
Abstract
Aims/hypothesis
Recent studies have suggested that proinsulin C-peptide improves vascular functions, possibly through nitric oxide (NO) production. To clarify the molecular mechanisms of vascular NO production induced by C-peptide, we examined the effects of C-peptide on NO production and NO synthase expression in rat aortic endothelial cells in connection with mitogen-activated protein kinase (MAPK) activation.
Methods
Aortic endothelial cells were isolated from female Wistar rats, cultured to confluence, and serum-starved for 24 h before treatment with C-peptide. Nitric oxide production was measured by the DAF-2 fluorescence dye method and relative amounts of endothelial nitric oxide synthase (eNOS) protein and its mRNA were semi-quantified by western blot and RT-PCR analyses respectively. Activation of MAPK was estimated by western blot detection of activity-related phosphorylation and in vitro kinase assay.
Results
Stimulation of cells with C-peptide for 3 h doubled NO production, which was suppressed by the NO synthase inhibitor, NG-nitro-L-arginine methyl ester (L-NAME). Stimulation also increased mRNA and protein contents of eNOS in a manner sensitive to the transcription inhibitor actinomycin D. It did not affect inducible NO synthase mRNA. C-peptide also induced rapid phosphorylation and activation of extracellular signal-regulated kinase (ERK, also known as p44/42MAPK), but not of p38MAPK. In cells pretreated with the ERK inhibitor PD98059 the C-peptide-elicited increase of NO production and eNOS was abrogated in a dose-dependent manner; suppression of ERK phosphorylation induced by C-peptide also occurred.
Conclusions/interpretation
Our results show that C-peptide increases NO production by increasing eNOS protein contents through ERK-dependent up-regulation of eNOS gene transcription. This could explain some actions of C-peptide on the vasculature, indicating a pivotal role for C-peptide in vascular homeostasis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
C-peptide is secreted, along with insulin, from pancreatic beta cells after cleavage of proinsulin. Recent studies in patients with Type 1 diabetes mellitus have shown that C-peptide causes circulatory responses, e.g. increased renal and skin blood flow [1, 2, 3, 4], accompanied by increased plasma cyclic GMP, which is an indicator of nitric oxide (NO) production [4]. When given to patients with Type 1 diabetes, C-peptide also increases forearm blood flow [5, 6, 7] with brachial artery dilatation [7]. In vitro studies have shown that C-peptide induces dilatation of skeletal muscle arterioles in the presence of insulin, an effect that is abolished by NG-nitro-L-arginine methyl ester (L-NAME), an inhibitor of NO synthesis [8]. These findings suggest that the circulatory effects of C-peptide are attributable to vasodilatation mediated by NO. Indeed, in aortic rings isolated from rats which have been injected with C-peptide, NO release is increased [9, 10]. However, the underlying mechanisms of how C-peptide induces NO production are poorly understood.
Formation of NO from L-arginine can be catalysed by three isoforms of nitric oxide synthase (NOS), two of which could be involved in vascular NO production [11, 12]. Endothelial nitric oxide synthase (eNOS) is constitutively expressed in vascular endothelial cells, and is activated by an agonist-induced increase of intracellular Ca2+, followed by a complex formation of Ca2+-calmodulin and eNOS [13]. The activity of eNOS is also controlled at the transcriptional and post-translational levels, e.g. through palmitoylation, protein to protein interactions, and phosphorylation by various protein kinases [11, 12, 14, 15]. In contrast, inducible nitric oxide synthase (iNOS) is regulated predominantly at the transcriptional level by endotoxins, inflammatory cytokines and growth factors in various types of cells including endothelial cells [16, 17]. It is also known to bind Ca2+-calmodulin even at resting cellular Ca2+ concentrations [18].
We recently found that C-peptide specifically activates mitogen-activated protein kinase (MAPK), also known as extracellular signal-regulated kinase (ERK), in Swiss3T3 and 3T3-F442A fibroblasts, the process being dependent on phosphoinositide 3-kinase and protein kinase C (PKC) [19]. Others have reported that in L6 myotubes C-peptide induces activation of ERK in a manner dependent on phosphoinositide 3-kinase [20]. More importantly, stimulation of LEII mouse lung capillary endothelial cells with C-peptide led to considerable activation of a different type of MAPK in endothelial cells, namely p38MAPK, and through that to activation of transcription factors such as cyclic AMP response element-binding protein and activating transcription factor 1 [21]. Thus it seems likely that C-peptide influences endothelial NO production by altering either eNOS or iNOS expression through MAPK-mediated transcriptional activation. Alternatively, C-peptide could directly stimulate eNOS activity by increasing intracellular concentrations of Ca2+ and/or by inducing phosphorylation by protein kinase B/Akt, a downstream effector of phosphoinositide 3-kinase. To determine the molecular mechanisms of increases in vascular NO production induced by C-peptide, we examined the effects of C-peptide on NO production and NOS expression in rat aortic endothelial cells (RAEC), focusing in particular on the involvement of MAPK.
Materials and methods
Materials
Human C-peptide was generously provided by Eli Lilly (Indianapolis, Ind., USA). Bovine insulin and human TNF-α were obtained from Sigma-Aldrich (St. Louis, Mo., USA) and Strathmann Biotech (Hamburg, Germany) respectively. Hydrogen peroxide (H2O2) and actinomycin D came from Wako Pure Chemical (Osaka, Japan). Antibodies against phospho-specific ERK1/2 (p44/42MAPK, Thr-202/Tyr-204), ERK1/2, phospho-specific p38MAPK (Thr-180/Tyr-182), p38MAPK, phospho-specific Akt (Thr-308) and Akt were bought from Cell Signalling Technology (Beverly, Mass., USA), while antibodies to eNOS and von Willebrand factor were from Santa Cruz Biotechnology (Santa Cruz, Calif., USA). The NO donor 3-(2-hydroxy-1-methyl-2-nitrosohydrazino)-N-methyl-1-propanamine (NOC7) was supplied by Alexis Biochemicals (San Diego, Calif., USA) and the NOS inhibitor L-NAME by Dojindo (Kumamoto, Japan).
Cell isolation, culture and treatments
Experimental procedure and care of animals were in accordance with the Guidelines of the Animal Care and Use of Hokkaido University, Japan, which follows the Guide for the Care and Use of Laboratory Animals (NIH publication No.85-23, revised 1985). Our study was approved by the Committee for the Care and Use of Laboratory Animals in the Graduate School of Veterinary Medicine, Hokkaido University.
Rat aortic endothelial cells were isolated from female Wistar rats (Nippon SLC, Shizuoka, Japan, 150–200 g) using the method described previously [22] and cultured on 100-mm or 35-mm type-I collagen-coated dishes in DMEM (Sigma) at 37 C and 5% CO2 under humidified conditions. The DMEM was supplemented with 20% fetal bovine serum(TRACE Scientific, Melbourne, Australia), 90 µg/ml heparin (Sigma), 4 mmol/l L-glutamine (Sigma) and 60 µg/ml endothelial cell growth supplement (ECGS, Harbor Bio-products, Norwood, Mass., USA). The isolated cells were confirmed to be endothelial cells by their cobblestone-like shape, by western blot detection of von Willebrand factor and eNOS proteins, and by their uptake of acetylated LDL, as described previously [22]. Rat lung microvascular endothelial cells (RLMEC) were also isolated from the peripheral edge of lungs of female Wistar rats [22] and cultured in 60-mm Primaria plates (Becton Dickinson Labware, Franklin Lakes, N.J., USA) in L-valine-free minimal essential medium (Sigma) supplemented with 15% fetal bovine serum, 90 µg/ml heparin, 4 mmol/l L-glutamine, 0.1 mmol/l non-essential amino acids (Gibco-BRL, Gaithersberg, Md., USA) and 60 µg/ml ECGS. They also had a cobblestone-like shape, sequestered acetylated LDL, and von Willebrand factor and eNOS protein expression. LEII mouse lung capillary endothelial cells [23] were cultured on 100-mm type-I collagen-coated dishes in DMEM supplemented with 10% fetal bovine serum.
After the cells were grown to confluence, they were cultured in serum-free DMEM for 24 h to render them quiescent. They were then stimulated with C-peptide (10 nmol/l), insulin (100 nmol/l), TNF-α (1 ng/ml) and H2O2 (200 µmol/l) at 37°C for 10 min (0.17 h) to 24 h. Before C-peptide was added to the RAEC, they were treated either with actinomycin D (4 µg/ml) or PD98059 (an ERK kinase inhibitor, Biomol Research Laboratories, Plymouth Meeting, Pa., USA) for 30 min or 2 h respectively.
Measurement of NO production
The RAEC were cultured in 24-well plates (1×105 cells per well), serum-starved for 24 h after confluence, and stimulated with PBS or C-peptide for 3 h. After two washes with PBS, the cells were incubated for 1 h at 37°C with 10 µmol/l diaminofluorescein diacetate (Daiichi Pure Chemicals, Tokyo, Japan), a NO-dependent fluorescent dye. They were then washed twice with PBS and the amount of fluorescent diaminofluorescein-2 triazole converted in a NO-dependent manner from diaminofluorescein diacetate was measured using a fluorescence plate reader (Labsystems, Helsinki, Finland) with excitation and emission wavelengths at 485 nm and 538 nm respectively. NOC-7 was used as a positive control to validate this measurement. In some experiments, the cells were incubated with diaminofluorescein diacetate before being washed and treated with C-peptide or insulin for 5 min.
RT-PCR analysis
Total RNA was extracted using TRIzol reagent (Gibco-BRL) and 2 µg RNA were reverse-transcribed by 60 U Moloney murine leukaemia virus reverse transcriptase (Gibco-BRL) in 20 µl of buffer solution. This was done for 1 h at 30°C. The buffer solution [50 mmol/l Tris/HCl (pH 8.3), 40 mmol/l KCl, 6 mmol/l MgCl2, 1 mmol/l dithiothreitol] contained 40 nmol dNTP and 10 pmol oligo(dT). After heating for 5 min at 94°C, 10 µl of the solution were subjected to PCR amplification with 5 µmol/l of forward and reverse primers, 1 U AmpliTaq DNA polymerase (Applied Biosystems, Brunchberg, N.J., USA) and 20 nmol dNTP in 50 µl of a reaction buffer [20 mmol/l Tris/HCl (pH 8.3), 50 mmol/l KCl, 1.5 mmol/l MgCl2]. Design of the oligonucleotide primers was based on the corresponding cDNA sequences of eNOS (GeneBank accession No.U53142), iNOS (M87039) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, M32599) as follows: (i) eNOS forward: 3049-GAC TGG CAT TGC ACC CTT CCG G-3070, eNOS reverse: 3381-CCG TGC AGA GAA TTC TGG CAA C-3402; (ii) iNOS forward: 493-AGG ATC AAA AAC TGG GGC AAT G-514, iNOS reverse: 1230-GGA GCA TCC CAA GTA CGA GTG G-1251; (iii) GAPDH forward: 566-ACC ACA GTC CAT GCC ATC AC-585, GAPDH reverse: 998-TAC AGC AAC AGG GTG GTG GA-1017. The PCR was done for 35 cycles (eNOS and iNOS) and 25 cycles (GAPDH), each consisting of denaturation for 30 s at 94 C, annealing for 30 s at 52°C and elongation for 30 s at 72°C. After electrophoresis in 2% agarose gel, the PCR products were stained with ethidium bromide.
Western-blot analysis
The cells were washed twice with PBS and lysed with 1 ml of ice-cold lysis buffer [50 mmol/l Hepes (pH 7.5), 150 mmol/l NaCl, 5 mmol/l EDTA, 10 mmol/l sodium pyrophosphate, 2 mmol/l NaVO3, protease inhibitor mixture (Complete, Boehringer Mannheim, Mannheim, Germany) and 1% Nonidet P-40]. The lysates were kept on ice for 30 min and centrifuged at 15 000 g for 20 min at 4°C. Having collected the supernatant, we measured the protein concentration using bovine serum albumin as a standard [24]. Aliquots of the supernatant (40 µg protein) were separated by SDS-PAGE and transferred onto PVDF membranes (Immobilon, Millipore, Bedford, Mass., USA). The membranes were incubated overnight in a blocking buffer [20 mmol/l Tris/HCl (pH 7.5), 150 mmol/l NaCl] containing 0.1% Tween 20 and 5% skimmed milk, and then in the buffer containing an antibody for 1 h. The bound antibody was made visible using horseradish peroxidase-linked goat anti-rabbit immunoglobulin (Zymed Laboratories, San Francisco, Calif., USA) and an enhanced chemiluminescence system (Amersham, Little Chalfont, Bucks, UK). The intensity of chemiluminescence for the corresponding bands was analysed by NIH Image, public-domain image processing and analysis program (US National Institutes of Health; available on the Internet at http://rsb.info.nih.gov/nih-image/).
MAPK assays
The activities of MAPK were measured using ERK1/2 and p38MAPK assay kits (Cell Signalling Technology) according to the manufacturer’s recommended protocols. In brief, cells were stimulated with PBS or C-peptide (10 nmol/l) for 5 min, washed with PBS and then lysed in 1 ml ice-cold lysis buffer [20 mmol/l Tris/HCl (pH 7.5), 150 mmol/l NaCl, 1 mmol/l EDTA, 1 mmol/l EGTA, 1 mmol/l β-glycerophosphate, 2.5 mmol/l sodium pyrophosphate, 1 mmol/l NaVO3, protease inhibitor mixture (Complete) and 1% Triton X-100]. After centrifugation for 10 min at 4°C, the supernatant was subjected to immunoprecipitation using immobilised phospho-ERK1/2 (Thr-202/Tyr-204) or phospho-p38MAPK (Thr-180/Tyr-182) monoclonal antibodies. Each immunoprecipitate was washed twice with ice-cold lysis buffer, twice with ice-cold kinase buffer [25 mmol/l Tris/HCl (pH 7.5), 5 mmol/l β-glycerophosphate, 2 mmol/l dithiothreitol, 0.1 mmol/l NaVO3 and 10 mmol/l MgCl2], then resuspended in kinase buffer containing 200 µmol/l ATP and 2 µg of a substrate for respective kinases. After incubation for 30 min at 30°C, the reaction mixtures were boiled in SDS-gel loading buffer. The samples were subjected to SDS-PAGE followed by western blot analysis using antibody to phospho-Elk1 (Ser-383) for ERK or phospho-ATF2 (Thr-71) for p38MAPK. MAPK activities were quantified by scanning the X-ray film as described above.
Statistical analysis
Data were expressed as means ± SEM. and analysed by ANOVA followed by Fisher’s protected least-squares difference or Student’s t test. A p value of less than 0.05 was considered to be statistically significant.
Results
As shown in Figure 1, stimulation of RAEC with C-peptide for 3 h increased fluorescence derived from diaminofluorescein-2 triazole (p<0.05) as in the case of cells treated with NOC7, an NO donor. The stimulatory effect of C-peptide was completely abrogated by L-NAME, a competitive inhibitor of NOS, indicating that C-peptide stimulates NOS-mediated NO production.

Effects of C-peptide on nitric oxide production in RAEC. RAEC were stimulated for 3 h with PBS and human C-peptide (10 nmol/l). NO production was then measured for 1 h in the presence (+) or absence (−) of the NOS inhibitor L-NAME (1 mmol/l). Measurement was by NO-dependent conversion of diaminofluorescein diacetate to diaminofluorescein-2 triazole. As a positive control for NO production RAEC were also treated for 5 min with NOC7 (200 µmol/l). The fluorescence of the cells is expressed relative to the autofluorescence of the diaminofluorescein diacetate added to the medium. Data are means ± SEM of three independent experiments. *p<0.05 vs PBS controls; † p<0.05 vs cells not treated with L-NAME. RAEC rat aortic endothelial cells
To find the NOS isoform involved in the C-peptide action, we examined the effects of C-peptide on eNOS and iNOS mRNA expression in RAEC. Stimulation of the cells with C-peptide increased eNOS mRNA expression within 10 min (p<0.05), an effect which lasted for at least 1 h before returning basal levels (Fig. 2). In contrast, C-peptide did not alter iNOS mRNA expression on any occasion. However, treatment with TNF-α greatly increased iNOS, but not eNOS mRNA expression (Fig. 2). We also examined eNOS protein content in cell lysate after C-peptide stimulation. In 30 min C-peptide produced larger amounts of eNOS protein (Fig. 3a). This effect lasted for 6 h, then gradually decreased. Pretreatment with a transcription inhibitor, actinomycin D, completely stopped the increase of eNOS protein induced by C-peptide (Fig. 3b).

Effects of C-peptide on eNOS mRNA expression in RAEC. Total RNA was obtained from RAEC stimulated for 10 min (0.17 h) to 24 h with C-peptide (10 nmol/l) or for 3 h with TNF-α (1 ng/ml). RT-PCR was done using the primer sets for eNOS, iNOS and GAPDH. Representative results of three independent experiments are shown. RAEC rat aortic endothelial cells

Effects of C-peptide on eNOS protein expression in RAEC. Cell lysates were obtained (a) from RAEC stimulated for 10 min (0.17 h) to 24 h with PBS (open circle) or with C-peptide (10 nmol/l, closed circle) and subjected to western blot analyses of eNOS protein. Representative blots of three independent experiments are shown. The amounts of eNOS protein were quantified and expressed relative to those of untreated control cells (0-time). Data are means ± SEM of three independent experiments. *p<0.05 vs controls. Cell lysates (b) were also obtained from RAEC stimulated for 3 h with PBS and C-peptide (10 nmol/l) plus and minus Actinomycin D (4 µg/ml) and analysed for eNOS protein. Representative blots of three independent experiments are shown. RAEC, rat aortic endothelial cells
To evaluate the role of ERK and p38MAPK signalling pathways in C-peptide-induced NO production, we examined the effects of C-peptide on the activity-related site-specific phosphorylation of MAPKs in RAEC. Stimulation with C-peptide produced ERK1/2 phosphorylation (Thr-202/Tyr-204) within 5 min. This lasted for 30 min then disappeared without noticeable effects on the total amount of ERK1/2 (Fig. 4a). In contrast, neither phosphorylation (Thr-183/Tyr-185) nor total protein of p38MAPK were influenced by C-peptide (Fig. 4a). In parallel, the kinase activity of ERK1/2 was 3.21±0.6-fold (n=3) higher after 5-min stimulation with C-peptide than in cells treated with PBS (p<0.05). The p38MAPK activity was unchanged after stimulation with C-peptide [1.1±0.1-fold (n=3)], whereas it increased 3.9±0.9-fold (n=3) in cells treated with H2O2 as positive control. To compare the effects of C-peptide on RAEC with those of C-peptide in other types of endothelial cell, C-peptide-induced MAPK activation was also examined in microvascular endothelial cells isolated from rat lung. Stimulation of RLMEC with C-peptide induced ERK1/2 and p38MAPK phosphorylation without altering their protein levels, in the same way as in LEII cells (Fig. 4b).

Effects of C-peptide on phosphorylation of ERK1/2 and p38MAPK in RAEC. Cell lysates were obtained from three types of vascular endothelial cells stimulated with PBS or C-peptide (10 nmol/l) and subjected to western blot analyses to determine activity-related site-specific phosphorylation of ERK1/2 and p38MAPK (phosphorylated) and their total protein contents. (a) Representative blots of RAEC stimulated with C-peptide for 1 to 180 min. (b) Representative blots of RAEC, rat lung microvascular endothelial cells and LEII lung capillary endothelial cells stimulated with (+) or without (−) C-peptide for 5 min. RAEC, rat aortic endothelial cells
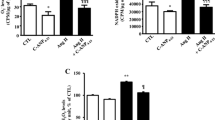
To identify the possible role of the ERK pathway in C-peptide-induced NO production in RAEC, we next examined the effects of an ERK kinase inhibitor, PD98059, on C-peptide-induced ERK phosphorylation, eNOS protein levels and NO production. Pretreatment with PD98059 reduced the C-peptide-induced increase of eNOS protein in a dose-dependent manner, while inhibiting ERK phosphorylation (Fig. 5a). Importantly, PD98059 also lowered the C-peptide-induced increase of NO production, this effect being parallel to the decrease in eNOS protein levels (Fig. 5b).

Involvement of ERK pathway in C-peptide-induced eNOS expression and NO production in RAEC. RAEC were pretreated for 2 h with or without increasing concentrations of the ERK kinase inhibitor PD98059. They were then stimulated for 5 min with PBS or C-peptide (10 nmol/l) and analysed for phospho-specific and total ERK1/2 as in Fig. 4. The RAEC pretreated with the inhibitor were also stimulated for 3 h with C-peptide. Production of NO and eNOS protein were measured as in Figs. 1 and 3. The representative results (a) of three independent experiments are shown. The amounts of eNOS protein are relative to those of control cells (black columns) and NO-dependent diaminofluorescein-2 triazole fluorescence (grey columns) (b). Data are means ± SEM of three independent experiments. *p<0.05 vs PBS controls; † p<0.05 vs cells not treated with PD98059. RAEC, rat aortic endothelial cells
With regard to the effect of protein kinases like Akt, we showed (Fig. 6) that when RAEC were stimulated with insulin for 5 min, activity-related site-specific phosphorylation (Thr-308) of Akt and NO production both increased (p<0.05). However, stimulation with C-peptide for 5 min had no effect on Akt phosphorylation and NO production (Fig. 6). It did, however, induce ERK phosphorylation (Fig. 4a).

Acute effects of C-peptide on nitric oxide production and Akt phosphorylation in RAEC. RAEC were stimulated for 5 min with PBS, C-peptide (10 nmol/l) and insulin (100 nmol/l), and NO-dependent diaminofluorescein-2 triazole fluorescence (a) and Akt phosphorylation (b) were measured. Data are means ± SEM (a) and representatives (b) of three independent experiments. *p<0.05 vs PBS treated cells. RAEC, rat aortic endothelial cells
Discussion
Our study showed that proinsulin C-peptide increases NO production in primary cultures of RAEC, after inducing eNOS mRNA expression and a consequent increase in eNOS protein but without affecting iNOS mRNA expression. As the C-peptide-induced increase of NO production was parallel to eNOS protein contents, the enhanced NO production is attributable to increases in eNOS protein, rather than to post-translational eNOS activation and iNOS induction.
In addition to C-peptide, other extracellular stimuli have been shown to induce eNOS protein and increase NO production in endothelial cells, possibly through two different mechanisms [14, 25, 26]. For example, increases of eNOS protein induced by lysophosphatidylcholine are due to activation of mRNA transcription [14]. In contrast, vascular endothelial growth factor-induced increases of eNOS mRNA are insensitive to a transcription inhibitor, suggesting some post-transcriptional regulation of eNOS mRNA [25]. Our finding that C-peptide-induced eNOS protein expression was completely abolished by actinomycin D suggests that C-peptide activates eNOS mRNA transcription rather than increase stability.
We also showed that C-peptide activates ERK in RAEC and that blockade of the ERK pathway by PD98059 suppresses both eNOS protein expression induced by C-peptide and NO production. These results suggest that C-peptide-induced up-regulation of eNOS transcription is mediated through the ERK signalling pathway. ERK has also been recognised to play a role, possibly through activation of SP1 transcription factor [15], in eNOS expression induced by lysophosphatidylcholine, basic fibroblast growth factor and epidermal growth factor [14, 26]. We have reported that C-peptide not only activates ERK, but also p38MAPK in LEII capillary endothelial cells, and that the activation of p38MAPK increases the binding of cyclic AMP response element-binding protein and activating transcription factor 1 to DNA [21]. The binding sites for other p38MAPK-regulated transcription factors, the Ets family of proteins, are known to be present in the promoter region of the eNOS gene [27]. However, involvement of p38MAPK in the effects of C-peptide is unlikely in aortic endothelial cells, because C-peptide failed to activate p38MAPK in the cells used. It remains to be determined which transcription factor(s) and/or mechanism(s) contribute to the ERK-mediated up-regulatory effect of C-peptide on eNOS.
Our study and unpublished observations by us have shown that activation of p38MAPK by C-peptide was limited in RLMEC and LEII cells derived from lung capillary blood vessels, but not in other cells which responded to C-peptide, including RAEC, Swiss3T3 and 3T3-F442A cells. Similar differences in p38MAPK activation have also been reported between lung microvascular and arterial endothelial cells treated with anti-intercellular adhesion molecule-1 antibody for cross-linking [28]. It is, therefore, likely that different machineries of p38MAPK activation are present in the two types of endothelial cell. Recently, it has been reported that C-peptide, when given to rats, increases NO release from isolated aortic rings and eNOS mRNA expression in the lung [9]. As noted above, increased NO release from aortic rings can be due to the direct action of C-peptide on aortic endothelial cells, i.e. the activation of ERK signalling to enhance eNOS transcription, with a consequent increase in eNOS protein and NO production. In the lung, however, the involvement of p38MAPK in C-peptide-induced eNOS mRNA expression cannot be ruled out.
There have been reports that eNOS activity is rapidly modulated by its site-specific phosphorylation by various protein kinases including Akt and PKC [11, 12]. For example, insulin induces Akt phosphorylation and activation, followed by eNOS phosphorylation and a consequent increase in intracellular NO concentrations [29]. Our study confirmed this in RAEC. C-peptide, however, failed to induce Akt phosphorylation and rapid NO production. The failure of C-peptide to activate Akt has also been reported in L6 myotubes [20]. Thus, our results suggest that Akt signalling leading to eNOS activation is independent of C-peptide signalling and that this dissociation could explain why C-peptide does not modulate rapid NO production. Protein kinase C is involved in various C-peptide actions, e.g. activation of Na+,K+-ATPase activity in rat medullary thick ascending limb [30] and activation of ERK in Swiss3T3 [19] and LEII cells (T. Kitamura and K. Kimura, unpublished observation). However, as phosphorylation by PKC decreased rather than increased eNOS activity [31], it is likely that PKC regulates eNOS activity in RAEC more by ERK-mediated induction than by direct phosphorylation of eNOS protein. Recently it was also reported that C-peptide and its analogues rapidly increase intracellular Ca2+ levels in human renal tubular cells [32]. Although it is well known that eNOS activity is regulated by intracellular Ca2+ [11, 12], the inability of C-peptide to increase rapid NO production implies that intracellular Ca2+ movement in RAEC after C-peptide stimulation is marginal or absent.
As noted above, C-peptide seems to activate different signalling pathways mediated by ERK, p38MAPK and/or Ca2+ in a way specific to cell types. As some of these signalling pathways were inactivated by pretreatment with pertussis toxin [19, 33], the putative C-peptide receptor(s) could be G-protein-coupled receptor(s) linked to Gi/Go proteins. At present little is known about the molecular structure of the receptor(s). However, recent studies have provided compelling evidence that G-protein-coupled receptor(s) can couple with multiple G-proteins [34, 35]. Thus an agonist for bradykinin B2 receptor activates Gi and Gq proteins to induce calcium influx [34]. It therefore seems possible that C-peptide activates cell-type-specific signals depending on the G-protein subtypes coupled with putative C-peptide receptor(s).
In conclusion, we showed that C-peptide increases NO production in aortic endothelial cells by enhancing eNOS expression through ERK-dependent transcriptional activation. This is one of the likely mechanisms of NO-mediated C-peptide action in vivo. Since endothelium-derived NO plays an important part in maintaining vascular tone and endothelial cell integrity [36], our observations increase understanding of the physiological and pathophysiological roles of C-peptide.
Abbreviations
- ECGS:
-
Endothelial cell growth supplement
- eNOS:
-
endothelial nitric oxide synthase
- ERK:
-
extracellular signal-regulated kinase
- GAPDH:
-
glyceraldehyde 3-phosphate dehydrogenase
- iNOS:
-
inducible nitric oxide synthase
- L-NAME:
-
NG-nitro-L-arginine methyl ester
- MAPK:
-
mitogen-activated protein kinase
- NO:
-
nitric oxide
- NOC-7:
-
3-(2-hydroxy-1-methyl-2-nitrosohydrazino)-N-methyl-1-propanamine
- PKC:
-
protein kinase C
- RAEC:
-
rat aortic endothelial cells
- RLMEC:
-
rat lung microvascular endothelial cells
References
Wahren J, Ekberg K, Johansson J et al. (2000) Role of C-peptide in human physiology. Am J Physiol Endocrinol Metab 278:E759–E768
Johansson BL, Sjoberg S, Wahren J (1992) The influence of human C-peptide on renal function and glucose utilization in type 1 (insulin-dependent) diabetic patients. Diabetologia 35:121–128
Forst T, Kunt T, Pohlmann T et al. (1998) Biological activity of C-peptide on the skin microcirculation in patients with insulin dependent diabetes mellitus. J Clin Invest 101:2036–2041
Forst T, De La Tour DD, Kunt T et al. (2000) Effects of proinsulin C-peptide on nitric oxide, microvascular blood flow and erythrocyte Na+,K+-ATPase activity in diabetes mellitus type I. Clin Sci 98:283–290
Johansson BL, Linde B, Wahren J (1992) Effects of C-peptide on blood flow, capillary diffusion capacity and glucose utilization in the exercising forearm of type 1 (insulin-dependent) diabetic patients. Diabetologia 35:1151–1158
Johansson BL, Pernow J (1999) C-peptide potentiates the vasoconstrictor effect of neuropeptide Y in insulin-dependent diabetic patients. Acta Physiol Scand 165:39–44
Fernqvist-Forbes E, Johansson BL, Erikson MJ (2001) Effects of C-peptide on forearm blood flow and brachial artery dilatation in patients with type 1 diabetes mellitus. Acta Physiol Scand 172:159–165
Jensen ME, Messina EJ (1999) C-peptide induces a concentration-dependent dilation of skeletal muscle arterioles only in presence of insulin. Am J Physiol 276:H1223–H1228
Scalia R, Coyle KM, Levine BJ, Booth G, Lefer AM (2000) C-peptide inhibits leukocyte-endothelium interaction in the microcirculation during acute endothelial dysfunction. FASEB J 14:2357–2364
Young LH, Ikeda Y, Scalia R, Lefer AM (2000) C-peptide exerts cardioprotective effects in myocardial ischemia-reperfusion. Am J Physiol Heart Circ Physiol 279:H1453–H1459
Govers R, Rabelink TJ (2001) Cellular regulation of endothelial nitric oxide synthase. Am J Physiol Renal Physiol 280:F193–F206
Fulton D, Gratton JP, Sessa WC (2001) Post-translational control of endothelial nitric oxide synthase: why isn’t calcium/calmodulin enough? J Pharmacol Exp Ther 299:818–824
Venema RC, Sayegh HS, Kent JD, Harrison DG (1996) Identification, characterization, and comparison of the calmodulin-binding domains of the endothelial and inducible nitric oxide synthases. J Biol Chem 271:6435–6440
Zembowicz A, Tang JL, Wu KK (1995) Transcriptional induction of endothelial nitric oxide synthase type III by lysophosphatidylcholine. J Biol Chem 270:17006–17010
Cieslik K, Lee CM, Tang JL, Wu KK (1999) Transcriptional regulation of endothelial nitric-oxide synthase by an interaction between casein kinase 2 and protein phosphatase 2A. J Biol Chem 274:34669–34675
Nathan C (1997) Inducible nitric oxide synthase: what difference does it make? J Clin Invest 100:2417–2423
Purdie KJ, Whitley GS, Johnstone AP, Cartwright JE (2002) Hepatocyte growth factor-induced endothelial cell motility is mediated by the upregulation of inducible nitric oxide synthase expression. Cardiovasc Res 54:659–668
Ruan J, Xie Q, Hutchinson N, Cho H, Wolfe GC, Nathan C (1996) Inducible nitric oxide synthase requires both the canonical calmodulin-binding domain and additional sequences in order to bind calmodulin and produce nitric oxide in the absence of free Ca2+. J Biol Chem 271:22679–22686
Kitamura T, Kimura K, Jung BD et al. (2001) Proinsulin C-peptide rapidly stimulates mitogen-activated protein kinases in Swiss 3T3 fibroblasts: requirement of protein kinase C, phosphoinositide 3-kinase and pertussis toxin-sensitive G-protein. Biochem J 355:123–129
Grunberger G, Qiang X, Li Z et al. (2001) Molecular basis for the insulinomimetic effects of C-peptide. Diabetologia 44:1247–1257
Kitamura T, Kimura K, Jung BD et al. (2002) Proinsulin C-peptide activates cAMP response element-binding proteins through the p38 mitogen-activated protein kinase pathway in mouse lung capillary endothelial cells. Biochem J 366:737–744
Magee JC, Stone AE, Oldham KT, Guice KS (1994) Isolation, culture, and characterization of rat lung microvascular endothelial cells. Am J Physiol 267:L433–L441
Curtis AS, Renshaw RM (1982) Lymphocyte-endothelial interactions and histocompatibility restriction. Adv Exp Med Biol 149:193–198
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the folin phenol reagent. J Biol Chem 193:265–275
Bouloumié A, Schini-Kerth VB, Busse R (1999) Vascular endothelial growth factor up-regulates nitric oxide synthase expression in endothelial cells. Cardiovasc Res 41:773–780
Zheng J, Bird IM, Melsaether AN, Magness RR (1999) Activation of the mitogen-activated protein kinase cascade is necessary but not sufficient for basic fibroblast growth factor- and epidermal growth factor-stimulated expression of endothelial nitric oxide synthase in ovine fetoplacental artery endothelial cells. Endocrinology 140:1399–1407
Karantzoulis-Fegaras F, Antoniou H, Lai SL et al. (1999) Characterization of the human endothelial nitric-oxide synthase promoter. J Biol Chem 274:3076–3093
Wang Q, Pfeiffer GR 2nd, Stevens T, Doerschuk CM (2002) Lung microvascular and arterial endothelial cells differ in their responses to intercellular adhesion molecule-1 ligation. Am J Respir Crit Care Med 166:872–877
Montagnani M, Chen H, Barr VA, Quon MJ (2001) Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser(1179). J Biol Chem 276:30392–30398
Tsimaratos M, Roger F, Chabardes D et al. (2003) C-peptide stimulates Na(+),K(+)-ATPase activity via PKC alpha in rat medullary thick ascending limb. Diabetologia 46:124–131
Michell BJ, Chen Zp, Tiganis T et al. (2001) Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. J Biol Chem 276:17625–17628
Shafqat J, Juntti-Berggren L, Zhong Z et al. (2002) Proinsulin C-peptide and its analogues induce intracellular Ca2+ increases in human renal tubular cells. Cell Mol Life Sci 59:1185–1189
Johansson J, Ekberg K, Shafqat J et al. (2002) Molecular effects of proinsulin C-peptide. Biochem Biophys Res Commun 295:1035–1040
Prado GN, Taylor L, Zhou X, Ricupero D, Mierke DF, Polgar P (2002) Mechanisms regulating the expression, self-maintenance, and signaling-function of the bradykinin B2 and B1 receptors. J Cell Physiol 193:275–286
Kimura K, White B, Sidhu A (1995) Coupling of human D-1 dopamine receptors to different guanine nucleotide binding proteins: Evidence that D-1 dopamine receptors can couple to both Gs and G(o). J Biol Chem 270:14672–14678
Honing ML, Morrison PJ, Banga JD, Stroes ES, Rabelink TJ (1998) Nitric oxide availability in diabetes mellitus. Diabetes Metab Rev 14:241–249
Acknowledgements
This work was supported by grants from the Ministry of Education, Science and Culture of Japan and by a PROBRAIN grant from the Bio-oriented Technology Research Advancement Institution, Japan. The work was also supported by Research Fellowships of the Japan Society for the Promotion of Science for Young Scientists (T. Kitamura) and of the Ministry of Education, Science and Culture of Japan for Foreign Students (K. Makondo).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kitamura, T., Kimura, K., Makondo, K. et al. Proinsulin C-peptide increases nitric oxide production by enhancing mitogen-activated protein-kinase-dependent transcription of endothelial nitric oxide synthase in aortic endothelial cells of Wistar rats. Diabetologia 46, 1698–1705 (2003). https://doi.org/10.1007/s00125-003-1232-3
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-003-1232-3