
Abstract
Aims/hypothesis
Vascular endothelial growth factor (VEGF) plays a pivotal role in the pathogenesis of diabetic retinopathy. We investigated whether transgenic mice with moderate VEGF expression in photoreceptors (trVEGF029) developed changes similar to diabetic retinopathy and whether retinopathy progressed with time.
Materials and methods
Human VEGF165 (hVEGF165) expression was analysed using ELISA and quantitative RT-PCR; serum glucose levels were also measured. Fundus fluorescein angiography (FA) was used to screen the degree of retinopathy from 6 weeks. Dynamic changes in the density of retinal microvasculature, as well as other changes similar to diabetic retinopathy, including retinal leucostasis, capillary endothelial cell and pericyte loss, and numbers of acellular capillaries, were quantified.
Results
trVEGF029 mice were normoglycaemic and showed a moderate, short-term hVEGF165 upregulation for up to 3 weeks. Changes in the retinal microvasculature not only mimicked those seen in diabetic retinopathy, but also showed similar pathological progression with time. FA at 6 weeks identified two phenotypes, mild and moderate, which were distinguished by the extent of vascular leakage. Quantitative analysis of diabetic retinopathy-like changes revealed that these parameters were tightly correlated with the initial degree of vascular leakage; low levels reflected slow and limited retinal microvascular changes in mild cases and high levels reflected more rapid and extensive changes in moderate cases.
Conclusions/interpretation
The data suggest that even an early short-term elevation in hVEGF165 expression might set a train of events that lead to progressive retinopathy. Induction of many features characteristic of diabetic retinopathy in trVEGF029 enables mechanisms leading to the disease state to be examined, and provides a relevant animal model for testing novel therapeutics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Damage to the retinal microvasculature is the hallmark of many eye diseases including diabetic retinopathy [1] a chronic disorder that is the leading cause of legal blindness and eventually develops in nearly all patients with diabetes mellitus [1, 2]. Diabetic retinopathy is characterised by readily identifiable and progressive retinal vascular changes, which have been extensively described clinically and experimentally [1–6]. Early and middle stages are classed as non-proliferative diabetic retinopathy (NPDR), being characterised by increased vessel permeability, microaneurysms and capillary loss. Ensuing retinal ischaemia triggers advanced stages, termed proliferative diabetic retinopathy (PDR), in which neovascularisation leads to fibrovascular changes, retinal traction, detachment and blindness [7].
Although hyperglycaemic animal models of types 1 and 2 diabetes exist, they do not present all features of NPDR or PDR. Alternative models focus on the many biochemical pathways in diabetic retinopathy, which stimulate the production of a variety of growth factors including IGF-1 and vascular endothelial growth factor (VEGF) [2]. VEGF is hypoxia-induced [8] and plays a role in ischaemia-induced retinal neovascularisation [3, 9–11]. However, it is also highly expressed at early stages in diabetic retinopathy, i.e. well before clinical signs of retinal non-perfusion and neovascularisation, suggesting that VEGF plays a pivotal role in the pathogenesis of diabetic retinopathy, rather than solely at the late ischaemic stages [12]. VEGF is also implicated in other features of diabetic retinopathy, since it (1) increases retinal vascular permeability either directly after intraocular injection [13] or indirectly in streptozotocin (STZ)-induced diabetic models [6, 14–18], and (2) triggers retinal leucostasis in STZ-treated rats, which in turn further increases vascular permeability [6, 16].
However, most VEGF-based animal models have focussed principally on retinal neovascularisation, which occurs in the most damaging, end-phase of diabetic retinopathy. Intravitreal injection, slow-release implants and transgenic VEGF models [11, 19] induce extensive, rapid neovascularisation and severe retinal damage which preclude examination of early changes and their progression [20–22]. By contrast, we have recently generated four transgenic VEGF lines, of which one, transgenic mice with moderate VEGF expression in photoreceptors (trVEGF029), demonstrated moderate elevation of human VEGF165, (hVEGF165), relatively limited neovascular changes and minimal retinal damage, thus providing an opportunity to examine early pathological changes [23]. Here we have further characterised trVEGF029 to determine whether moderate hVEGF165 protein elevation triggers early features of diabetic retinopathy such as vascular leakage, damage to, and remodelling of the retinal vasculature including capillary dropout, leucostasis, microaneurysms and loss of endothelial cells and pericytes with an accompanying increase in capillary acellularity. In addition, we undertook longitudinal studies to determine whether such changes progressed with time as occurs in diabetic retinopathy.
Materials and methods
VEGF transgenic mice
The work was approved by the Animal Ethics Committee, The University of Western Australia. Mice were generated through microinjection of pcDNA.opsin.VEGF, a DNA construct containing hVEGF165 driven by a truncated mouse rhodopsin promoter that confines transgene expression to the eye [23]. Fifth and sixth generation heterozygous trVEGF029 and age-matched wildtype littermates (wt) were produced through backcrossing with C57BL/6J mice.
Quantitation of hVEGF165 mRNA transcripts by real-time PCR
mRNA levels of pcDNA.opsin.VEGF were assayed at 3 (n=6) and 6 (n=6) weeks in wt and trVEGF029. Due to small eye size at 3 weeks, fundus fluorescein angiography (FA) could not be performed, precluding identification of mild and moderate phenotypes (see below). However, at 6 weeks, mild and moderate cases were distinguished by FA prior to mRNA extraction and subsequent cDNA synthesis [24]. Semi-quantitative real-time PCR was performed using primers specific for pcDNA.opsin.VEGF and the housekeeping gene peptidylprolyl isomerase A (Ppia), with hVEGF165 expression normalised against Ppia. pcDNA.opsin.VEGF transcript was calculated using a standard curve.
Quantitation of hVEGF165 protein
Ocular hVEGF165 protein was quantified in wt and trVEGF029 at 3 weeks (n=4), and in those having distinguished mild and moderate phenotypes at 6, 9 and 19 weeks (n≥7 per phenotype per age). Intact eyes were enucleated and hVEGF165 quantified by ELISA (Quantikine R&D Systems, Minneapolis, MN, USA) [23].
VEGF immunostaining
Eyes from wt and trVEGF029 (1.5, 3 and 6 weeks) were enucleated, processed and immunostained with anti-VEGF antibody as described previously [25], using donkey anti rabbit IgG AlexaFluor 555 (Molecular Probes, Eugene, OR, USA) as the secondary antibody. Sections were examined by fluorescence microscopy.
Fundus fluorescein angiography
In all mice in this study, the retinal microvasculature was examined by fundus fluorescein angiography (FA) at 6 weeks (n=244), using a modified portable small animal fundus camera (Genesis; Kowa, Tokyo, Japan). Vascular leakage was graded as described previously [23] and two phenotypes, mild and moderate, identified. To monitor retinal vascular leakage longitudinally within each phenotype, 30 additional mice were examined at 6 weeks and followed up every 2 to 3 weeks until 14 weeks and every 5 weeks until 24 weeks.
Measurement of glucose levels
Eyes were enucleated from 8 to 12-week-old trVEGF029 (n=3) and wt (n=4), homogenised in deionised water (500 μl), centrifuged (20,000 g) and the clarified homogenate (40 μl) incubated with 60 μl of enzyme (AMES glucose test packs; Bayer Diagnostics, Basingstoke, UK). Complex formation was measured using a mini photometer (AMES; Bayer) and the amount of glucose determined using glucose standards (0–50.00 mmol/l).
Retinal perfusion with rhodamine-coupled concanavalin A for quantitative topological and fractal analysis of the retinal vasculature and quantitative analysis of leucostasis
Mild and moderate trVEGF029 (n=43) and wt (n=12) were perfused with rhodamine-coupled concanavalin A at 6, 9, 19 and 24 weeks [16]. Eyes were enucleated, fixed in 2% paraformaldehyde for 30 min, flat-mounted for fluorescence microscopy and digital images captured (BX60, DP70, respectively; both from Olympus Optical, Tokyo, Japan).
Low-power (×4) images were used to chart dynamic changes in the microvasculature across the entire retina, i.e. globally. Two to four 4–6 mm2 samples were taken for each retina and results averaged. Images were processed and analysed using computer-based image analysis software (QWin; Leica Imaging Systems, Cambridge UK) with customised macroroutines [26] to determine the following topological parameters: (1) percentage of retinal area covered by vessels per field of view; (2) ‘crossing points’ of retinal vessels; and (3) a related metric, the number of ‘vessel segments’. Mean values for each parameter were calculated per square millimeter. However, the above topological parameters do not fully characterise structural complexity, and the fractal parameters ‘fractal dimension, (D f)’ and ‘lacunarity’ were also calculated [26]. Leucocyte adhesion in the retinal veins and capillaries was quantified from high-power images (×10) by counting numbers of adherent leucocytes using NIH ImageJ software [27] with the cell counter plugin.
Retinal trypsin digestion for quantification of endothelial cells and pericytes
Retinal trypsin digests were prepared as described previously [28] for mild and moderate trVEGF029 (n=35) and wt (n=14) at 9, 19 and 24 weeks. Endothelial cells and pericytes were identified using standard criteria [29], counted in four to six randomly selected fields at ×50 magnification using NIH ImageJ software with the cell counter plugin, and data averaged. Acellular capillaries were simultaneously identified and quantitated.
Statistics
Results are expressed as mean±SE. Data were analysed using ANOVA for multiple comparisons (GB-STAT v9.0, Dynamic Microsystems, Silver Spring, MD, USA) with a p value of <0.05 accepted as statistically significant.
Results
hVEGF165 protein expression
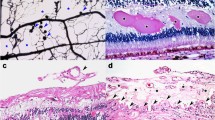
At 3 weeks, hVEGF165 protein levels were 1.8±1.0 pg hVEGF/mg protein. By 6 weeks, when mild and moderate phenotypes could be distinguished, values had fallen and did not differ between phenotypes (mild: 0.8±0.4 pg/mg; moderate: 0.7±0.1 pg/mg). Thereafter, values remained similarly low in both phenotypes (9 weeks mild: 0.7±0.1 pg/mg; 9 weeks moderate: 0.6±0.1 pg/mg; 19 weeks mild: 0.7±0.1 pg/mg; 19 weeks moderate 0.8±0.2 pg/mg). Analysis of hVEGF165 mRNA in 3- and 6-week-old trVEGF029 demonstrated a drop in transgene expression between 3 and 6 weeks, but no significant difference between ages or between mild and moderate phenotypes at 6 weeks (3 weeks: 79±21 copies/μl; 6 weeks mild: 56±11 copies/μl; 6 weeks moderate: 64±1 copies/μl). The high variance of protein and mRNA levels at 3 weeks presumably reflects the inclusion of mild and moderate phenotype in the samples, since, at this early age, FA could not be used to distinguish phenotypes. Wild-type littermates did not have detectable levels of hVEGF165 transcript or protein at any age. Immunohistochemistry revealed VEGF-positive cell bodies primarily in the inner and outer nuclear layers in trVEGF029 at 1.5 weeks with comparatively minimal staining at 3 and 6 weeks; wt lacked immunopositive cells (Fig. 1a–d).

VEGF immunohistochemistry. trVEGF029 at 1.5 weeks (a) and 3 weeks (c); wt at 1.5 weeks (b). Control panel (d) shows trVEGF029 at 1.5 weeks without added primary antibody. VEGF was observed primarily in the inner and outer nuclear layers in trVEGF029 at 1.5 weeks (a) with only background staining in the wt (b); staining in trVEGF029 at 3 weeks (c) was also at background levels. Insets are adjacent haemotoxylin- and eosin-stained sections for orientation. Single-headed arrows, retinal pigment epithelium; double-headed arrows, photoreceptor layer. Scale bars: 60 μm
Glucose levels in wt and trVEGF029
Glucose levels did not differ significantly between wt and trVEGF029 (wt: 5.36±0.99 mmol/l; trVEGF029: 5.42±0.90 mmol/l; p>0.05) and were similar to those in the aqueous humour of an IGF-1 transgenic mouse [30].
Retinal vascular leakage in trVEGF029
In wt, retinal vessels had an even calibre and radiated from a well-defined optical nerve head; fluorescein leakage was not observed (Fig. 2a, normal). At 6 weeks, trVEGF029 demonstrated increased retinal vascular permeability and we identified two phenotypes. Mild cases (75%, n=206) were characterised by small numbers of readily identifiable and isolated leaky lesions with the retention of large areas of normal-appearing retinal capillaries (scores 1–3). Moderate cases (25%, n=68) had extensive vascular changes characterised by heavy fluorescein leakage (scores 4 and 5).

Retinal vascular leakage was assessed after FA. Leakage grading (a): normal, normal pattern of fluorescein filling with no vascular leakage; 0, sclerotic vessels with no vascular leakage; 1, less than five isolated, leaky lesions; 2, five to ten isolated, leaky lesions; 3, more than ten evenly distributed, but not confluent, leaky lesions; 4, confluent leaky lesions and normal-appearing retinal capillaries rarely visible; 5, massive vascular leakage and retinal capillary destruction. Mild cases (Mi): scores 1–3. Moderate case (Mo): scores 4 and 5. b–i Longitudinal dynamic changes in retinal vascular leakage revealed by FA. Note the slight regression (arrows) and progression (open arrows) in some lesions in the same eye with mild changes (b, c); note also: dramatic cessation of fluorescein leakage from 19 weeks postnatal in mild cases (d, e, j), but cessation of fluorescein leakage appears earlier, at 9 weeks postnatal, in moderate cases (f–i, k). Semi-quantitative analysis of graded images of fluorescein leakage in mild (j; n=10) and moderate (k; n=10) cases over time. *p<0.01, compared with 6 weeks; § p<0.01 and ‡ p<0.05, compared with mild cases, respectively
Longitudinal FA revealed that mild and moderate phenotypes remained distinct and showed characteristic changes over time. In mild cases, vascular leakage remained relatively stable for up to 12 weeks, although slight progression (Fig. 2b,c) and regression (Fig. 2b,c) were observed in individual lesions. Most lesions had stopped leaking by 19 weeks and retinal capillaries were relatively well preserved with only minor capillary drop-out and few microaneurysms (Fig. 2d,e). Semi-quantitative analysis revealed a significant decrease in the grade of fluorescein leakage at 19 and 24 weeks compared with 6 and 9 weeks (Fig. 2j, p<0.01), with some evidence of capillary dropout. By contrast, leakage was more severe in moderate cases at early stages (Fig. 2f), and semi-quantitative analysis revealed that most retinal vessels had stopped leaking by as early as 9 weeks (Fig. 2g). In addition, and in contrast to the mild cases, the cessation of leakage and more pronounced neovascularisation were related to widespread destruction of retinal capillaries (Fig. 2f–i; Fig. 2k, p<0.01 and p<0.05).
Topological and fractal changes in the retinal vasculature
Transformation from digital to binary and skeletonised images is shown in Fig. 3. The retinal vasculature in wt was characterised by a fine and dense meshwork of well-defined retinal capillaries (Fig. 4a). Mild trVEGF029 showed some evidence for tortuous retinal vessels, a limited number of proliferating, or neovascular, microaneurysm-like lesions and a reduction in the density of the microvasculature over time (Fig. 4b–d). In moderate cases, such changes were considerably more pronounced and occurred earlier (compare Fig. 4b,e). A large number of neovascular lesions appeared at 9 weeks with a more rapid and dramatic progressive loss of retinal capillaries (Fig. 4e,f).

Schematic illustration of the parameters used to characterise the morphology of the retinal vasculature. A gradient detection algorithm (Sobel filter) was applied to the original digital image (a) and binary thresholding performed on the gradient image by selecting its mean grey value as the threshold. The procedure allowed good identification of vascular profiles to calculate the first topological parameter, the percentage of retinal area covered by the retinal vessels per field of view (b). A second binary image (c) showing the skeleton of the retinal vessels was derived from the vascular profiles in (b) using binary thinning procedures. The skeletonised binary image (c) was used to measure total length of vessels in the field of view and two other topological parameters, namely (1) the crossing points of retinal vessels (black dots), i.e. all points at which vessels crossed regardless of the level of the vascular bed and a related metric, and (2) the number of resultant vessel segments (some are indicated by arrows), i.e. all segments that were identified by two adjacent crossing points. However, the above topological parameters do not fully characterise vascular structure and fractal analysis of the image was also undertaken [26]. The fractal parameters D f and lacunarity are inversely related and are measures of the degree of structural variance; high values for D f and low lacunarity indicate a high level of structure, or homogeneity, whereas low values indicate lack of structure or heterogeneity

Topological and fractal analysis of retinal vasculature in trVEGF029. Representative micrographs of rhodamine-coupled concanavalin A-perfused retinal vasculature from wt (a) and trVEGF029 (TG) (b–f) with mild (Mi) (b, c, d) and moderate (Mo) (e, f) vascular changes at different ages. a 1– f 1 , Images of the boxed areas (a–f) used for topological and fractal analysis. a 2 –f 2 Binary images of (a 1 –f 1 ) used for calculation of the percentage of area covered by vessels. a 3 –f 3 Skeletonised images of (a 2 –f 2 ) after application of the binary thinning procedure, which was used to calculate the length of vessels, the number of crossing points and crossed segments, fractal dimension and lacunarity. g–l Quantitative analysis of the retinal vasculature based on topological and fractal parameters. *p<0.01, compared with wt (n=10); † p<0.01, compared with TG-Mi-9 weeks (n=7); ‡ p<0.01 compared with TG-Mo-9 weeks (n=7); § p<0.01, compared with TG-Mi-19 weeks (n=5). TG-Mi-24 weeks, n=6. TG-Mo-19 w, n=8. TG-Mi, trVEGF029 mild cases. TG-Mo, trVEGF029 moderate cases. WT, wild-type. Scale bars=200 μm
Quantitative topological and fractal analysis in wt did not reveal any differences at either 9, 19 or 24 weeks (data not shown). The percentage of area covered by vessels showed no significant differences between the wt and mild trVEGF029 (Fig. 4g, p>0.05). However, compared with wt, vessel length, the number of crossing points and vessel segments per mm2 were significantly different in mild and moderate trVEGF029, indicating that topological analysis based on the skeletonised retinal vasculature is more sensitive than measurement of the area covered by vessels. In wt, the length of vessels was 36.92±8.44 mm/mm2, with 781±200 per mm2 crossing points and 1122±199 per mm2 vessel segments. In mild trVEGF029, these parameters were significantly increased at 9 weeks (length of vessels: 47.58±9.67 mm/mm2; crossing points: 950±219 per mm2; crossed segments: 1300±226 per mm2) indicating a mild neovascular response (Fig. 4h–j: p<0.01 vs wt). By 24 weeks in mild trVEGF029, there was a significant reduction in vessel length, numbers of crossing points and crossed segments compared to 9 weeks, indicating capillary dropout and the progression of retinopathy (Fig. 4h–j: p<0.01 vs trVEGF029 with mild retinal vascular changes [Mi] at 9 weeks and p<0.01 vs Mi at 19 weeks, respectively). Compared to mild cases, changes in moderate cases were more pronounced and occurred earlier. Significant reductions in the percentage of area covered by vessels, vessel length, numbers of crossing points and crossed segments were observed as early as 9 weeks (Fig. 4g–j: p<0.01 vs wt; p<0.01 vs Mi at 9 weeks; p<0.01 vs Mi at 19 weeks and p<0.01 vs trVEGF029 with moderate retinal vascular changes [Mo] at 9 weeks; respectively).
In mild trVEGF029, the fractal dimension D f was 1.73±0.02 at 9 and 19 weeks, a value that did not differ from wt (p>0.05). However, by 24 weeks, D f was significantly reduced to 1.70±0.02 (Fig. 4k: p<0.01 vs wt, p<0.01 vs Mi at 9 weeks, respectively). In moderate cases, the reduction in D f was more dramatic and occurred by 9 weeks (D f=1.60±0.08) (Fig. 4k: p<0.01 vs wt, p<0.01 vs Mi at 9 weeks, p<0.01 vs Mi at 19 weeks, respectively). An inverse relationship was observed between D f and lacunarity (Fig. 4l). In mild cases, lacunarity was 0.19±0.02 and 0.19±0.01 at 9 and 19 weeks, respectively, and did not differ from wt (p>0.05); however, by 24 weeks, lacunarity had increased to 0.22±0.02 (p<0.01 vs wt). In moderate cases, lacunarity was significantly increased to 0.31±0.07 and 0.36±0.09 at 9 and 19 weeks, respectively (Fig. 4l: p<0.01 vs wt, p<0.01 vs Mi-9 weeks and p<0.01 vs Mi-19 weeks, respectively).
Leucostasis and vascular damage
Leucocyte adhesion was occasionally observed in the veins of wt (Fig. 5a,b) but was more frequent in the retinal capillaries of mild trVEGF029, particularly in the periphery where large areas of retinal non-perfusion and microaneurysms accompanied by vascular proliferation were seen (Fig. 5c,d). Adherent leucocytes were also frequently found in capillaries at the posterior retinal pole (Fig. 5d, inset). Leucostasis was most common in moderate trVEGF029 and, as in mild cases, was found in large areas of retinal non-perfusion with prominent vascular proliferation (Fig. 5e–g). Adherent leucocyte numbers correlated well with the severity of retinal vascular abnormality (Fig. 5h: p<0.01 vs wt and p<0.01 and p<0.05 vs mild).

Retinal wholemounts and adherent leucocytes after perfusion with rhodamine-coupled concanavalin A (Rho-Con A). Mice were perfused with Rho-Con A (50 μg/ml in PBS; Vector Labs, Birmingham, CA, USA) at 6 weeks as previously described [16]. a, b Wild-type littermates (WT). trVEGF029 with mild (Mi) (c, d) and moderate (Mo) (e–g) vascular changes. a, c, e Overall changes in the retinal vasculature. b, d, f, g Leucocyte adhesion (arrows) in retinal veins and capillaries (cp). Inset (d), leucocyte plugging in retinal capillaries in the periphery where a cluster of aneurysm-like vascular proliferation (*) was apparent. Note the decrease in retinal capillary density with increased severity of vascular changes in WT (a), Mi (c) and Mo (e), and the correlation between leucocyte adhesion in retinal capillaries with capillary non-perfusion and aneurysm-like vascular proliferation (*) (d, e, g). Scale bars=25 μm. h Quantitation of retinal leucostasis in retinal veins and capillaries at 6 weeks. The number of adherent leucocytes in the retinal veins and capillaries increased with the severity of retinal vascular abnormalities. Open bars, WT; hatched bars, Mi; filled bars, Mo. *p<0.01 vs WT; † p<0.01 and ‡ p<0.05 vs Mi. WT, n=9; mild trVEGF029, n=19; moderate trVEGF029, n=9
Cellular changes in the retinal capillaries
Cellular changes in the retinal capillaries were not observed in wt at 9, 19 and 24 weeks (Fig. 6a,b). In mild cases at 9 weeks, most regions of the trypsin digests were generally indistinguishable from wt when observed with low (×10) magnification (Fig. 6c). However, higher power microscopy revealed occasional microaneurysm-like structures (inset, Fig. 6d), slightly tortuous retinal vessels (Fig. 6d) and acellular capillaries (Fig. 6d), although, compared with wt, there were no differences in the number of endothelial cells, pericytes or acellular capillaries and the ratio of endothelial cells : pericytes remained unchanged (Fig. 6k–n; p>0.05). By 19 and 24 weeks, however, the retinal vasculature had changed dramatically (Fig. 6e–h). Retinal vessels were more tortuous and vascular density reduced (Fig. 6e,g). Both endothelial cells and pericytes were sparse and acellular capillaries commonly observed (Fig. 6f,h). At 24 weeks, there was a significant loss of pericytes (Fig. 6l; p<0.05 vs Mo at 9 weeks), but there was no significant change in the ratio of endothelial cells : pericytes (Fig. 6m; p>0.05). The number of acellular capillaries was significantly increased at 24 weeks (Fig. 6n; p<0.01 vs wt).

Retinal digest preparations of wild-type (WT) and trVEGF029. Digests were prepared as described previously [28]. Digests were mounted vitreous side uppermost, air-dried and stained with periodic acid Schiff and haematoxylin. Endothelial cells and pericytes were identified using standard criteria [29] with endothelial cells (black arrow heads, inset [b]) having elongated, flattened nuclei adjacent to the vessel walls, and pericytes (white arrow heads, inset [b]) having smaller, rounded nuclei that protruded somewhat from the vessel walls. Magnification: low-power (a, c, e, g, i), high-power (b, d, f, h, j). Mild cases trVEGF029 (Mi) at 9 weeks (c, d), 19 weeks (e, f) and 24 weeks (g, h). Moderate (Mo) cases trVEGF029 at 9 weeks (i, j). Note the dynamic loss of vascular cells and the increase in acellular capillaries (d, f, h); compare also differences in the number of aneurysm-like clusters, retinal vascular cell loss and capillary density reduction in age-matched eyes (d, j). Inset (d) shows an aneurysm-like cluster. Arrows (d, f, h, j) point to acellular capillaries. Scale bars=100 μm. k–n Quantitation of the retinal vascular cell loss in trVEGF029. WT, n=14. Mi-9 weeks, n=13. Mi-24 weeks, n=8. Mo-9 weeks, n=10. *p<0.05 vs Mi-9 weeks; **p<0.01 vs Mi-9 weeks; † p<0.01 vs WT
In moderate cases at 9 weeks, clusters of vascular proliferation were commonly detected indicating active retinal neovascularisation (Fig. 6i,j). Reduced numbers of capillaries with fewer vascular cells as well as acellular capillaries were frequently observed adjacent to the proliferating clusters, indicating capillary closure and retinal non-perfusion (Fig. 6j). Loss of endothelial cells and pericytes was more advanced than that in age-matched eyes with mild vascular changes (Fig. 6k,l; p<0.05 vs Mi at 9 weeks, p<0.01 vs Mi at 9 weeks, and p<0.01 vs wt, respectively). Retinal vascular deterioration was accompanied by significant increases in the ratio of endothelial cells: pericytes and the number of acellular capillaries (Fig. 6m,n, *p<0.05 vs Mi at 9 weeks, p<0.01 vs Mi at 9 weeks, and p<0.01 vs wt, respectively).
Discussion
We have previously demonstrated that, compared with the other lines generated, the moderate hVEGF165 protein levels at 1.5 weeks (35.5 pg hVEGF/mg total protein) in trVEGF029 were sufficient to produce pathological changes consistent with those in NPDR and early stages of PDR [23]. We now show that sustained hVEGF165 protein overexpression is not required to develop vascular changes. Rather, a cascade mimicking diabetic retinopathy can be triggered by the presence of early moderate levels of hVEGF165 protein at around 1.5 weeks, followed by a rather rapid decline to a low but steady level (0.6–0.8 pg hVEGF/mg total protein) at between 6 to 19 weeks.
The pattern of hVEGF165 expression observed was unexpected, but advantageous for developing a model with neither too severe nor too rapid vascular damage. Several explanations of the observed, short-term hVEGF165 protein expression are possible. Loss, due to cell death, of the cell type that expresses the transgene is unlikely, since, in contrast to previous VEGF transgenics [20, 21], photoreceptors to which the transgene was targeted are not reduced significantly before 15 weeks [23]. Evidence of hVEGF mRNA production at 3 and 6 weeks also excludes the possibility of complete promoter shut down. The anti-angiogenic factor pigment epithelium-derived factor down-regulates VEGF in vitro and is a key factor in angiogenic control [31–33]. However, any upregulation of pigment epithelium-derived factor in trVEGF029 is presumably insufficient to prevent the progression of vascular damage that we describe, despite its ability to curtail aberrant retinal vessel growth in retinopathy of prematurity [33]. Although the opsin promoter is assumed to be eye-specific, opsin transcripts have been detected in the thymus [34], which might underpin a possible state of tolerance in trVEGF029, in agreement with the small number of infiltrating cells (data not shown). Increased hVEGF165 levels might also have triggered a negative feed-back mechanism to dampen VEGF translation. Further studies are necessary to elucidate the mechanism(s) responsible for hVEGF165 regulation in trVEGF029 and to investigate the signalling events that result in vascular changes similar to those seen in diabetic retinopathy.
The vascular changes in trVEGF029 differ from two other models in which VEGF was upregulated via a transgene or via intravitreal or subretinal injection of a recombinant adeno-associated virus carrying the gene encoding VEGF (AAV.VEGF) [21, 35] with high protein levels leading to severe neovascularisation, an immune reaction and photoreceptor destruction [21, 36]. In comparison, moderate VEGF expression in another model induced by AAV.VEGF gene transfer also produced a mild phenotype with features similar to trVEGF029 and with no immune reaction elicited [37]. trVEGF029 thus provides an opportunity for detailed examination of vascular changes without the confounding factors of severe and rapid vascular and retinal damage in other models.
Our techniques for visualising the retinal microvasculature throughout its depth did not, for the most part, distinguish different retinal vascular beds, but are similar to clinical examination of retinal microvascular changes in diabetic retinopathy [23]. FA of the same mice over time revealed two phenotypes. Mild cases had initial vessel leakage at 6 weeks, which was minor and confined to a limited number of locations. Although individual lesions either worsened or resolved with time, overall leakage remained similar until 12 weeks and by 19 to 24 weeks had virtually ceased. By contrast, in moderate cases, leakage was more severe at the earliest stage examined and also declined earlier, that is, by 9 weeks. A precedent for the severity of the initial damage giving rise to distinct phenotypes within the retinal vasculature has also been observed in IGF-1 transgenic mice [30].
The decline in vascular leakage in both mild and moderate cases is initially counterintuitive, but could be partially explained by the decreasing hVEGF165 levels. However, while hVEGF165 expression was significantly reduced as early as 3 weeks, decline in vascular leakage only occurred at 9 (moderate) or 19 (mild) weeks. We propose that the decline in vascular leakage is primarily due to the loss of capillaries, a phenomenon seen in clinical diabetic retinopathy [4, 38]. In mild cases, the initial relatively limited vessel leakage was correlated with a minor loss of capillaries that mostly maintained good structural integrity. Leucostasis was limited, with no apparent endothelial cell loss, minimal pericyte loss and only a small number of acellular capillaries. Nevertheless, damage progressed and capillary dropout led to reduced vascular leakage. Mild cases also displayed a neovascular response in the formation of microaneurysms, although these were sparse at early stages. A strikingly different scenario was observed in the moderate phenotype, in which initial leakage was severe. The ensuing extensive damage to the retinal microvasculature, as evidenced by a considerable loss of capillary beds, declining structural integrity, extensive leucostasis, loss of both endothelial cells and pericytes and the appearance of extensive acellular capillaries, would also have led to a decline in vascular leakage. In addition, moderate cases displayed a pronounced neovascular response with large numbers of microaneurysms appearing at early stages. Since mild cases remain mild and moderate cases remain moderate, the extent of initial damage appears to dictate long-term consequences with mild cases having better outcomes than moderate ones.
Regardless of the intensity of microvascular changes in mild and moderate phenotypes, trVEGF029 shared many features of diabetic retinopathy. Increased vascular permeability is an early sign of diabetic retinopathy [2, 4, 14, 39–41]. As in diabetic animal models in which the retinal vasculature becomes permeable shortly after the onset of diabetes [6, 14], early increases in vascular permeability are consistently observed in trVEGF029 [20, 21, 23]. While increased retinal vascular permeability is caused by the elevated production of VEGF [9, 16, 42], we now show that vascular leakage can result from an early, short-term increase in VEGF165 expression. In addition to vascular leakage, other early changes in clinical diabetic retinopathy include the formation of spatio-temporally dynamic microaneurysms [40, 41]. Our longitudinal FA analysis in individual trVEGF029 provides evidence of individual microaneurysms that also improve or progress with time. In addition, we provide histological confirmation of the formation of microaneurysms.
Leucostasis, another early event seen in clinical diabetic retinopathy and in experimental diabetes [5, 6, 16, 43, 44], is causally linked to VEGF-induced inflammation [5, 6, 16, 43]. In trVEGF029, leucostasis was correlated with the severity of retinal vascular damage. In moderate cases, leucostasis occurred more frequently where vascular proliferation and capillary non-perfusion were significantly developed. In STZ-induced diabetic rats, patent capillaries become occluded downstream from static leucocytes [5, 6]. Although adhesive leucocytes can become dislodged and occluded capillaries re-open, repeated plugging and un-plugging can also lead to blockage and non-perfusion. A major consequence of capillary non-perfusion is endothelial cell and pericyte damage and loss, readily detectable features of early diabetic retinopathy, which culminate in acellular capillaries and capillary dropout with a resultant decline in vascular permeability [40, 45]. Finally, as diabetic retinopathy progresses, capillary loss and retinal non-perfusion lead to ischaemia [45] with associated ischaemia-induced neovascularisation. Neovascularisation also occurs in trVEGF029, but to a lesser extent than the major lesions often described in other transgenic VEGF models [10, 21, 22].
A number of features seen in diabetic retinopathy have yet to be characterised in trVEGF029. For example, capillary basement membrane thickening, as assessed ultrastructurally, occurs at early and late stages [43, 46]. Furthermore, profound neuronal and glial degeneration also occurs in diabetic retinopathy [47, 48]. A notable difference between VEGF transgenic models and diabetic retinopathy is that neovascular changes occur in the outer retina, presumably reflecting the source of transgene, rather than in the inner retina with epiretinal membranes extending into the vitreous, as occurs clinically [3]. Interestingly, although vascular changes in trVEGF029 result from a short-term elevation in VEGF expression, they are similar to those in NPDR and the early stages of PDR, which are presumed to be associated with persistent VEGF expression [9–12]. Although vitreous haemorrhage and macular oedema with subsequent fibrosis, retinal traction and detachment are frequently seen in diabetic retinopathy and also in some animal models including others and our own [2, 20, 21, 23], we did not observe these severe changes in trVEGF029. Further studies are required to determine the exact relevance and limitations of the trVEGF029 model.
Similar to an IGF-1 transgenic mouse line, trVEGF029 lacks a hyperglycaemic background, yet both display many features of diabetic retinopathy [30]. Nevertheless, hyperglycaemia is a major trigger in diabetic retinopathy, with factors such as sorbitol and advanced glycation end products producing reactive oxygen species, which leads to pericyte and endothelial cell loss and ensuing vascular damage [2, 42]. However, both clinically and experimentally, good glycaemic control does not necessarily stop the progression of diabetic retinopathy [49]; indeed, and there is some evidence for a worsening [50]. Our demonstration that many pathological features of diabetic retinopathy could result from an early short-term, moderate increase in VEGF levels occurring in the absence of hyperglycaemia provides an opportunity to examine individual factors that contribute to early stages of this devastating disease, as well as offering the potential to test early therapeutic interventions.
Abbreviations
- AAV.VEGF:
-
adeno-associated virus carrying the gene encoding VEGF
- Df :
-
fractal dimension
- FA:
-
fundus fluorescein angiography
- hVEGF165 :
-
human VEGF165
- Mi:
-
trVEGF029 with mild retinal vascular changes
- Mo:
-
trVEGF029 with moderate retinal vascular changes
- NPDR:
-
non-proliferative diabetic retinopathy
- PDR:
-
proliferative diabetic retinopathy
- Ppia :
-
peptidylprolyl isomerase A
- trVEGF029:
-
transgenic mice with moderate VEGF expression in photoreceptors
- VEGF:
-
vascular endothelial growth factor
- wt:
-
wild type littermates
References
Aiello LP, Gardner TW, King GL et al (1998) Diabetic retinopathy. Diabetes Care 21:143–156
Fong DS, Aiello LP, Ferris FL 3rd, Klein R (2004) Diabetic retinopathy. Diabetes Care 27:2540–2553
Campochiaro PA (2000) Retinal and choroidal neovascularization. J Cell Physiol 184:301–310
Frank RN (2004) Diabetic retinopathy. N Engl J Med 350:48–58
Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP (2001) Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol 158:147–152
Miyamoto K, Khosrof S, Bursell SE et al (1999) Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci USA 96:10836–10841
Tolentino MJ, McLeod DS, Taomoto M, Otsuji T, Adamis AP, Lutty GA (2002) Pathologic features of vascular endothelial growth factor-induced retinopathy in the nonhuman primate. Am J Ophthalmol 133:373–385
Forsythe JA, Jiang BH, Iyer NV et al (1996) Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol 16:4604–4613
Adamis AP, Miller JW, Bernal MT et al (1994) Increased vascular endothelial growth factor levels in the vitreous of eyes with proliferative diabetic retinopathy. Am J Ophthalmol 118:445–450
Miller JW, Adamis AP, Aiello LP (1997) Vascular endothelial growth factor in ocular neovascularization and proliferative diabetic retinopathy. Diabetes Metab Rev 13:37–50
Tolentino MJ, Adamis AP (1998) Angiogenic factors in the development of diabetic iris neovascularization and retinopathy. Int Ophthalmol Clin 38:77–94
Amin RH, Frank RN, Kennedy A, Eliott D, Puklin JE, Abrams GW (1997) Vascular endothelial growth factor is present in glial cells of the retina and optic nerve of human subjects with nonproliferative diabetic retinopathy. Invest Ophthalmol Vis Sci 38:36–47
Aiello LP, Bursell SE, Clermont A et al (1997) Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective beta-isoform-selective inhibitor. Diabetes 46:1473–1480
Barber AJ, Antonetti DA (2003) Mapping the blood vessels with paracellular permeability in the retinas of diabetic rats. Invest Ophthalmol Vis Sci 44:5410–5416
Gilbert RE, Kelly DJ, Cox AJ et al (2000) Angiotensin converting enzyme inhibition reduces retinal overexpression of vascular endothelial growth factor and hyperpermeability in experimental diabetes. Diabetologia 43:1360–1367
Ishida S, Usui T, Yamashiro K et al (2003) VEGF164 is proinflammatory in the diabetic retina. Invest Ophthalmol Vis Sci 44:2155–2162
Murata T, Ishibashi T, Khalil A, Hata Y, Yoshikawa H, Inomata H (1995) Vascular endothelial growth factor plays a role in hyperpermeability of diabetic retinal vessels. Ophthalmic Res 27:48–52
Qaum T, Xu Q, Joussen AM et al (2001) VEGF-initiated blood-retinal barrier breakdown in early diabetes. Invest Ophthalmol Vis Sci 42:2408–2413
Ozaki H, Hayashi H, Vinores SA, Moromizato Y, Campochiaro PA, Oshima K (1997) Intravitreal sustained release of VEGF causes retinal neovascularization in rabbits and breakdown of the blood-retinal barrier in rabbits and primates. Exp Eye Res 64:505–517
Ohno-Matsui K, Hirose A, Yamamoto S et al (2002) Inducible expression of vascular endothelial growth factor in adult mice causes severe proliferative retinopathy and retinal detachment. Am J Pathol 160:711–719
Okamoto N, Tobe T, Hackett SF et al (1997) Transgenic mice with increased expression of vascular endothelial growth factor in the retina: a new model of intraretinal and subretinal neovascularization. Am J Pathol 151:281–291
Tobe T, Okamoto N, Vinores MA et al (1998) Evolution of neovascularization in mice with overexpression of vascular endothelial growth factor in photoreceptors. Invest Ophthalmol Vis Sci 39:180–188
Lai CM, Dunlop SA, May LA et al (2005) Generation of transgenic mice with mild and severe retinal neovascularisation. Br J Ophthalmol 89:911–916
Binz N, Graham CE, Simpson K et al (2006) Long-term effect of therapeutic laser photocoagulation on gene expression in the eye. Faseb J 20:383–385
Shen WY, Yu MJ, Barry CJ, Constable IJ, Rakoczy PE (1998) Expression of cell adhesion molecules and vascular endothelial growth factor in experimental choroidal neovascularisation in the rat. Br J Ophthalmol 82:1063–1071
Guidolin D, Vacca A, Nussdorfer GG, Ribatti D (2004) A new image analysis method based on topological and fractal parameters to evaluate the angiostatic activity of docetaxel by using the Matrigel assay in vitro. Microvasc Res 67:117–124
Abramoff M, Magalhaes P, Ram S (2004) Image processing with ImageJ. Biophoton Int 11:36–42
Su EN, Alder VA, Yu DY, Yu PK, Cringle SJ, Yogesan K (2000) Continued progression of retinopathy despite spontaneous recovery to normoglycemia in a long-term study of streptozotocin-induced diabetes in rats. Graefes Arch Clin Exp Ophthalmol 238:163–173
Kuwabara T, Cogan DG (1960) Studies of retinal vascular patterns. I. Normal architecture. Arch Ophthalmol 64:904–911
Ruberte J, Ayuso E, Navarro M et al (2004) Increased ocular levels of IGF-1 in transgenic mice lead to diabetes-like eye disease. J Clin Invest 113:1149–1157
Gao G, Li Y, Gee S et al (2002) Down-regulation of vascular endothelial growth factor and up-regulation of pigment epithelium-derived factor: a possible mechanism for the anti-angiogenic activity of plasminogen kringle 5. J Biol Chem 277:9492–9497
Gao G, Li Y, Zhang D, Gee S, Crosson C, Ma J (2001) Unbalanced expression of VEGF and PEDF in ischemia-induced retinal neovascularization. FEBS Lett 489:270–276
Stellmach V, Crawford SE, Zhou W, Bouck N (2001) Prevention of ischemia-induced retinopathy by the natural ocular antiangiogenic agent pigment epithelium-derived factor. Proc Natl Acad Sci USA 98:2593–2597
Ham DI, Kim SJ, Chen J et al (2004) Central immunotolerance in transgenic mice expressing a foreign antigen under control of the rhodopsin promoter. Invest Ophthalmol Vis Sci 45:857–862
Lebherz C, Maguire AM, Auricchio A et al (2005) Nonhuman primate models for diabetic ocular neovascularization using AAV2-mediated overexpression of vascular endothelial growth factor. Diabetes 54:1141–1149
Lebherz C, Auricchio A, Maguire AM et al (2005) Long-term inducible gene expression in the eye via adeno-associated virus gene transfer in nonhuman primates. Hum Gene Ther 16:178–186
Rakoczy PE, Brankov M, Fonceca A, Zaknich T, Rae BC, Lai CM (2003) Enhanced recombinant adeno-associated virus-mediated vascular endothelial growth factor expression in the adult mouse retina: a potential model for diabetic retinopathy. Diabetes 52:857–863
No authors listed (1981) Diabetic retinopathy study. Report Number 6. Design, methods, and baseline results. Report Number 7. A modification of the Airlie House classification of diabetic retinopathy. Prepared by the Diabetic Retinopathy. Invest Ophthalmol Vis Sci 21:1–226
Antonetti DA, Lieth E, Barber AJ, Gardner TW (1999) Molecular mechanisms of vascular permeability in diabetic retinopathy. Semin Ophthalmol 14:240–248
Cunha-Vaz J, Bernardes R (2005) Nonproliferative retinopathy in diabetes type 2. Initial stages and characterization of phenotypes. Prog Retin Eye Res 24:355–377
Hellstedt T, Immonen I (1996) Disappearance and formation rates of microaneurysms in early diabetic retinopathy. Br J Ophthalmol 80:135–139
Caldwell RB, Bartoli M, Behzadian MA et al (2003) Vascular endothelial growth factor and diabetic retinopathy: pathophysiological mechanisms and treatment perspectives. Diabetes Metab Res Rev 19:442–455
Joussen AM, Poulaki V, Le ML et al (2004) A central role for inflammation in the pathogenesis of diabetic retinopathy. Faseb J 18:1450–1452
Joussen AM, Poulaki V, Qin W et al (2002) Retinal vascular endothelial growth factor induces intercellular adhesion molecule-1 and endothelial nitric oxide synthase expression and initiates early diabetic retinal leukocyte adhesion in vivo. Am J Pathol 160:501–509
Early Treatment Diabetic Retinopathy Study Research Group (1991) Grading diabetic retinopathy from stereoscopic color fundus photographs—an extension of the modified Airlie House classification. ETDRS report number 10. Ophthalmology 98:786–806
Dosso AA, Rungger-Brandle E, Leuenberger PM (2004) Ultrastructural alterations in capillaries of the diabetic hypertensive rat retina: protective effects of ACE inhibition. Diabetologia 47:1196–1201
Barber AJ (2003) A new view of diabetic retinopathy: a neurodegenerative disease of the eye. Prog Neuropsychopharmacol Biol Psychiatry 27:283–290
Gardner TW, Antonetti DA, Barber AJ, LaNoue KF, Levison SW (2002) Diabetic retinopathy: more than meets the eye. Surv Ophthalmol 47(Suppl 2):S253–S262
Ramsay RC, Goetz FC, Sutherland DE et al (1988) Progression of diabetic retinopathy after pancreas transplantation for insulin-dependent diabetes mellitus. N Engl J Med 318:208–214
No authors listed (1998) Early worsening of diabetic retinopathy in the Diabetes Control and Complications Trial. Arch Ophthalmol 116:874–886
Acknowledgements
This study, a joint research effort of the Diabetic Research Consortium (Perth), was funded by the Juvenile Diabetic Retinopathy Foundation (USA), the National Health and Medical Research Council (Australia) and Westpac Foundation (Australia). We thank R. Himbeck for the glucose measurements.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shen, WY., Lai, C.M., Graham, C.E. et al. Long-term global retinal microvascular changes in a transgenic vascular endothelial growth factor mouse model. Diabetologia 49, 1690–1701 (2006). https://doi.org/10.1007/s00125-006-0274-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-006-0274-8