
Abstract
Data derived from several recent studies implicate peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) in the pathogenesis of type 2 diabetes. Lacking DNA binding activity itself, PGC-1α is a potent, versatile regulator of gene expression that co-ordinates the activation and repression of transcription via protein-protein interactions with specific, as well as more general, factors contained within the basal transcriptional machinery. PGC-1α is suggested to play a pivotal role in the control of genetic pathways that result in homeostatic glucose utilisation in liver and muscle, beta cell insulin secretion and mitochondrial biogenesis. This review focuses on the role of PGC-1α in glucose metabolism and considers how PGC-1α links cellular glucose metabolism, insulin sensitivity and mitochondrial function, and why defects in PGC-1α expression and regulation may contribute to the pathophysiology of type 2 diabetes in humans.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The incidence of non-insulin dependent type 2 diabetes—a disease once mildly prevalent in older, overweight adults—is increasing worldwide at an alarming rate in progressively younger populations [1]. By 2025, epidemiologists have predicted that one in every three American children born in the year 2000 will carry a significant lifetime risk of developing type 2 diabetes [2], and therefore become prone to premature cardiovascular disease, blindness, kidney failure and amputations. Pathophysiologically characterised by insulin resistance, pancreatic beta cell dysfunction and enhanced hepatic gluconeogenesis, the exact aetiology of type 2 diabetes is still unknown. However, obesity (particularly that centred in the abdominal region), a sedentary lifestyle, advancing age and alterations in genetic programmes that control glucose homeostasis are key contributing factors [3].
Peroxisome proliferator-activated receptor (PPAR)-γ coactivator-1α (PGC-1α, also known as PPARGC1A), a multifunctional transcriptional protein, acts as a ‘molecular switch’ in pathways controlling glucose homeostasis, and may be a critical link in the pathogenesis of type 2 diabetes. This review highlights the role of PGC-1α in fuel metabolism and considers how both large differences in PPARGC1A expression (such as ablation, knockdown by RNA interference [RNAi] or overexpression in mice) and sequence substitutions at the PPARGC1A locus and their haplotype structure contribute to glucose intolerance in vivo.
PGC-1α: a versatile coactivator
Eukaryotic gene regulation is co-ordinated by a multitude of factors that exist in dynamic equilibrium to control mRNA transcription from the approximately 2.0-m-long tightly coiled DNA within the nucleus in human cells. Chromatin remodelling enzymes such as histone acetyltransferases (HATs)/deacetylases enable access of transcription factors to cis-acting promoter or enhancer regions, and supplementary factors termed coregulators (coactivators and corepressors), which do not bind DNA directly, are recruited by, and alter the transactivation potency of, gene-specific transcription factors [4]. Coactivators typically stimulate transcription factor activity and target gene transcription by remodelling of chromatin and by forming complexes with HAT and proteins contained within the basal transcriptional apparatus. Corepressors disrupt these interactions or recruit enzymes (i.e. deacetylases) that inhibit transcription [5]. Coregulators, therefore, have a pivotal role in the regulation of transcription and assist in the exquisite spatio-temporal control of eukaryotic gene expression in vivo.
PGC-1α, originally identified as a coactivator of PPARγ [6], has since been shown to increase the transcriptional activity of PPARα and many additional nuclear receptor families, including members of the oestrogen, retinoid X, mineralocorticoid, glucocorticoid (GR), liver X (LXR), pregnane X, the constitutive androstane (CAR), vitamin D and thyroid hormone receptor families [6–17]. PGC-1α can also bind unliganded nuclear receptors, as in the case of the orphan hepatocyte nuclear factor (HNF) 4α, farnesoid X receptor (FXR), and oestrogen-related receptor (ERR) α, suggesting that their conformations are conducive to ligand-independent mechanisms of gene regulation [18–20]. PGC-1α targets are not confined to the nuclear receptor superfamily, however, and this versatile coactivator associates with a diverse array of other transcription factors involved in the insulin and glucagon signalling pathway, including the forkhead/winged helix protein family member FOXO1 [21].
PGC-1α, like few other known coregulators, also influences downstream events in mRNA biogenesis, such as pre-mRNA elongation and splicing via domain-specific protein-RNA interactions [22, 23]. Although the specific RNA targets of PGC-1α await identification, this function may aid the orchestration of complex genetic pathways such as glucose homeostasis.
Cofactors: coactivators and corepressors
Gene expression is a complex process that involves the binding of transcription factors to specific cis-regulatory DNA elements and the amplification or repression of transcription factor activity by coactivators or corepressors, respectively. Cofactors do not bind to DNA. Because of functional differences, two classes of cofactors may be distinguished:
Class I cofactors possess enzymatic activities resulting in histone modification or alterations in DNA tertiary structure. Coactivators of this class loosen the tightly coiled DNA or modify histones by acetylation or methylation, thereby allowing access of other proteins to the DNA. Conversely, class I corepressors make DNA less accessible and often possess histone deacetylase activity.
Class II cofactors lack enzymatic activities that modify histones. After their recruitment by transcription factors, coactivators of this class interact with RNA polymerase II and other accessory proteins of the transcription apparatus, thereby amplifying the transcriptional activity of the respective transcription factor. As a result of their lack of histone acetylase activity, such coactivators potentiate transfactor activity from naked DNA used in transfection experiments, but require interactions with class I coactivators for amplification of transcriptional responses from chromosomes in vivo. Class II corepressors abolish productive interaction with the transcription apparatus.
PGC-1α is a class II coactivator, but also interacts with factors involved in RNA splicing and transcript elongation, and may therefore play a role in mRNA maturation.
PGC-1α protein structure and interactions
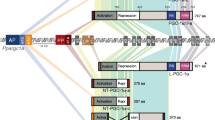
Lacking HAT activity, PGC-1α associates via an acidic N-terminal activation domain with other coregulators that acetylate chromatin, including cAMP response element-binding protein (CREB)-binding protein (CBP/p300) and steroid receptor coactivator 1 [24]. Located within the N-terminal domain is the first of three LXXLL (L1–L3) motifs (Fig. 1). These aptly named nuclear receptor boxes [25] are present in the close relatives of PGC-1α, PGC-1β (PERC, PPARGC1B) and PGC-1-related coactivator (PRC, PPRC1). Combined with the N-terminal activation domain, the L2 motif is sufficient for most, but not all PGC-1α-nuclear receptor interactions. ERRα binds PGC-1α via the L3 motif, whereas ERRγ requires both the L2 and L3 motifs for its coactivation [26, 27].

Schematic representation of PGC-1α protein structure and alignment of conserved functional domains in orthologues. In humans, the N-terminal activation domain (AD) harbours a two-amino acid insertion (light blue box) that is absent in other species. Three LXXLL motifs (L1–L3) are completely conserved, as is a host cell factor binding site (HCB) within a region shown to bind MEF2C. Three p38 MAPK phosphorylation sites (red ovals) are located within a negative regulatory region. A novel DEAD box, present in human PGC-1α and conserved in Canis familiaris lies proximal to the putative casein kinase (CK) 1 (yellow oval) and CK2 (grey oval) phosphorylation sites. RS protein interaction domains, the highly conserved nuclear localisation (NL) signal and the RNA recognition motif (RRM) are indicated. The C-terminal region has been shown to bind the TRAP220 mediator complex, splicing factors (U1-70K) and several transcription factors. Functional domains and interacting proteins are discussed in the main text. CAR constitutive androstane receptor, ER oestrogen receptor, RXR retinoid X receptor, SRC-1 steroid receptor coactivator 1, USF upstream stimulatory factor, MYBBP1a
The L3 motif marks the upstream boundary of a negative regulatory region that aids the docking of PPARγ, FXR and the nuclear respiratory factors NRF-1 and NRF-2. The central hinge region (amino acids 400–500) harbours a tetrapeptide (DHDY) representing a host cell factor docking site [HCB, HBM, (D/E)HXY] that is also present in PGC-1β and PRC [28]. Although highly conserved in mammals and implicated in cell cycle regulation and viral infection, the function of this domain in PGC-1α awaits clarification. The region just distal to this site is required for coactivation of the insulin-sensitive GLUT4 (now known as SLC2A4) via MADS box transcription enhancer factor (MEF) 2C [29].
The C-terminal region is particularly relevant for type 2 diabetes. As shown in mice, it is required for the interaction of PGC-1α with FOXO1 and HNF4α, both of which contribute to the regulation of gluconeogenic target genes [18, 21]. Harbouring two arginine/serine-rich (RS) domains and an RNA recognition motif (RRM), it is not yet clear whether the C-terminal region requires RNA binding for its interaction with members of the forkhead transcription family. RS domains interact with components of the spliceosome [30] and with TRAP220, required for post-chromatin-remodelling transcription initiation events [31], whereas the RRM domain is implicated in the control of transcriptional elongation. In addition to the intervening nuclear localisation signal, the RS and RRM domains were indispensable for translocation of PGC-1α to the nucleus and for altering the splicing pattern of a fibronectin minigene placed under the control of a PGC-1α-coactivated transcription factor [22]. The RS domains in PGC-1α, like those in selected other RNA-binding proteins, contain several Akt/protein kinase (PK) B consensus sites (RXRXXS/T). Phosphorylation of such motifs in SRp40 is associated with alternative splicing of PKCβII in an insulin dependent fashion [32] and may therefore be critical for PGC-1α-mediated splicing effects. Furthermore, occupation of amino acids 209–213 by ERRα abrogated the pattern of PGC-1α nuclear localisation normally associated with splicing [26]. Therefore, co-operative associations among proteins that bind to different regions of PGC-1α may regulate its RNA-related functions. Moreover, these studies support recent concepts suggesting that pre-mRNA processing occurs co-transcriptionally rather than post-transcriptionally and may depend on transcriptional cofactors, including PGC-1α [33–35].
Taxonomic comparison of PGC-1α proteins
Attesting to their functional importance, all major PGC-1α interacting domains, including those involved in RNA binding, are highly conserved across recently diverged mammalian species (Fig. 1). Key interacting motifs, especially the nuclear receptor boxes, are conserved in lower, genetically more distant eukaryotes, such as the green-spotted puffer fish (Tetraodon nigroviridis), implicating similar nuclear functions. However, what is different in higher chordates is the presence of sequence modifications in motifs and/or phosphorylation sites that may adapt PGC-1α to more complex cellular pathways.
An N-terminal insertion of two amino acids (ES) in the activation domain is exclusively found in humans and may influence the binding to, and the coactivation of, target proteins. Furthermore, by aligning the PGC-1α proteins taxonomically (using protein sequences obtained from http://www.ensembl.org, last accessed in April 2006, and aligned with Clustal W 1.83 software, available from http://www.ebi.ac.uk/clustalw/, last accessed in April 2006), we have identified a DEAD box motif, just proximal to the RS and RNA binding motifs, that is contained only in higher eukaryotes. Proteins with this domain typically possess RNA helicase activity and aid in RNA processing, splicing and nuclear export of nascent RNA molecules [36]. DEAD box RNA helicases may also disrupt RNA-protein interactions [37] and repress or coactivate nuclear receptors [38]. Although PGC-1α lacks signature motifs associated with RNA helicase activity, its DEAD box may facilitate docking of proteins that are critical for RNA regulatory functions. Mutational analyses will be required to address this possibility.
Regulation of PPARGC1A expression and PGC-1α activity
To prevent its uncontrolled and untimely activation, PGC-1α activity is regulated at several levels. Phosphorylation of three conserved Thr and Ser residues within the negative regulatory domain by p38 mitogen-activated protein kinase (MAPK) relieves transcriptional repression and affects the turnover of PGC-1α and its ability to interact with transcription factors and/or accessory proteins [39]. A MAPK-sensitive repressor mechanism is implicated in the coactivation of the glucocorticoid receptor (GR) by PGC-1α [40]. Such a mechanism also controls the coactivation of PPARα at a multipartite response element within the promoter region of the gene encoding uncoupling protein (UCP) 1 [11]. Conversely, this mechanism does not regulate coactivation of PPARγ, RAR or LXR by PGC-1α [10]. The MYB binding protein (P160) 1a was identified as a PGC-1α repressor and interacts with the inhibitory domain between amino acids 170 and 350 to negatively regulate mitochondrial respiration [41], while upstream stimulatory factor represses PGC-1α at a more N-terminal region [42].
Acetylation/deacetylation is another post-translational mechanism that affects specific PGC-1α functions. SIRT1 and SIRT3, two homologues of the Saccharomyces cerevisiae silencing information regulator 2 (Sir2), which delay ageing in several species, deacetylate PGC-1α [43, 44]. Like Ppargc1a, both Sirt1 and Sirt3 display tissue-specific expression and are induced by caloric restriction. While deacetylation of PGC-1α by SIRT1 enhances hepatic gluconeogenesis without affecting mitochondrial proliferation [43], deacetylation by SIRT3 regulates mitochondrial function, at least in brown adipocytes [45].
Notwithstanding the importance of post-translational mechanisms for its activity, the regulation of PPARGC1A expression itself appears to be central to specific transcription programmes, including the fasted liver response (see below) and metabolic adaptions to exercise in skeletal muscle.
PGC-1α activity may also be regulated by alternative splicing. In rats, a single bout of exercise induced a smaller PGC-1α protein translated from a transcript lacking exon 8 [46]. Distinct human PGC-1α isoforms have been identified that lack specific domains and motifs (unpublished data). Although their functional significance and method of control await further analyses, such isoforms may allow for a wider spectrum of PGC-1α function.
PGC-1α and glucose homeostasis
Cold-induced upregulation of PGC-1α levels provided the first clue that this coactivator acts as a nuclear sensor of adverse environmental stimuli [6]. PGC-1α integrates metabolic pathways that support mammalian survival during prolonged starvation or hibernation [47]. Such pathways include increased hepatic gluconeogenesis and β-oxidation, more effective mitochondrial function, insulin-independent glucose uptake and metabolism in muscle and reduced insulin secretion (thereby reducing glucose entry into adipose tissue and providing glucose for brain and kidney).
Skeletal muscle
In myocytes (Fig. 2), glucose uptake is chiefly regulated by the transmembrane transporter GLUT4 which, in response to Akt/PKB-mediated insulin signalling and contractile-induced activation of the AMP-activated kinase (AMPK), is redirected from its sequestered cytoplasmic location to the cell membrane, thereby stimulating glucose entry [48]. In type I skeletal muscle fibres of rodents, Ppargc1a expression is rapidly upregulated by AMPK and calcium/calmodulin-dependent protein kinase IV [49, 50]. The latter enzyme phosphorylates CREB and aids the release of MEF2 proteins from repressors of class II histone deacetylases [51]. MEF2C binds to two consensus elements within, and activates the Ppargc1a promoter. Since PGC-1α coactivates MEF2C, a positive-feedback loop generates strong expression of Ppargc1a in cardiac and skeletal muscle [52]. Elegant in vivo studies in mice, using bioluminescence imaging during motor nerve stimulation, demonstrated that both MEF2C binding sites and the CREB response element were necessary for Ppargc1a promoter activation in skeletal muscle [53]. PGC-1α not only facilitates glucose entry, but also ensures effective glucose utilisation. Ectopic expression of Ppargc1a in C2C12 cells upregulates nuclear genes required for mitochondrial biogenesis and proliferation, including those encoding NRF-1 and NRF-2 and mitochondrial transcription factors A, B1M and B2M [54, 55]. In addition, PGC-1α induces oxidative phosphorylation gene expression via coactivation of ERRα and GA-repeat binding protein α/β [56, 57]. With increased physical activity, the number of, and expression of Ppargc1a in, type I skeletal myofibres increases in animals. In fact, studies in transgenic mice suggest that robust levels of PGC-1α may induce a fibre-type switch from fast-twitch, type II muscle fibres to slow-twitch, type I fibres [58].

Model of PGC-1α interactions that are implicated in transcriptional amplification and mRNA maturation. CAR constitutive androstane receptor, NR nuclear receptor, SRC-1 steroid receptor coactivator 1
Liver
PGC-1α integrates the metabolic adaptions of the rodent liver to fasting (Fig. 3). Hepatic Ppargc1a expression is induced by glucagon and further enhanced by the synergistic effects of glucagon with glucocorticoids via a CREB response element in its promoter [59]. In combination with CREB, FOXO1 enhances Ppargc1a promoter activity at three insulin response sequences [60]. Elevations of PGC-1α induce gluconeogenic enzymes such as phosphenolpyruvate carboxykinase (PCK) and glucose-6-phosphatase (G6PC), thereby enhancing glucose output. Efficient stimulation of the Pck1 promoter requires coactivation by both the GR and HNF4α [18]. In livers of Ppargc1a −/− mice, the programme of hormone-stimulated gluconeogenesis is defective, while constitutively activated gluconeogenesis is maintained by elevated levels of CAAT/enhancer-binding protein β [61]. The gluconeogenic function of PGC-1α is also regulated at the post-translational level via SIRT1-induced deacetylation [43]. Importantly, both hepatic Ppargc1a expression and gluconeogenesis are aberrantly induced in mouse models of insulin resistance and type 2 diabetes [62]. Insulin abolishes gluconeogenesis by disrupting the interaction of PGC-1α with FOXO1, as phosphorylation of FOXO1 in response to insulin/Akt signalling results in its nuclear exclusion [21, 63].

Regulation of glucose metabolism in skeletal muscle by PGC-1α. Ca2+ signalling and kinases induced by muscle contraction release MEF2 proteins from class II histone deacetylases (HDACs). PGC-1α coactivates the MEF2C-dependent transcription of GLUT4. Sustained exercise induces PPARGC1A transcription via a positive-feedback mechanism. As a result, GLUT4 transcription is further amplified, and coactivation of genes involved in oxidative phosphorylation ensures effective glucose utilisation. GABAb GA-repeat binding protein α/β
Enhanced fatty acid oxidation, supporting hepatic ATP production during fasting, is induced by PGC-1α via PPARα coactivation [7, 64]. The mammalian tribbles homologue TRIB3 is a downstream target of PPARα that binds to, and prevents activation of, Akt/PKB [65]. PGC-1α-deficient mice, generated by Ppargc1a RNAi virus delivery to the liver, showed hypoglycaemia, enhanced hepatic insulin sensitivity and reduced expression of Trib3. Similarly, glucose tolerance was improved in mice transduced with Trib3 RNAi virus. Conversely, overexpression of Trib3 in liver abrogated the enhanced insulin sensitivity in PGC-1α-deficient mice. Thus, induction of Trib3 via PGC-1α mediated coactivation of PPARα induces hepatic insulin resistance [66].
Adipose tissue
White adipose tissue is essential for energy storage, while brown adipose tissue is specialised in adaptive thermogenesis [67]. These functional differences are reflected at the level of expression of genes controlling mitochondrial proliferation and function, including PPARGC1A. Ectopic expression of Ppargc1a in white adipocytes increases the expression of Ucp1 and genes encoding respiratory chain proteins and fatty acid oxidation enzymes [6, 68], and causes white adipocytes to acquire features of brown adipocytes. In white adipose tissues of ob/ob mice, the expression of transcripts encoding mitochondrial proteins decreases with the onset of obesity. Thiazolidinedione treatment of ob/ob mice induces expression of Ppargc1a and increases mitochondrial mass and energy expenditure [69]. Induction of PGC-1α enhances expression of the gene for glycerol kinase by releasing corepressors [70]. These data imply an insulin-sensitising role for PGC-1α in white adipose tissue. Two models of Ppargc1a −/− mice showed variations in body weight homeostasis. While both PGC-1α-deficient lines exhibited cold intolerance, one line showed an age-related increase in body fat [71], whereas the other line was lean [61]. Differences in genetic backgrounds, neurological phenotypes (reduced locomotor activity in the former, hyperactivity in the latter) or gene targeting methods may have contributed to the phenotypical differences.
Beta cells
Ppargc1a expression is upregulated in beta cells from animal models of type 2 diabetes [72]. Ectopic Ppargc1a expression in rat islets and INS-1 cells reduced both early and delayed glucose-stimulated insulin secretion and suppressed membrane depolarisation without affecting the basal secretory apparatus. Concomitantly, G6pc mRNA increased several-fold, while expression of genes encoding glucokinase, glycerol-3-phosphate dehydrogenase, GLUT2 and the transcription factors HNF4α, HNF1α and insulin promoting factor 1 were downregulated. The effects of PGC-1α on insulin secretion were mimicked by ectopic expression of G6pc, but partially corrected by ectopic glucokinase gene expression. Hence, futile cycling of glucose may have reduced the insulin response [70]. Ucp2 could have been another effector of PGC-1α upregulation, since enhanced beta cell UCP2 level/activity diminishes glucose-induced insulin secretion in both humans and animal models [73–75]. The upregulation of Ppargc1a expression in diabetic animal models is not understood, but fatty acids enhance Ppargc1a expression and impair beta cell function in rat islets [76]. Also, incomplete inactivation of FOXO1 may have contributed, as FOXO1 haploinsufficiency rescued beta cell failure in mice lacking the gene for insulin receptor substrate 2 (Irs2 −/−) [77] and a gain-of-function FOXO1 mutation targeted to liver and beta cells enhanced hepatic gluconeogenesis and impaired beta cell compensation [78].
Functional studies in humans
Studies in humans are consistent with the functions of PGC-1α defined in animal and cell culture models. PPARGC1A is strongly expressed in human skeletal muscle, but was surprisingly found to predominate in the moderately oxidative, glycolytic type IIa muscle fibres as opposed to the highly oxidative type I fibres [79]. Although 60 min of cycling did not increase PGC-1α levels, such exercise augmented GLUT4 expression by an early non-transcriptional response, whereby MEF2C is released from histone deacetylase 5 and coactivated by PGC-1α to enhance GLUT4 expression and glucose uptake [80]. Upon moderate- to high-intensity endurance training, however, PGC-1α levels are robustly increased and effects on GLUT4 expression are further amplified [79, 81]. Elevations of plasma fatty acids resulting from infusion of triglyceride emulsions caused downregulation of PPARGC1A and several nuclear-encoded mitochondrial genes in skeletal muscle of healthy human subjects [82]. As fatty acids induce PGC-1α in rat pancreatic islets [76], tissue-specific regulation of PPARGC1A expression by fatty acids can be implied. PPARGC1A and PPARGC1B expression was compared in muscle biopsies from young and elderly dizygotic and monozygotic twins, before and after insulin stimulation [83]. Insulin increased, and older age reduced, the expression of both genes. Sex, birthweight and aerobic capacity also modulated PPARGC1A expression. Furthermore, PPARGC1A expression correlated with insulin-stimulated glucose uptake and oxidation, whereas PPARGC1B expression correlated with fat oxidation and non-oxidative glucose metabolism. In type 2 diabetes, coordinated downregulation of PPARGC1A and its downstream targets involved in oxidative phosphorylation was observed in comparison with controls [84]. Consistent with these results are studies showing reduced expression of PPARGC1A and PPARGC1B in muscle tissues of non-diabetic subjects with a positive family history [85]. Reduced expression of PPARGC1A would be expected to impair mitochondrial function, thereby reducing insulin sensitivity. Associations of insulin resistance with increased levels of triglycerides and decreased mitochondrial oxidative activity and ATP production in skeletal muscle of healthy, lean elderly subjects have been reported [86]. Impaired mitochondrial function was also observed in insulin-resistant offspring of patients with type 2 diabetes [87], and may result in the accumulation of intracellular metabolites that impede insulin signalling [88].
Genetic studies in humans
Since transcriptional coregulators act at the amplification step of gene expression, minor imbalances in coregulator expression or activity levels may contribute to the pathogenesis of multifactorial diseases such as type 2 diabetes. Thus, functional sequence substitutions in PPARGC1A may affect one or more of the three pathophysiological hallmarks of type 2 diabetes, insulin sensitivity, insulin secretion and hepatic gluconeogenesis (Fig. 4). PPARGC1A has been mapped to chromosome 4p15.1–2 [89]. This chromosomal region has been associated with basal insulin levels in Pima Indians [90], abdominal subcutaneous fat in the Quebec Family Study [91], high BMI in women of Utah pedigrees [92], obesity indices in Mexican Americans [93] and systolic blood pressure in families from the Netherlands [94]. Diabetes-related phenotypes have also been associated with single-nucleotide polymorphisms or haplotypes at the PPARGC1A locus [95–100].

Regulation of hepatic gluconeogenesis by PGC-1α. In the fasted state, PPARGC1A expression is stimulated by glucagon- and glucocorticoid-mediated activation of CREB and transactivation by FOXO1. Increased PPARGC1A mRNA levels and PGC-1α activity (the latter enhanced by SIRT1 deacetylation), promote gluconeogenic gene expression by coactivation of HNF4α and FOXO1. PGC-1α also aids the induction and FXR-mediated transactivation of the gene encoding PPARα, which contributes to fatty acid oxidation and inhibits Akt/PKB phosphorylation of FOXO1, via TRIB3, thereby ensuring FOXO1 nuclear entry. In the postprandial state, FOXO1 is phosphorylated by Akt/PKB and excluded from the nucleus, resulting in decreased gluconeogenesis
Associations of a Gly482Ser polymorphism with diabetes and related traits were studied in several populations. The Ser variant increased the relative risk of type 2 diabetes in a Danish population. The association was confirmed in a replication study (p=0.0007 for the studies combined) [95]. In a Japanese population, the distribution of two-loci haplotypes comprising the Gly482Ser and the Thr394Thr polymorphisms differed between 834 type 2 diabetic subjects and 1074 controls (p=0.00003) [101]. Carriers of the Ser variant, in comparison with the Gly482Gly subjects, showed a 1.6-fold higher risk of conversion from IGT to type 2 diabetes in the Study to Prevent Non-Insulin-Dependent Diabetes Mellitus (STOP-NIDDM) trial [96]. In twins, the Ser variant was associated with a greater age-dependent reduction in muscle PPARGC1A expression, suggesting an explanation for the age dependence of type 2 diabetes susceptibility [83]. No associations of Gly428Ser with type 2 diabetes were noted in French and Austrian populations or in Pima Indians [102–104]. However, in all European populations studied, the frequency of the Ser variant tended to be higher in patients with type 2 diabetes than in controls. Among non-diabetic Pima Indians, carriers of the Ser variant displayed higher insulin secretory responses to intravenous and oral glucose, higher rates of lipid oxidation, lower plasma free fatty acid concentrations and smaller subcutaneous adipocytes [104]. No associations between the Gly482Ser SNP and diabetes-related traits, such as insulin resistance, antilipolysis, insulin secretion, maximal oxygen consumption or intramyocellular lipid content, were observed in non-diabetic German and Dutch populations [105]. Possible explanations for the lack of replication across studies are numerous and may include statistical fluctuations, insufficient power of some studies, genetic or phenotypical heterogeneity within and among study samples and differences in environmental factors.
The Gly482Ser polymorphism is located two amino acids downstream of the DEAD box motif, yet its functionality has not been proven (Fig. 5). In humans, the substitution of Ser—present in the wild-type protein of mice and rats—by the more common Gly results in the loss of a consensus phosphorylation site (in silico probability of 97%, NetPhos 2.0 analysis, program available from http://www.cbs.dtu.dk/services/NetPhos, last accessed in April 2006). However, humans possess a new putative phosphorylation site, which results from a Gly to Ser substitution at amino acid 487. pSer487, in turn, is predicted to be part of a highly conserved casein kinase (CK) 1 site (pS-X-X-S/T). Putative CK2 and glycogen synthase kinase 3 sites are located several amino acids downstream. Such multisite phosphorylation domains targeted by CK1 and other kinases have been shown to be critical for nuclear exclusion of transcription factors, including FOXO1 [106], and may therefore play a role in the subcellular localisation of PGC-1α. Moreover, their close proximity to the DEAD box motif may affect protein binding to this region. Clearly, detailed studies are warranted to define possible functional and phenotypical consequences of the variant site located at amino acid 482.

Amino acid alignment of sequences spanning a newly identified DEAD box domain in the human PGC-1α protein. A Gly482Ser substitution and adjacent polymorphic sites are indicated as are a putative CK1 [phospho(P)Ser-X-X-Ser] and overlapping CK2 [Ser/Thr-X-X-D/E] /glycogen synthase kinase (GSK) 3 sites
The Gly482Ser polymorphism may not be the main or sole causative site, but may be part of a haplotype harbouring other functional sites. By characterising and typing sequence substitutions across the entire PPARGC1A locus, we identified two distinct haplotype blocks, each containing five common haplotypes. Haplotype block 1 comprised the promoter and extended into intron 2, while haplotype block 2 extended from intron 2 beyond the polyA signals. Several promoter SNPs were located in transcription factor binding sites and affected transactivation of reporter constructs in an allele-specific manner. Score testing revealed moderate associations of specific block 1 haplotypes with beta cell indices. However, a common block 2 haplotype (containing Gly at codon 482) was associated with the strongest insulin secretory response to glucose in 405 glucose-tolerant subjects (p<0.01) and conferred the lowest risk of type 2 diabetes (p=0.009), as determined by comparison of 494 cases and 1,478 controls. Thus, effects of PGC-1α on beta cell function added to the risk of type 2 diabetes in our population. Surprisingly, no associations with indices of insulin resistance were observed, but errors in measurement may have contributed [103]. These results are consistent with animal studies showing an inhibitory effect of PGC-1α on insulin secretion [72]. That increased expression of PPARGC1A in beta cells and liver plays a role in type 2 diabetes is supported by experiments showing reversal of diabetes and hepatic steatosis by Ppargc1a antisense oligonucleotides in mice fed a saturated fat-rich diet [107].
Two recent studies also reported two haplotype blocks at the PGC-1α locus and associations with type 2 diabetes. In a Korean population comprising 762 cases and 303 controls, two-loci promoter haplotypes showed associations with early-onset type 2 diabetes [108]. In 159 non-diabetic offspring of type 2 diabetic Finns, three common block 2 haplotypes accounting for 80% of haplotypes defined by six SNPs were identified, and associations of specific haplotypes with diabetes-related traits were observed [109]. One SNP with a minor allele frequency of 49% in Austrians was not considered (Electronic Supplementary Material [ESM] Fig. 1). Hence, comparison of haplotype effects among studies are limited. Nevertheless, the haplotype carrying minor alleles in codons 482 (Ser) and 528 was associated with a high glucose AUC in OGTTs in Finns and the lowest or highest scores for beta cell function or type 2 diabetes, respectively, in Austrians, suggesting some consistency of results. Thus, haplotypes comprising Ser at codon 482 showed associations with diabetes-related traits in Japanese, Finnish and Austrian populations.
Further fine-mapping of haplotypes and use of such haplotypes in association studies, along with functional studies, should identify the SNP(s) underlying the associations of PPARGC1A with type 2 diabetes and related traits. Such studies should be greatly facilitated by data emerging from the International HapMap Project [110]. Currently available results of 90 samples from a Utah population with ancestry from northern and western Europe would be consistent with one haplotype block extending from intron 2 beyond exon 13, and one or several smaller blocks in intron 2 and the promoter region. Data from 45 unrelated Japanese individuals in Tokyo, Japan, are more consistent with a single block in the promoter region extending into intron 2 (ESM Fig. 2), but a higher SNP density will be required for haplotype fine mapping.
Human haplotypes and utility of the HapMap Project
No two individuals selected at random will have a completely identical genetic make-up, but will instead possess either subtle (SNPs) or more extensive (duplications, inversions) structural alterations that may (or may not) affect the risk of developing disease. The human genome possesses about ten million polymorphisms that occur with a frequency >1% in populations. Time and cost prohibit the ascertainment of associations between all individual variants and diseases. Most SNPs result from a single mutational event on a specific chromosomal background. The set of alleles observed within a chromosomal region is termed a haplotype. New haplotypes result from new mutations or from crossing over during meiosis. Theoretically, within a genomic stretch harbouring n SNPs in a population, 2n haplotypes may be found, but empirical studies show that nearby SNPs are strongly correlated. As a result, in many chromosomal regions, only few haplotypes are observed that are inherited as blocks comprising 5 to >100 kb. Typing of the few discriminatory SNPs will therefore provide enough information to identify all common haplotypes within a block. The genomic region shown below harbours eight SNPs (red) and may theoretically contain 256 SNP combinations or haplotypes. However, phased chromosome analysis may reveal only five common haplotypes that can be unambiguously identified by the typing of four SNPs (bold red).
AGTG..TATG..GCAG..GTAC..ATGC..GTGA..ATAC..GATT
AGTG..TATG..GCAG..GTAC..ATGC..GTGA..ATAC..GACT
AGTG..TATG..GCAG..GTCC..ATGC..GTGA..ATGC..GATT
AGTG..TAGG..GCAG..GTAC..ATGC..GTGA..ATAC..GACT
AGCG..TATG..GCGG..GTAC..ATCC..GTAA..ATAC..GATT
Simple in theory, haplotype typing has been successfully used to study single gene disorders wherein the disease causing mutation(s) has been identified in one of several haplotypes of known structure. However, haplotype tagging has proved more challenging in analyses of complex diseases such as type 2 diabetes, since the complete haplotype structure of all possible disease-causing genes has not been fully defined. The International HapMap Consortium provides a public database that compares common variations in the human genome among four defined populations [CEU: CEPH (Utah residents with ancestry from northern and western Europe); CHB: Han Chinese in Beijing, China; JPT: Japanese in Tokyo, Japan; YRI: Yoruba in Ibadan, Nigeria] (http://www.hapmap.org/, last accessed in April 2006). The consortium has defined a fine-scale genetic map of the human genome that is based on genotyping at least one common SNP every 5 kb of DNA (Phase I). Phase II is attempting to genotype an additional 4.6 million SNPs in each of the HapMap samples to further refine haplotype structures and to ultimately allow for comprehensive genome-wide association studies.
Future prospects
Considerable progress has been made in our understanding of how coregulators affect gene expression at promoters or enhancers. A number of transcriptional programmes have been elucidated that are integrated by PGC-1α. Multiple transcription factors and an even greater number of target genes are activated by PGC-1α. While signalling cascades and post-translational modifications that mediate the interactions of PGC-1α with other factors have been identified, additional studies will be required to define the specificity of interactions and the respective signal transduction pathways. Much of the current knowledge about PGC-1α interactions and functions stems from studies in model systems or in vitro assays. More direct experimentation, including chromatin immunoprecipitation and visualisation of specific interactions in living cells, will be required to address the human situation, which, in comparison with cell and animal models, is most likely associated with much smaller variations in PPARGC1A expression. Very little is known about the role of PGC-1α in mRNA maturation and the physiological relevance of PGC-1α isoforms. These questions must be addressed to rationalise the full spectrum of PGC-1α activities. The functionality of sequence substitutions and their tissue-specific effects warrant further characterisation. Thus, further study of the PPARGC1A locus in the context of its downstream targets and other genetic and environmental factors promises new insight into the pathogenesis of complex human disorders, including type 2 diabetes.
Abbreviations
- AMPK:
-
AMP-activated kinase
- CK:
-
casein kinase
- CREB:
-
cAMP response element-binding protein
- CBP/p300:
-
CREB-binding protein
- ERR:
-
oestrogen-related receptor
- ESM:
-
Electronic Supplementary Material
- FXR:
-
farnesoid X receptor
- G6PC:
-
glucose-6-phosphatase
- GR:
-
glucocorticoid receptor
- HAT:
-
histone acetyltransferase
- HNF:
-
hepatocyte nuclear factor
- LXR:
-
liver X receptor
- MAPK:
-
mitogen-activated protein kinase
- MEF:
-
MADS box transcription enhancer factor
- NRF:
-
nuclear respiratory factor
- PCK:
-
phosphoenolpyruvate carboxykinase
- PK:
-
protein kinase
- PPAR:
-
peroxisome proliferator-activated receptor
- PGC-1α:
-
PPARγ coactivator-1α
- PRC:
-
PGC-1-related coactivator
- RNAi:
-
RNA interference
- RRM:
-
RNA recognition motif
- RS:
-
arginine/serine-rich
- SNP:
-
single-nucleotide polymorphism
- UCP:
-
uncoupling protein
References
Dietz WH (2004) Overweight in childhood and adolescence. N Engl J Med 350:855–857
Narayan KM, Boyle JP, Thompson TJ, Sorensen SW, Williamson DF (2003) Lifetime risk for diabetes mellitus in the United States. JAMA 290:1884–1890
O’Rahilly S, Barroso I, Wareham NJ (2005) Genetic factors in type 2 diabetes: the end of the beginning? Science 307:370–373
McKenna NJ, O’Malley BW (2002) Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 108:465–474
Freedman LP (1999) Increasing the complexity of coactivation in nuclear receptor signaling. Cell 97:5–8
Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM (1998) A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92:829–839
Vega RB, Huss JM, Kelly DP (2000) The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol 20:1868–1876
Knutti D, Kaul A, Kralli A (2000) A tissue-specific coactivator of steroid receptors, identified in a functional genetic screen. Mol Cell Biol 20:2411–2422
Tcherepanova I, Puigserver P, Norris JD, Spiegelman BM, McDonnell DP (2000) Modulation of estrogen receptor-alpha transcriptional activity by the coactivator PGC-1. J Biol Chem 275:16302–16308
Oberkofler H, Schraml E, Krempler F, Patsch W (2003) Potentiation of liver X receptor transcriptional activity by peroxisome-proliferator-activated receptor gamma co-activator 1 alpha. Biochem J 371:89–96
Oberkofler H, Esterbauer H, Linnemayr V, Strosberg AD, Krempler F, Patsch W (2002) Peroxisome proliferator-activated receptor (PPAR) gamma coactivator-1 recruitment regulates PPAR subtype specificity. J Biol Chem 277:16750–16757
Delerive P, Wu Y, Burris TP, Chin WW, Suen CS (2002) PGC-1 functions as a transcriptional coactivator for the retinoid X receptors. J Biol Chem 277:3913–3917
Huss JM, Kopp RP, Kelly DP (2002) Peroxisome proliferator-activated receptor coactivator-1alpha (PGC-1alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1alpha. J Biol Chem 277:40265–40274
Schreiber SN, Knutti D, Brogli K, Uhlmann T, Kralli A (2003) The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor alpha (ERRalpha). J Biol Chem 278:9013–9018
Wu Y, Delerive P, Chin WW, Burris TP (2002) Requirement of helix 1 and the AF-2 domain of the thyroid hormone receptor for coactivation by PGC-1. J Biol Chem 277:8898–8905
Shiraki T, Sakai N, Kanaya E, Jingami H (2003) Activation of orphan nuclear constitutive androstane receptor requires subnuclear targeting by peroxisome proliferator-activated receptor gamma coactivator-1 alpha. A possible link between xenobiotic response and nutritional state. J Biol Chem 278:11344–11350
Bhalla S, Ozalp C, Fang S, Xiang L, Kemper JK (2004) Ligand-activated pregnane X receptor interferes with HNF-4 signaling by targeting a common coactivator PGC-1alpha. Functional implications in hepatic cholesterol and glucose metabolism. J Biol Chem 279:45139–45147
Rhee J, Inoue Y, Yoon JC et al (2003) Regulation of hepatic fasting response by PPARgamma coactivator-1alpha (PGC-1): requirement for hepatocyte nuclear factor 4alpha in gluconeogenesis. Proc Natl Acad Sci USA 100:4012–4017
Zhang Y, Castellani LW, Sinal CJ, Gonzalez FJ, Edwards PA (2004) Peroxisome proliferator-activated receptor-gamma coactivator 1alpha (PGC-1alpha) regulates triglyceride metabolism by activation of the nuclear receptor FXR. Genes Dev 18:157–169
Kallen J, Schlaeppi JM, Bitsch F et al (2004) Evidence for ligand-independent transcriptional activation of the human estrogen-related receptor alpha (ERRalpha): crystal structure of ERRalpha ligand binding domain in complex with peroxisome proliferator-activated receptor coactivator-1alpha. J Biol Chem 279:49330–49337
Puigserver P, Rhee J, Donovan J et al (2003) Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 423:550–555
Monsalve M, Wu Z, Adelmant G, Puigserver P, Fan M, Spiegelman BM (2000) Direct coupling of transcription and mRNA processing through the thermogenic coactivator PGC-1. Mol Cell 6:307–316
Kornblihtt AR (2004) Shortcuts to the end. Nat Struct Mol Biol 11:1156–1157
Puigserver P, Adelmant G, Wu Z et al. (1999) Activation of PPARgamma coactivator-1 through transcription factor docking. Science 286:1368–1371
Heery DM, Kalkhoven E, Hoare S, Parker MG (1997) A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 387:733–736
Ichida M, Nemoto S, Finkel T (2002) Identification of a specific molecular repressor of the peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1alpha). J Biol Chem 277:50991–50995
Liu D, Zhang Z, Teng CT (2005) Estrogen-related receptor-gamma and peroxisome proliferator-activated receptor-gamma coactivator-1alpha regulate estrogen-related receptor-alpha gene expression via a conserved multi-hormone response element. J Mol Endocrinol 34:473–478
Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM (2002) Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta ), a novel PGC-1-related transcription coactivator associated with host cell factor. J Biol Chem 277:1645–1648
Michael LF, Wu Z, Cheatham RB et al (2001) Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc Natl Acad Sci USA 98:3820–3825
Shen H, Kan JL, Green MR (2004) Arginine–serine-rich domains bound at splicing enhancers contact the branchpoint to promote prespliceosome assembly. Mol Cell 13:367–376
Ge K, Guermah M, Yuan CX et al (2002) Transcription coactivator TRAP220 is required for PPAR gamma 2-stimulated adipogenesis. Nature 417:563–567
Patel NA, Kaneko S, Apostolatos HS et al (2005) Molecular and genetic studies imply Akt-mediated signaling promotes protein kinase CbetaII alternative splicing via phosphorylation of serine/arginine-rich splicing factor SRp40. J Biol Chem 280:14302–14309
Auboeuf D, Honig A, Berget SM, O’Malley BW (2002) Coordinate regulation of transcription and splicing by steroid receptor coregulators. Science 298:416–419
Proudfoot NJ, Furger A, Dye MJ (2002) Integrating mRNA processing with transcription. Cell 108:501–512
Orphanides G, Reinberg D (2002) A unified theory of gene expression. Cell 108:439–451
Rocak S, Linder P (2004) DEAD-box proteins: the driving forces behind RNA metabolism. Nat Rev Mol Cell Biol 5:232–241
Fairman ME, Maroney PA, Wang W et al (2004) Protein displacement by DExH/D ‘RNA helicases’ without duplex unwinding. Science 304:730–734
Rajendran RR, Nye AC, Frasor J, Balsara RD, Martini PG, Katzenellenbogen BS (2003) Regulation of nuclear receptor transcriptional activity by a novel DEAD box RNA helicase (DP97). J Biol Chem 278:4628–4638
Puigserver P, Rhee J, Lin J et al (2001) Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol Cell 8:971–982
Knutti D, Kressler D, Kralli A (2001) Regulation of the transcriptional coactivator PGC-1 via MAPK-sensitive interaction with a repressor. Proc Natl Acad Sci USA 98:9713–9718
Fan M, Rhee J, St Pierre J et al (2004) Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1alpha: modulation by p38 MAPK. Genes Dev 18:278–289
Moore ML, Park EA, McMillin JB (2003) Upstream stimulatory factor represses the induction of carnitine palmitoyltransferase-Ibeta expression by PGC-1. J Biol Chem 278:17263–17268
Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P (2005) Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434:113–118
Nemoto S, Fergusson MM, Finkel T (2005) SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1alpha. J Biol Chem 280:16456–16460
Shi T, Wang F, Stieren E, Tong Q (2005) SIRT3, a mitochondrial Sirtuin deacetylase, regulates mitochondrial function and thermogenesis in Brown adipocytes. J Biol Chem 280:13560–13567
Baar K, Wende AR, Jones TE et al (2002) Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J 16:1879–1886
Storey KB (2003) Mammalian hibernation. Transcriptional and translational controls. Adv Exp Med Biol 543:21–38
Bryant NJ, Govers R, James DE (2002) Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol 3:267–277
Wu H, Kanatous SB, Thurmond FA et al (2002) Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science 296:349–352
Terada S, Goto M, Kato M, Kawanaka K, Shimokawa T, Tabata I (2002) Effects of low-intensity prolonged exercise on PGC-1 mRNA expression in rat epitrochlearis muscle. Biochem Biophys Res Commun 296:350–354
Czubryt MP, McAnally J, Fishman GI, Olson EN (2003) Regulation of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1 alpha) and mitochondrial function by MEF2 and HDAC5. Proc Natl Acad Sci USA 100:1711–1716
Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM (2003) An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1alpha expression in muscle. Proc Natl Acad Sci USA 100:7111–7116
Akimoto T, Sorg BS, Yan Z (2004) Real-time imaging of peroxisome proliferator-activated receptor-gamma coactivator-1alpha promoter activity in skeletal muscles of living mice. Am J Physiol Cell Physiol 287:C790–C796
Gleyzer N, Vercauteren K, Scarpulla RC (2005) Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol Cell Biol 25:1354–1366
Wu Z, Puigserver P, Andersson U et al (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98:115–124
Mootha VK, Handschin C, Arlow D et al (2004) Errα and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc Natl Acad Sci USA 101:6570–6575
Schreiber SN, Emter R, Hock MB et al (2004) The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc Natl Acad Sci USA 101:6472–6477
Lin J, Wu H, Tarr PT et al (2002) Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 418:797–801
Herzig S, Long F, Jhala US et al (2001) CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413:179–183
Daitoku H, Yamagata K, Matsuzaki H, Hatta M, Fukamizu A (2003) Regulation of PGC-1 promoter activity by protein kinase B and the forkhead transcription factor FKHR. Diabetes 52:642–649
Lin J, Wu PH, Tarr PT et al (2004) Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 119:121–135
Yoon JC, Puigserver P, Chen G et al (2001) Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413:131–138
Nakae J, Park BC, Accili D (1999) Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive pathway. J Biol Chem 274:15982–15985
Louet JF, Hayhurst G, Gonzalez FJ, Girard J, Decaux JF (2002) The coactivator PGC-1 is involved in the regulation of the liver carnitine palmitoyltransferase I gene expression by cAMP in combination with HNF4 alpha and cAMP-response element-binding protein (CREB). J Biol Chem 277:37991–38000
Du K, Herzig S, Kulkarni RN, Montminy M (2003) TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 300:1574–1577
Koo SH, Satoh H, Herzig S et al (2004) PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat Med 10:530–534
Himms-Hagen J (1990) Brown adipose tissue thermogenesis: interdisciplinary studies. FASEB J 4:2890–2898
Tiraby C, Tavernier G, Lefort C et al (2003) Acquirement of brown fat cell features by human white adipocytes. J Biol Chem 278:33370–33376
Wilson-Fritch L, Nicoloro S, Chouinard M et al (2004) Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J Clin Invest 114:1281–1289
Guan HP, Ishizuka T, Chui PC, Lehrke M, Lazar MA (2005) Corepressors selectively control the transcriptional activity of PPARgamma in adipocytes. Genes Dev 19:453–461
Leone TC, Lehman JJ, Finck BN et al (2005) PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol 3:e101
Yoon JC, Xu G, Deeney JT et al (2003) Suppression of beta cell energy metabolism and insulin release by PGC-1alpha. Dev Cell 5:73–83
Zhang CY, Baffy G, Perret P et al (2001) Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 105:745–755
Krempler F, Esterbauer H, Weitgasser R et al (2002) A functional polymorphism in the promoter of UCP2 enhances obesity risk but reduces type 2 diabetes risk in obese middle-aged humans. Diabetes 51:3331–3335
Krauss S, Zhang CY, Scorrano L et al (2003) Superoxide-mediated activation of uncoupling protein 2 causes pancreatic beta cell dysfunction. J Clin Invest 112:1831–1842
Zhang P, Liu C, Zhang C et al (2005) Free fatty acids increase PGC-1alpha expression in isolated rat islets. FEBS Lett 579:1446–1452
Kitamura T, Nakae J, Kitamura Y et al (2002) The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest 110:1839–1847
Nakae J, Biggs WH III, Kitamura T et al (2002) Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet 32:245–253
Russell AP, Feilchenfeldt J, Schreiber S et al (2003) Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor-gamma coactivator-1 and peroxisome proliferator-activated receptor-alpha in skeletal muscle. Diabetes 52:2874–2881
McGee SL, Hargreaves M (2004) Exercise and myocyte enhancer factor 2 regulation in human skeletal muscle. Diabetes 53:1208–1214
Pilegaard H, Saltin B, Neufer PD (2003) Exercise induces transient transcriptional activation of the PGC-1alpha gene in human skeletal muscle. J Physiol 546:851–858
Richardson DK, Kashyap S, Bajaj M et al (2005) Lipid infusion decreases the expression of nuclear encoded mitochondrial genes and increases the expression of extracellular matrix genes in human skeletal muscle. J Biol Chem 280:10290–10297
Ling C, Poulsen P, Carlsson E et al (2004) Multiple environmental and genetic factors influence skeletal muscle PGC-1alpha and PGC-1beta gene expression in twins. J Clin Invest 114:1518–1526
Mootha VK, Lindgren CM, Eriksson KF et al (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34:267–273
Patti ME, Butte AJ, Crunkhorn S et al (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci USA 100:8466–8471
Petersen KF, Befroy D, Dufour S et al (2003) Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 300:1140–1142
Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI (2004) Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 350:664–671
Lowell BB, Shulman GI (2005) Mitochondrial dysfunction and type 2 diabetes. Science 307:384–387
Esterbauer H, Oberkofler H, Krempler F, Patsch W (1999) Human peroxisome proliferator activated receptor gamma coactivator 1 (PPARGC1) gene: cDNA sequence, genomic organization, chromosomal localization, and tissue expression. Genomics 62:98–102
Pratley RE, Thompson DB, Prochazka M et al (1998) An autosomal genomic scan for loci linked to prediabetic phenotypes in Pima Indians. J Clin Invest 101:1757–1764
Perusse L, Rice T, Chagnon YC et al (2001) A genome-wide scan for abdominal fat assessed by computed tomography in the Quebec Family Study. Diabetes 50:614–621
Stone S, Abkevich V, Hunt SC et al (2002) A major predisposition locus for severe obesity, at 4p15-p14. Am J Hum Genet 70:1459–1468
Arya R, Duggirala R, Jenkinson CP et al (2004) Evidence of a novel quantitative-trait locus for obesity on chromosome 4p in Mexican Americans. Am J Hum Genet 74:272–282
Allayee H, de Bruin TW, Dominguez KM, Cheng LS et al (2001) Genome scan for blood pressure in Dutch dyslipidemic families reveals linkage to a locus on chromosome 4p. Hypertension 38:773–778
Ek J, Andersen G, Urhammer SA et al (2001) Mutation analysis of peroxisome proliferator-activated receptor-gamma coactivator-1 (PGC-1) and relationships of identified amino acid polymorphisms to Type II diabetes mellitus. Diabetologia 44:2220–2226
Andrulionyte L, Zacharova J, Chiasson JL, Laakso M (2004) Common polymorphisms of the PPAR-gamma2 (Pro12Ala) and PGC-1alpha (Gly482Ser) genes are associated with the conversion from impaired glucose tolerance to type 2 diabetes in the STOP-NIDDM trial. Diabetologia 47:2176–2184
Andersen G, Wegner L, Jensen DP et al (2005) PGC-1{alpha} Gly482Ser polymorphism associates with hypertension among Danish whites. Hypertension 45:565–570
Oberkofler H, Holzl B, Esterbauer H et al (2003) Peroxisome proliferator-activated receptor-gamma coactivator-1 gene locus: associations with hypertension in middle-aged men. Hypertension 41:368–372
Esterbauer H, Oberkofler H, Linnemayr V et al (2002) Peroxisome proliferator-activated receptor-gamma coactivator-1 gene locus: associations with obesity indices in middle-aged women. Diabetes 51:1281–1286
Franks PW, Barroso I, Luan J et al (2003) PGC-1alpha genotype modifies the association of volitional energy expenditure with VO2max. Med Sci Sports Exerc 35:1998–2004
Hara K, Tobe K, Okada T et al (2002) A genetic variation in the PGC-1 gene could confer insulin resistance and susceptibility to Type II diabetes. Diabetologia 45:740–743
Lacquemant C, Chikri M, Boutin P, Samson C, Froguel P (2002) No association between the G482S polymorphism of the proliferator-activated receptor-gamma coactivator-1 (PGC-1) gene and Type II diabetes in French Caucasians. Diabetologia 45:602–603
Oberkofler H, Linnemayr V, Weitgasser R et al (2004) Complex haplotypes of the PGC-1alpha gene are associated with carbohydrate metabolism and type 2 diabetes. Diabetes 53:1385–1393
Muller YL, Bogardus C, Pedersen O, Baier L (2003) A Gly482Ser missense mutation in the peroxisome proliferator-activated receptor gamma coactivator-1 is associated with altered lipid oxidation and early insulin secretion in Pima Indians. Diabetes 52:895–898
Stumvoll M, Fritsche A, t’Hart LM et al (2004) The Gly482Ser variant in the peroxisome proliferator-activated receptor gamma coactivator-1 is not associated with diabetes-related traits in non-diabetic German and Dutch populations. Exp Clin Endocrinol Diabetes 112:253–257
Rena G, Bain J, Elliott M, Cohen P (2004) D4476, a cell-permeant inhibitor of CK1, suppresses the site-specific phosphorylation and nuclear exclusion of FOXO1a. EMBO Rep 5:60–65
De Souza CT, Araujo EP, Prada PO, Boschero AD, Velloso LA (2005) Short-term inhibition of peroxisome proliferator-activated receptor-γ coactivator-1α expression reverses diet-induced diabetes mellitus and hepatic steatosis in mice. Diabetologia 48:1860–1871
Kim JH, Shin HD, Park BL et al (2005) Peroxisome proliferator-activated receptor gamma coactivator 1 alpha promoter polymorphisms are associated with early-onset type 2 diabetes mellitus in the Korean population. Diabetologia 48:1323–1330
Pihlajamäki J, Kinnunen M, Ruotsalainen E et al (2005) Haplotypes of PPARGC1A are associated with glucose tolerance, body mass index and insulin sensitivity in offspring of patients with type 2 diabetes. Diabetologia 48:1331–1334
The International HapMap Consortium (2003) The International HapMap Project. Nature 426:789–796
Acknowledgements
This work was supported by Jubilaeumsfondsprojekt No. 10932 and No. 10678 from the Oesterreichische Nationalbank and by grants from the Bundesland Salzburg and the Medizinische Forschungsgesellschaft Salzburg.
Duality of interest
None of the authors state any duality of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s00125-006-0341-1
Electronic supplementary materials
Electronic Supplementary Material Fig. 1
Comparison of single-nucleotide polymorphisms used for haplotype construction in Austrian, Korean and Finnish populations [103, 108, 109]. Linear map of PPARGC1A. Exons are identified by boxes labelled 1–13. SNPs identified and typed in >10 Austrians are shown. SNPs typed in 494 patients with type 2 diabetes and 1478 controls are in bold. Promoter SNPs not typed in all subjects but found to be in perfect linkage disequilibrium with promoter SNPs typed in all subjects are shown in bold italic font. SNPs affecting allele-specific expression in transfection studies are indicated by **. SNP qualifiers refer to database entries (http://www.ncbi.nlm.nih.gov/projects/SNP/, last accessed in March 2006). Positions relative to the transcriptional start site are given in parentheses. SNPs typed in subjects with type 2 diabetes and controls from a Korean population [109] or in non-diabetic Finns [109] are shown below the Austrian data. Haplotype blocks 1 and 2 are displayed at the bottom of the figure

Rights and permissions
About this article
Cite this article
Soyal, S., Krempler, F., Oberkofler, H. et al. PGC-1α: a potent transcriptional cofactor involved in the pathogenesis of type 2 diabetes. Diabetologia 49, 1477–1488 (2006). https://doi.org/10.1007/s00125-006-0268-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-006-0268-6