3)GIP-treated ob/ob mice
Effects of (Pro3)GIP on circulating triacylglycerol and cholesterol
As shown in Fig. 5a, in ob/ob mice, (Pro3)GIP treatment for 60 days significantly reduced (p < 0.05) circulating triacylglycerol concentrations. Total cholesterol levels were not significantly different between the two groups of ob/ob mice (Fig 5b). More detailed assessment revealed significantly reduced (p < 0.05) levels of LDL-cholesterol in (Pro3)GIP-treated ob/ob mice (Fig 5d). However, concentrations of the various lipids in the two groups were consistently greater than those observed in normal lean mice.

Effects of daily (Pro3)GIP administration on lipid profile in (Pro3)GIP-treated ob/ob mice (black bars), saline-treated ob/ob mice (white bars) and untreated lean control mice (hatched bars). Parameters were measured after daily treatment with (Pro3)GIP (25 nmol/kg body weight) or saline for 60 days. Values are means±SEM for groups of 7–8 mice. *p < 0.05 vs saline-treated ob/ob mice; †† p < 0.01, ††† p < 0.001 vs (Pro3)GIP-treated ob/ob mice
Effects of (Pro3)GIP on circulating glucagon, corticosterone and adipokines
Once daily injection of ob/ob mice with (Pro3)GIP did not significantly affect circulating glucagon or corticosterone concentrations (Fig. 6a and b). Plasma adiponectin was also unchanged, but a small decrease (p < 0.05) of circulating resistin was observed following treatment with (Pro3)GIP (Fig. 6c and d). Concentrations of glucagon and corticosterone in both groups of ob/ob mice were substantially raised (p < 0.001) compared with those in normal lean mice.

Effects of daily (Pro3)GIP administration on circulating glucagon (a), corticosterone (b), adiponectin (c) and resistin (d) in (Pro3)GIP-treated ob/ob mice (black bars), saline-treated ob/ob mice (white bars) and untreated lean control mice (hatched bars). Parameters were measured after daily treatment with (Pro3)GIP (25 nmol/kg) or saline for 60 days. Values are means±SEM for groups of 7–8 mice. *p < 0.05, ***p < 0.001 vs saline-treated ob/ob mice; ††† p < 0.001 vs (Pro3)GIP-treated ob/ob mice
Discussion
It has been speculated for decades that GIP plays a role in the aetiology of obesity [39]. More recently it has been shown that obese diabetic (ob/ob) mice have intestinal K cell hyperplasia, markedly elevated concentrations of intestinal (2.8-fold increase) and circulating GIP (15.1-fold increase) [29, 40, 41] and display a diminished insulinotropic response to native GIP [11]. Additionally, ob/ob mice genetically manipulated to knock out GIP receptor function displayed significant amelioration of adiposity [30]. These key observations, together with increasing awareness of the extrapancreatic actions of GIP, such as effects on lipid metabolism, have led to speculation that GIP receptor antagonism may offer a novel and potentially useful therapeutic approach to the treatment of obesity and associated metabolic abnormalities [11, 35].
In the present study, (Pro3)GIP, a specific and stable GIP receptor antagonist [31], was utilised to assess whether chemical ablation of GIP signalling for 60 days in young prediabetic ob/ob mice could prevent the development of diabetes and associated metabolic features of the obesity-driven syndrome. The ob/ob mouse model represents an extreme phenotype, being noted for marked hyperphagia, gross obesity and particularly severe insulin resistance, driven by leptin deficiency [42]. As such, the ob/ob model represents an appropriate but strong challenge to potential antihyperglycaemic agents. Consistent with other observations [30, 32, 34], GIP blockade had no effect on food intake, and mice continued to thrive throughout the study period. Obesity was already well established in the ob/ob mice, which were 5–7 weeks of age at commencement of the study, and body weights increased progressively compared with lean controls. In contrast to the results of Miyawaki et al., obtained in a study that used GIP receptor knockout mice [30], interference of GIP signalling by daily injections of (Pro3)GIP did not significantly arrest the development of obesity as assessed by simple body weight or body weight gain measurements. Perhaps such a comparison is unfair given the narrow window of the present treatment compared with an 8 month study in ob/ob mice with lifelong knockout of the GIP receptor. Nevertheless, (Pro3)GIP did show an obvious and progressive trend towards decreased body weights over the present 60 day study. Further research is required to clarify such an effect, possibly involving more protracted observations with larger experimental groups and exploitation of dual-energy X-ray absorptiometry (DEXA) to accurately measure body fat mass. Notably, similar (Pro3)GIP treatment of mice fed high-fat and cafeteria diets resulted in significant decreases in body weight, tissue fat deposition and adipocyte size (unpublished results). Initiation of treatment in preweanling ob/ob mice before adipocyte proliferation, or consumption of a high-fat diet as opposed to the present carbohydrate-rich diet are also potentially interesting avenues for exploration.
Despite the absence of significant effects on the progression of obesity, the early administration of (Pro3)GIP to prediabetic ob/ob mice had many notable effects on metabolic features of the obesity-related diabetes syndrome. The GIP receptor antagonist significantly improved non-fasting glucose, HbA1c, i.p. glucose tolerance, meal tolerance and insulin sensitivity in ob/ob mice. Similar actions have also been noted with 11 days of (Pro3)GIP treatment in adult ob/ob mice [32]. However, more remarkably, GIP receptor blockade prevented the age-related development of diabetes in the present study, and none of the above mentioned parameters differed significantly between (Pro3)GIP-treated ob/ob mice and normal age-matched lean control mice. Furthermore, the present observations suggest that GIP antagonism may be particularly effective therapeutically in the early stages of diabetes.
Although (Pro3)GIP-treated ob/ob mice did not develop diabetes as judged against relevant parameters in normal lean mice, chemical GIP receptor blockade did not prevent the emergence of other characteristic features of the ob/ob syndrome. Plasma and pancreatic insulin concentrations were lower in treated than in untreated ob/ob mice, but plasma insulin concentrations were still substantially raised compared with those in lean control mice. This suggests the persistence of abnormalities in counter-regulatory mechanisms, such as those manifested by the observed elevations of glucagon and corticosterone, which were offset by moderate levels of beta cell hyperactivity. For example, although there is no doubt that insulin sensitivity was greatly improved by (Pro3)GIP administration, it is unlikely to have been totally normalised in ob/ob mice as judged by simple tests based on hypoglycaemic activity of injected insulin. Circulating adipokines were not appreciably affected by (Pro3)GIP, with only a small possibly beneficial decrease in resistin observed. This does not indicate a significant GIPergic adipokine generation in ob/ob mice and questions their recently postulated role in alleviating insulin sensitivity in GIP receptor knockout mice [43]. The abnormal lipid profile also persisted in ob/ob mice treated with (Pro3)GIP, but there were significant improvements in triacylglycerol and LDL-cholesterol. The former is in accordance with the reported elevation of GIP concentrations in hypertriglyceridaemic subjects [44].
Given that GIP is classically recognised as an important insulin-releasing hormone and a major component of the enteroinsular axis [2], the beneficial antihyperglycaemic actions of GIP receptor blockade reported here and elsewhere [30, 32] are quite notable. In normal mice, genetic knockout or chemical antagonism of the GIP receptor clearly results in mild impairments of glucose homeostasis and insulin secretion [34, 45]. Similar effects were produced by acute as opposed to longer-term administration of (Pro3)GIP to adult ob/ob mice [36]. Viewed in simple terms, ablation of GIP receptor function in normal mice resulted in modest detrimental deficits in insulin release, as also observed when ob/ob mice were administered (Pro3)GIP acutely. However, in situations of obesity and insulin resistance, as exemplified by ob/ob mice, longer-term GIP receptor blockade resulted in significant amelioration of diabetes and associated metabolic features [30, 32]. Thus, substantial differences exist between GIP actions in obesity as opposed to normal physiology, emphasising an important pathophysiological role of GIP. Adipocyte GIP receptors may be especially important, as GIP has been shown to promote fatty acid uptake and lipogenesis [25, 26]. Since ob/ob mice exhibit leptin deficiency and low sympathetic activity [42], we cannot rule out additional effects, such as increased sympathetic activity and fatty acid oxidation. However, it is important to note that similar metabolic actions of (Pro3)GIP were observed in mice fed high-fat and cafeteria diets to induce obesity and insulin resistance (unpublished results). These various observations suggest that changes in fat deposition in tissues that influence insulin sensitivity, such as liver and muscle, may be particularly important.
Although the above conclusions are necessarily based on experimental studies in animal models, there are strong parallels with recent studies in humans undergoing surgical bypass for treatment of gross obesity and associated diabetes [46–49]. The similarity with our results of GIP receptor blockage in ob/ob mice suggest that GIP is key in this type of surgery and links obesity to insulin resistance and diabetes in man. Accordingly, antagonism of GIP action appears to offer considerable promise for the development of a new and effective therapeutic approach to obesity-related diabetes through alleviation of insulin resistance.
Abbreviations
- DPPIV:
-
dipeptidyl-peptidase IV
- GIP:
-
gastric inhibitory polypeptide
- GLP-1:
-
glucagon-like peptide 1
References
Pederson RA, Brown JC (1976) The insulinotropic action of gastric inhibitory polypeptide in the perfused isolated rat pancreas. Endocrinology 99:780–785
Creutzfeldt W (2001) The entero-insular axis in type 2 diabetes-incretins as therapeutic agents. Exp Clin Endocrinol Diabetes 109:S288–S303
Holst JJ (2004) On the physiology of GIP and GLP-1. Horm Metab Res 36:747–754
Trumper A, Trumper K, Horsch D (2002) Mechanisms of mitogenic and anti-apoptotic signaling by glucose-dependent insulinotropic polypeptide in beta(INS-1)-cells. J Endocrinol 174:233–246
Ehses JA, Casilla VR, Doty T et al (2003) Glucose-dependent insulinotropic polypeptide promotes beta-(INS-1) cell survival via cyclic adenosine monophosphate-mediated caspase-3 inhibition and regulation of p38 mitogen-activated protein kinase. Endocrinology 144:4433–4445
Baggio LL, Drucker DJ (2006) Therapeutic approaches to preserve islet mass in type 2 diabetes. Annu Rev Med 57:265–281
Schirra J, Wank U, Arnold R, Goke B, Katschinski M (2002) Effects of glucagon-like peptide-1(7–36)amide on motility and sensation of the proximal stomach in humans. Gut 50:341–348
Green BD, Gault VA, O’Harte FP, Flatt PR (2004) Structurally modified analogues of glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) as future antidiabetic agents. Curr Pharm Des 10:3651–3662
Drucker DJ (2003) Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care 26:2929–2940
Deacon CF (2004) Circulation and degradation of GIP and GLP-1. Horm Metab Res 36:761–765
Gault VA, O’Harte FP, Flatt PR (2003) Glucose-dependent insulinotropic polypeptide (GIP): anti-diabetic and anti-obesity potential? Neuropeptides 37:253–263
Holst JJ, Gromada J (2004) Role of incretin hormones in the regulation of insulin secretion in diabetic and nondiabetic humans. Am J Physiol Endocrinol Metab 287:E199–E206
Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W (1993) Preserved incretin activity of glucagon-like peptide 1 [7–36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest 91:301–307
Meier JJ, Gallwitz B, Kask B et al (2004) Stimulation of insulin secretion by intravenous bolus injection and continuous infusion of gastric inhibitory polypeptide in patients with type 2 diabetes and healthy control subjects. Diabetes 53:S220–S224
Meier JJ, Nauck MA, Siepmann N et al (2003) Similar insulin secretory response to a gastric inhibitory polypeptide bolus injection at euglycemia in first-degree relatives of patients with type 2 diabetes and control subjects. Metabolism 52:1579–1585
Meier JJ, Gallwitz B, Askenas M et al (2005) Secretion of incretin hormones and the insulinotropic effect of gastric inhibitory polypeptide in women with a history of gestational diabetes. Diabetologia 48:1872–1881
Almind K, Ambye L, Urhammer SA et al (1998) Discovery of amino acid variants in the human glucose-dependent insulinotropic polypeptide (GIP) receptor: the impact on the pancreatic beta cell responses and functional expression studies in Chinese hamster fibroblast cells. Diabetologia 41:1194–1198
Kjems LL, Holst JJ, Volund A, Madsbad S (2003) The influence of GLP-1 on glucose-stimulated insulin secretion: effects on beta-cell sensitivity in type 2 and nondiabetic subjects. Diabetes 52:380–386
Porte D Jr, Kahn SE (1995) The key role of islet dysfunction in type II diabetes mellitus. Clin Invest Med 18:247–254
Yip RG, Wolfe MM (2000) GIP biology and fat metabolism. Life Sci 66:91–103
Brown JC, Pederson RA, Jorpes JE, Mutt V (1969) Preparation of a highly active enterogastrone. Can J Physiol Pharmacol 47:113–114
Andersen DK, Putnam WS, Hanks JB, Wise JE, Lebovitz HE, Jones RS (1980) Gastric inhibitory polypeptide (GIP) suppression of hepatic glucose production. Regul Pept 1:4–9
Rudovich NN, Rochlitz HJ, Pfeiffer AF (2004) Reduced hepatic insulin extraction in response to gastric inhibitory polypeptide compensates for reduced insulin secretion in normal-weight and normal glucose tolerant first-degree relatives of type 2 diabetic patients. Diabetes 53:2359–2365
O’Harte FPM, Gray AM, Flatt PR (1998) Gastric inhibitory polypeptide and effects of glycation on glucose transport and metabolism in isolated mouse abdominal muscle. J Endocrinol 156:237–243
Oben J, Morgan L, Fletcher J, Marks V (1991) Effect of the entero-pancreatic hormones, gastric inhibitory polypeptide and glucagon-like polypeptide-1(7–36) amide, on fatty acid synthesis in explants of rat adipose tissue. J Endocrinol 130:267–272
Knapper JM, Puddicombe SM, Morgan LM, Fletcher JM (1995) Investigations into the actions of glucose-dependent insulinotropic polypeptide and glucagon like peptide-1(7–36)amide on lipoprotein lipase activity in explants of rat tissue. J Nutr 125:183–188
Ross SA, Dupre J (1978) Effects of ingestion of triglyceride or galactose on secretion of gastric inhibitory polypeptide and on responses to intravenous glucose in normal and diabetic subjects. Diabetes 27:327–333
Creutzfeldt W, Ebert R, Willms B, Frerichs H, Brown JC (1978) Gastric inhibitory polypeptide (GIP) and insulin in obesity: increased response to stimulation and defective feedback control of serum levels. Diabetologia 14:15–24
Flatt PR, Bailey CJ, Kwasowski P, Swanston-Flatt SK, Marks V (1983) Abnormalities of GIP in spontaneous syndromes of obesity and diabetes in mice. Diabetes 32:433–435
Miyawaki K, Yamada Y, Ban N et al (2002) Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med 8:738–742
Gault VA, O’Harte FPM, Harriott P, Flatt PR (2002) Characterization of the cellular and metabolic effects of a novel enzyme-resistant antagonist of glucose-dependent insulinotropic polypeptide. Biochem Biophys Res Commun. 290:1420–1426
Gault VA, Irwin N, Green BD et al (2005) Chemical ablation of gastric inhibitory polypeptide receptor action by daily (Pro3)GIP administration improves glucose tolerance and ameliorates insulin resistance and abnormalities of islet structure in obesity-related diabetes. Diabetes 54:2436–2446
Bailey CJ, Flatt PR, Atkins TW (1982) Influence of genetic background and age on the expression of the obese hyperglycaemic syndrome in Aston ob/ob mice. Int J Obes 6:11–21
Irwin N, Gault VA, Green BD et al (2004) Effects of short-term chemical ablation of the GIP receptor on insulin secretion, islet morphology and glucose homeostasis in mice. Biol Chem 385:845–852
Gault VA, Flatt PR, O’Harte FPM (2003) Glucose-dependent insulinotropic polypeptide analogues and their therapeutic potential for the treatment of obesity-diabetes. Biochem Biophys Res Commun 308:207–213
Gault VA, O’Harte FP, Harriott P, Mooney MH, Green BD, Flatt PR (2003) Effects of the novel (Pro3)GIP antagonist and exendin(9–39)amide on GIP- and GLP-1-induced cyclic AMP generation, insulin secretion and postprandial insulin release in obese diabetic (ob/ob) mice: evidence that GIP is the major physiological incretin. Diabetologia 46:222–230
Flatt PR, Bailey CJ (1981) Abnormal plasma glucose and insulin responses in heterozygous lean (ob/+) mice. Diabetologia 20:573–577
Friedewald WT, Levy RI, Fredrickson DS (1972) Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem 18:499–502
Brown JC, Dryburgh JR, Frost JL, Otte SC, Pederson RA (1978) Physiology and pathophysiology of GIP. Adv Exp Med Biol 106:169–171
Flatt PR, Bailey CJ, Kwaskowski P, Page T, Marks V (1984) Plasma immunoreactive gastric inhibitory polypeptide in obese hyperglycaemic (ob/ob) mice. J Endocrinol 101:249–256
Bailey CJ, Flatt PR, Kwasowski P, Powell CJ, Marks V (1986) Immunoreactive gastric inhibitory polypeptide and K cell hyperplasia in obese hyperglycaemic (ob/ob) mice fed high fat and high carbohydrate cafeteria diets. Acta Endocrinol (Copenh) 112:224–229
Bailey CJ, Flatt PR (2003) Animal syndromes resembling type 2 diabetes. In: Pickup JC, Williams G (eds) Textbook of diabetes, 3rd edn. Blackwell Science, Oxford, pp 25.1–25.30
Hansotia T, Maida A, Flock G et al (2007) Extrapancreatic incretin receptors modulate glucose homeostasis, body weight, and energy expenditure. J Clin Invest 117:143–152
Gama R, Norris F, Morgan L, Hampton S, Wright J, Marks V (1997) Elevated post-prandial gastric inhibitory polypeptide concentrations in hypertriglyceridaemic subjects. Clin Sci (Lond) 9:343–347
Miyawaki K, Yamada Y, Yano H et al (1999) Glucose intolerance caused by a defect in the entero-insular axis: a study in gastric inhibitory polypeptide receptor knockout mice. Proc Nat Acad Sci USA 96:14843–14847
Clements RH, Gonzalez QH, Long CI, Wittert G, Laws HR (2004) Hormonal changes after Roux-en Y gastric bypass for morbid obesity and the control of type II diabetes mellitus. Am Surg 70:1–5
Rubino F, Gagner M, Gentileschi P et al (2004) The early effect of the Roux-en-Y gastric bypass on hormones involved in body weight regulation and glucose metabolism. Ann Surg 240:236–242
Guidone C, Manco M, Valera-Mora E et al (2006) Mechanisms of recovery from type 2 diabetes after malabsorptive bariatric surgery. Diabetes 55:2025–2031
Mari A, Manco M, Guidone C et al (2006) Restoration of normal glucose tolerance in severely obese patients after bilio-pancreatic diversion: role of insulin sensitivity and beta cell function. Diabetologia 49:2136–2143
Acknowledgements
These studies were supported by University of Ulster Research Strategy Funding and Diabetes UK. The authors would also like to acknowledge C. J. Bailey for donation of original ob/+ mice breeding pairs.
Conflict of interest
N. Irwin, F. P. M. O’Harte, V. A. Gault, P. Harriott and P. R. Flatt are shareholders in Diabetica.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Irwin, N., McClean, P.L., O’Harte, F.P.M. et al. Early administration of the glucose-dependent insulinotropic polypeptide receptor antagonist (Pro3)GIP prevents the development of diabetes and related metabolic abnormalities associated with genetically inherited obesity in ob/ob mice. Diabetologia 50, 1532–1540 (2007). https://doi.org/10.1007/s00125-007-0692-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-007-0692-2