
Abstract
Key message
cqSW.A03-2, one of the six identified quantitative trait loci associated with thousand-seed weight in rapeseed, is mapped to a 61.6-kb region on chromosome A03 and corresponds to the candidate gene BnaA03G37960D.
Abstract
Seed weight is an important factor that determines the seed yield of oilseed rape (Brassica napus L.). To elucidate the genetic mechanism of thousand-seed weight (TSW), quantitative trait locus (QTL) mapping was conducted using a double haploid population derived from the cross between an elite line ZY50 and a pol cytoplasmic male sterility restorer line 7-5. The genetic basis of TSW was dissected into six major QTLs. One major QTL denoted as cqSW.A03-2, which explained 8.46–13.70% of the phenotypic variation, was detected across multiple environments. To uncover the genetic basis of cqSW.A03-2, a set of near-isogenic lines were developed. Based on the test of self-pollinated progenies, cqSW.A03-2 was identified as a single Mendelian factor and the ZY50 allele at cqSW.A03-2 showed a positive effect on TSW. Fine mapping delimited the cqSW.A03-2 locus into a 61.6-kb region, and 18 genes within this region were predicted. Candidate gene association analysis and expression analysis indicated that a histidine kinase gene (BnaA03G37960D) is likely to be the candidate gene for the cqSW.A03-2 locus. Our results may contribute to a better understanding of the molecular mechanism of seed weight regulation and promote the breeding program for yield improvement in rapeseed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As the second largest oilseed crop worldwide, rapeseed is used as edible oil and a renewable energy resource (Weir 2008; Basunanda et al. 2010), as well as for feeding livestock because of the high content of protein in the oil extracted meal (Fattahi et al. 2018). Owing to the increasing demand for vegetable oil and the declining acreage used for rapeseed production, improving the yield of rapeseed has always been a major goal of rapeseed breeding (Sun et al. 2018). Seed weight, one of the three direct components (siliques per plant, seed number per silique and seed weight) of rapeseed yield, is a determinant factor in the improvement in rapeseed productivity (Chen et al. 2007; Fan et al. 2010). Furthermore, increasing seed weight and the number of seeds per silique should be effective for improving rapeseed production, while it is difficult to increase siliques per plant under the high-density planting system in China (Zhu et al. 2011). Therefore, dissection of the genetic basis of seed weight will not only deepen our understanding of seed development, but also increase the possibility to improve the breeding efficiency of rapeseed.
Quantitative trait loci (QTLs) analysis has been proved to be a powerful tool to reveal the genetic mechanism of seed weight regulation in B. napus (Brassica napus L.) (Quijada et al. 2006; Chen et al. 2007; Radoev et al. 2008; Shi et al. 2009; Basunanda et al. 2010; Fan et al. 2010; Zhang et al. 2011; Yang et al. 2012; Li et al. 2014b; Fu et al. 2015; Dhaka et al. 2017; Sun et al. 2018). Currently, a total of 168 QTLs for thousand-seed weight (TSW) have been identified in 22 QTL mapping studies, and the power of QTL detection varies in different experiments (Raboanatahiry et al. 2018). The complex genetic basis of TSW in rapeseed indicates that a number of genes are involved in seed weight regulation.
Genome-wide association study (GWAS) provides an alternative way to identify genes involved in seed weight regulation in rapeseed. A total of nine, two, and nine loci were identified to be associated with seed weight in three different studies, respectively (Cai et al. 2014; Li et al. 2014a; Lu et al. 2017). Nevertheless, the resolution of GWAS is largely affected by several issues including population structure, cryptic familial relatedness, markers with rare alleles and rare variants (Gupta et al. 2014), as well as depends on the linkage disequilibrium (LD) decay distance in B. napus, which is about 250–1500 kb (Zhou et al. 2017).
Although a number of loci for TSW have been identified in rapeseed genome, the inconsistent mapping results and different traditional markers used in different studies impede the integration of the identified QTLs (Raman et al. 2013). The inconsistent mapping results can be partly attributed to the genetic background of parental lines, the type and size of the population and the number of environments used in different studies (Zhu and Zhao 2007). Recently, high-density consensus linkage maps have been established via meta-analysis, allowing the integration of linkage maps and comparison between QTLs (Wang et al. 2013; Zhou et al. 2014; Zhao et al. 2016). Furthermore, QTL alignment maps were proposed to combine QTLs from different genetic backgrounds and environments based on B. napus ‘Darmor-bzh’ reference genome (Chalhoub et al. 2014; Raboanatahiry et al. 2017).
Although many efforts have been made to identify the natural variations for seed weight, only a few studies have gone further to fine map and identify the genes underlying QTLs in rapeseed. Up to date, only two genes controlling seed weight have been cloned in B. napus using map-based cloning strategy (Liu et al. 2015; Shi et al. 2019). Firstly, a number of factors impede the comprehensive dissection of the genetic basis of TSW in linkage mapping, such as soil fertility, co-located genes with opposite contributions, genotype-by-environment interactions and QTL mapping methods (Mackay et al. 2009; Cowling and Balazs 2010; Wurschum 2012; Elsoda et al. 2014; Luo et al. 2017). Secondly, the resolution of conventional linkage mapping is low owing to the limited number of recombination events exploited, and large populations segregating at the target loci are required for fine mapping and cloning of the causal genes underlying QTLs (Xiao et al. 2016; Bazakos et al. 2017; Kumar et al. 2017). Moreover, the complexity of allopolyploid rapeseed genome makes the map-based cloning of genes difficult.
In this study, we constructed a high-density linkage map using DH population and analyzed the genetic basis of TSW in B. napus. Fine mapping delimited a major QTL, cqSW.A03-2, into a 61.6-kb region. With the aid of candidate gene association analysis and gene expression analysis, the candidate gene encoding a histidine kinase was identified. The results provide new insights into seed weight regulation in rapeseed.
Materials and methods
Plant materials
A DH population was developed by microspore culture from the cross between male parent ZY50, a semi-winter B. napus variety, and female parent 7-5, a pol CMS restorer line. A random subset of 189 DH lines was used for linkage map construction and QTL analysis. In addition, a panel with 505 rapeseed inbred lines showing considerable variation in TSW (data unpublished) was used for candidate gene association analysis of TSW.
Linkage analysis revealed that cqSW.A03-2 was a major QTL for TSW. Then, a series of NILs were developed by successive backcrossing to validate this QTL. The F1 plants derived from a cross between 7-5 and ZY50 were backcrossed with ZY50. Among the 223 BC1F1 plants, three individuals with the highest recovery rates selected based on marker-assisted background selection (MBS) were used for further crossing. By self-pollination of the BC2F1 plants, BC2F2 progeny were produced. One BC2F2 family, CW57, was selected as the starting material for NIL development and fine mapping of the QTL cqSW.A03-2. During the backcrossing process, the progeny of each backcross, in which the target QTL region was heterozygous, were selected by flanking markers. For fine mapping of cqSW.A03-2, 2732 BC5F3 individuals were employed for screening of recombinants and a series of BC5F4 substitution lines containing recombination breakpoints across the target region were developed (Fig. S1).
Field trial and trait evaluation
The DH population developed by microspore culture and the parents were grown in Wuhan and Jingzhou, Hubei Province from September to May for 1 year (2012–2013). A random subset of 189 DH lines and the two parents were then grown in Wuhan and Jingzhou for two consecutive years (2013–2014 and 2014–2015). A randomized complete block design was conducted with two replications in all environments. Each line was planted in two rows, with 10–12 plants in each row, a distance of 15 cm between plants within each row and 30 cm between rows. At the mature stage, 8–10 open-pollinated plants from each DH line and parental line were selected and the seeds were threshed by hand. The seed weight of each individual was measured based on 500 fully developed seeds with three replications. The average seed weight was then converted to TSW. The TSW was calculated as the average dry weight (g) of 1000 well-filled seeds from 8 to 10 sampled plants. Components of phenotypic variance and broad-sense heritability were estimated with the linear mixed model in the lme4 package (Merk et al. 2012).
For the NIL populations, 274 BC5F2 plants were grown during the rapeseed growing season of 2015–2016. In 2016, 26 BC5F3 families (2732 individuals) were used for recombinant screening and BC5F4 populations were produced by self-pollination of informative recombinants. In the 2017–2018 growing season, the genotype of each recombinant for the target QTL region was determined based on the status of segregation of TSW in the progeny line. With a spacing pattern of 30 cm (between rows) × 15 cm (within rows), all the materials were grown in the experimental plots at Huazhong Agricultural University, Wuhan, China. At maturity, well-developed siliques in the middle of main inflorescence were collected from each plant. The siliques were air-dried for about 2 weeks. Seed weight was initially obtained by weighing 500–1000 dry seeds, and was then converted to TSW value.
The Wuhan and Jingzhou sites were of semi-winter-type rapeseed growing environments. Field management was conducted under standard agricultural procedures. All plots in different trials were harvested at the same time after maturity for all lines.
DNA extraction and genotyping of linkage population
Young leaf tissues were collected from the seedlings of DH population and the parents. Genomic DNA was extracted according to a modified CTAB method (Doyle and Doyle 1987) and preserved at − 20 °C. The DH population and their parents were genotyped using three groups of markers from different sources. The first group consisting of single nucleotide polymorphism (SNP) markers (prefixed “SNP”) were developed from the Brassica 60 K Illumina Infinium SNP array (Clarke et al. 2016) according to the manufacturer’s instructions (Infinium HD Array Ultra Protocol Guide). The automatic allele calling for each locus was accomplished using the Genome Studio software (Illumina Inc, SanDiego, USA). A bi-filtering method for processing SNP array data was applied (Cai et al. 2015), and high-quality SNP markers were filtered. The second group comprising simple sequence repeat (SSR) markers (prefixed “SSR”) were selected from public sources described previously (Zhang et al. 2011). The third group including SSR markers (prefixed “SSW”) were developed from publicly available B. rapa reference genome sequence (Cheng et al. 2011; Wang et al. 2011), following a previously described method (Cheng et al. 2009). The sequence information of all SSR markers is provided in Table S1. A total of 2302 markers, including 2237 SNP markers (prefixed by “SNP”) and 65 SSR markers (prefixed by “SSR” and “SSW”), were selected for linkage map construction.
Linkage map construction and QTL analysis
Linkage analysis was performed using MSTmap software (Wu et al. 2008). The parameters were set as follows: the cut-off p value was 1E−6, the no-map-dist was 30.0, and the no-map-size was 2. The kosambi mapping function was used to calculate the genetic map distance (Kosambi 1943).
QTL analysis was conducted by composite interval mapping (CIM) method (Zeng 1994) using WinQTL cartographer 2.5 software (http://statgen.ncsu.edu/qtlcart/WQTLCart.htm). The experiment-wise logarithm of the odds (LOD) threshold was determined by permutation analysis with 1000 times (Churchill and Doerge 1994). LOD scores corresponding to P = 0.05 (3.08–3.30) were adopted to identify significant QTLs. The number of control markers, window size and walking speed were set to 5, 10 centimorgan (cM) and 2 cM, respectively. Significant QTLs repeatedly detected in different environments were integrated into consensus QTLs by meta-analysis (Goffinet and Gerber 2000; Arcade et al. 2004).
Fine mapping of cqSW.A03-2
The location and effects of cqSW.A03-2 were determined and validated in the F2–F4 generations of BC5 populations. NIL populations were genotyped with markers in the target region and divided into three genotypic groups (cqSW.A03-2/cqSW.A03-2, cqSW.A03-2/cqsw.a03-2, cqsw.a03-2/cqsw.a03-2). The phenotypic means of these genotypic groups were then compared to measure the gene action of cqSW.A03-2. Meanwhile, informative recombinants were selected based on the genotype of each marker in the target region. The phenotypic means of different genotypic groups were compared in the progenies of recombinants to conduct a high-resolution mapping of cqSW.A03-2. New insertion and deletion (InDel) markers were developed from the re-sequencing of ZY50 and 7-5 by our lab (Table S1). The software Primer Premier 6 (Premier Biosoft, Palo Alto, CA, USA) was used to design primers flanking each InDel. Linkage analysis was performed by the software JoinMap4.0 (http://www.kyazma.nl/index.php/mc.JoinMap). The Kosambi mapping function was used for genetic distance calculation (Kosambi 1943).
Association analysis of TSW in natural population
Regional association analysis and candidate gene association analysis were conducted based on phenotypic and genotypic data collected from a panel of 505 inbred lines (data unpublished). Totally, 1554 and 153 SNPs were used for the regional and candidate gene association mapping of TSW, respectively. Principal component analysis (PCA) and kinship evaluation were performed by TASSEL 5.0 (Bradbury et al. 2007). The generalized linear model (GLM) and mixed linear model (MLM) were adapted for calculating trait-SNP association in regional and candidate gene association analysis, respectively. The threshold for significant SNP was set as P < 6.64 × 10−6 (P < 0.01/n, n = total marker number) for regional association analysis, while the threshold of P < 3.27 × 10−4 (P < 0.05/n, n = total marker number) was set for the candidate gene association analysis.
Sequence analysis of candidate genes
The whole-genome re-sequencing data of the parental lines were obtained using Illumina HiSeq X-Ten sequencer with paired-end reads of 151 bp in length. High-quality reads were mapped to the B. napus ‘Darmor-bzh’ reference genome (Chalhoub et al. 2014) by Bowtie 2 v2.1.0 (Langmead and Salzberg 2012). SNP and InDel calling were performed by GATK v2.5.2 (McKenna et al. 2010). Sequence alignments and comparisons of candidate genes were conducted using Geneious (Kearse et al. 2012).
Sequencing primer pairs covering the full length of BnaA03g37960D were designed according to the B. napus ‘Darmor-bzh’ reference genome sequence (Chalhoub et al. 2014). Primer sequences for candidate gene sequencing are listed in Table S1. The PCR products were gel purified with E.Z.N.A Gel DNA Extraction Kit (Omega, USA) and cloned into the pMD-18T vector (TaKaRa, Dalian, China) for sequencing. Coding sequence of BnaA03g37960D was confirmed by sequencing the clones, followed by alignment of the sequencing results with the corresponding genomic sequences.
Expression analysis of candidate genes
Ovaries of different lengths were collected from buds before flowering. After flowering, individual bud was tagged on the day of flowering, then the siliques and seeds were collected at five developmental stages, namely 2, 5, 10, 15 and 20 days after pollination (DAP). For quantitative reverse transcription-PCR (qRT-PCR), we isolated the total RNA from various tissues using a RNeasy Plant Mini Kit (Qiagen) according to the manufacturer’s instructions. RNA samples of 4 μg were reverse transcribed to cDNA using GoScript™ Reverse transcriptase (Promega, USA). qRT-PCR was performed with the GoTaq qPCR Master Mix (Promega, USA) and Bio-Rad CFX96 Real-time system (Bio-Rad). The relative expression levels were calculated by the 2−△△Ct method based on three biological samples and three replicates for each sample (Livak and Schmittgen 2001). The BnACTIN7 (AF111812) gene was used as the internal control.
Results
Phenotypic variation of TSW in the DH population
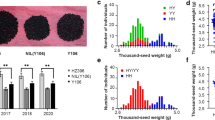
In this work, two B. napus lines 7-5 and ZY50, which showed significant differences in TSW, were used as the parents (Fig. 1a). The average TSW of ZY50 was nearly two times that of 7-5 in each investigated environment (Fig. 1b). Based on the cross between the two parents, DH population was constructed and a subset of 189 fertile DH lines was selected due to the segregation for fertility. Phenotypic data were collected from 189 DH lines in eight experiments conducted over 2 years and in two locations. Measurement of TSW showed a pattern of continuous and nearly normal distribution across all environments, indicating the quantitative inheritance of TSW in rapeseed (Fig. 1c and Table S2). The genetic component of variance was moderate for TSW, while the proportions of variance resulting from genetic interaction effects were relatively low (Table S3). The broad-sense heritability of TSW was 87.30%.

Variations in TSW of DH lines and parents. a Seeds of ZY50 and 7-5 showed considerable variations in seed weight. n = 200, Bar = 5 mm. b Comparison of TSW of ZY50 and 7-5 in different environments. Error bars represent standard deviations; **P < 0.01. c Violin and box plots depicting the phenotypic distribution of DH lines in eight experiments
Linkage map construction
Of the 52,157 SNP loci, 2237 non-redundant SNPs were filtered according to previously described procedures (Cai et al. 2015). In addition, SSR markers were also used for genome-wide polymorphism survey in the DH population. In total, 2302 markers were mapped onto 19 linkage groups, and a high-density linkage map covering 1951.5 cM of genetic distance with a mean of 0.85 cM between the adjacent markers was constructed (Fig. S2). The length of linkage groups varied from 41.04 to 186.32 cM and the number of markers on each linkage group varied from 38 to 216. To validate the quality of linkage map, comparative mapping between the physical map of B. napus and the linkage map was performed. A total of 1643 SNP markers were aligned to the pseudochromosomes of B. napus using basic local alignment search tool (BLAST) to identify the highest-scoring significant hits. The genetic and physical positions of the aligned markers showed high collinearity (Fig. S3). Following the previously described criteria (Shi et al. 2015), we calculated the coverage ratio of the linkage map based on the covered physical distance of linkage groups. As a result, two linkage groups, A09 and C09, which had low coverage ratios of 34.33% and 25.31%, were identified (Table S4).
QTL mapping
A total of 29 QTLs were identified in eight experiments with LOD values ranging from 3.62 to 9.23 (Fig. 2 and Table S5). The fraction of phenotypic variation explained by individual QTL varied between 4.39 and 13.70%. The identified QTLs were distributed on four linkage groups, and there was overlapping of the confidence intervals of QTLs in different experiments. Among the 29 QTLs, 24 could be integrated into six repeatable consensus QTLs by meta-analysis, which were designated as cqSW.A03-1, cqSW.A03-2, cqSW.A07-1, cqSW.A07-2, cqSW.A08-1 and cqSW.C02-1 (Table 1). Among these consensus QTLs, cqSW.A03-2 was consistently detected in six experiments and cqSW.A03-1 was commonly detected in five experiments. Two QTLs (cqSW.A08-1 and cqSW.C02-1) were detected in four experiments, while cqSW.A07-1 and cqSW.A07-2 were consistently found in three and two experiments, respectively (Table S5). The positive alleles for TSW were dispersed between two parents. For all the QTLs except for those located on the C02 linkage group, the positive alleles were inherited from ZY50.

Genetic linkage map and locations of QTLs for TSW. Different environments are indicated by different backgrounds on the cycle. From inside to outside, eight cycles represent eight experiments, 14WH1, 14WH2, 14JZ1, 14JZ2, 15WH1, 15WH2, 15JZ1, 15JZ2, respectively. The green cycle represents consensus QTLs identified in different environments. The two outmost cycles represent comparison of linkage map and physical map of B. napus. Bars with colors within the cycles indicate QTL regions on linkage groups. Blue bars, QTLs detected in 2013–2014 growing season; red bars, QTLs detected in 2014–2015 growing season; black bars, consensus QTLs identified by meta-analysis (color figure online)
Fine mapping of a major QTL
The QTL cqSW.A03-2 showed a main effect on TSW and accounted for 11.02% of the phenotypic variation on average. To further validate cqSW.A03-2, we developed NIL populations using a BC2F2 line, CW57, as the source material. Segregating populations of BC5F2, BC5F3, and BC5F4 generations were developed through three generations of backcross and a series of selfing. The mean TSW of the homozygous 7-5 genotype (cqsw.a03-2/cqsw.a03-2) was compared with that of the homozygous ZY50 genotype (cqSW.A03-2/cqSW.A03-2) in each line. The results showed significant differences in TSW between the two genotypes, suggesting that the gene action of cqSW.A03-2 was stable and significant across different generations and environments (Fig. S4). The TSW of heterozygous genotype (cqSW.A03-2/cqsw.a03-2) was intermediate between that of homozygous ZY50 and 7-5 genotypes, suggesting that cqSW.A03-2 has a partial-dominance effect on TSW.
To narrow down the candidate region of cqSW.A03-2, a segregating population of 2732 BC5F3 individuals was genotyped for informative recombinants, which were further confirmed by four newly developed InDel markers flanked by In1016 and In1170 (Fig. 3a). Based on the allelic composition and recombination breakpoints, the recombinants were grouped into seven genotypes, and progeny testing was performed in each recombinant-derived population. Considering the partial-dominance effect of cqSW.A03-2, comparison of the mean TSW was conducted between the two homozygous recombinants in each line. As a result, cqSW.A03-2 locus was narrowed down to a 61.6-kb region defined by In1202 and In1191 (Fig. 3b).

Fine mapping of cqSW.A03-2 on chromosome A03. a Genotype of the recombinants in the BC5F3 populations. NIL (ZY50) indicates the homozygous ZY50 segments, NIL (Het) indicates the heterozygous segments. b Progeny testing of seven representative recombinants narrowed cqSW.A03-2 to a region of 61.6 kb. Red bars indicate the phenotype of ZY50 homozygous segments and green bars indicate the phenotype of 7-5 homozygous segments. Data represent mean ± SD. *P < 0.05; **P < 0.01; NS not significant. c Annotated genes identified in the cqSW.A03-2 locus. Arrows indicate the direction and position of transcription. The solid arrows indicate the exonic SNP-located genes, in which the red arrow indicates the most likely candidate gene for cqSW.A03-2. Small red stars, diamonds and triangles present genes harboring non-synonymous SNPs, frameshift mutants and exonic InDels, respectively. d Association analysis of TSW with SNPs in the fine-mapped candidate region on chromosome A03. The X axis indicates the physical positions of markers. The Y axis indicates the − log base 10 P value. The most significantly associated SNP is shown in red (color figure online)
Identification of the candidate genes underlying cqSW.A03-2
Within the 61.6-kb region, 18 predicted coding sequences (CDS) were identified based on the annotation of the ‘Darmor-bzh’ reference genome (Chalhoub et al. 2014). The putative functions of the identified genes were predicted by searching the corresponding orthologous genes in Arabidopsis (Arabidopsis thaliana) annotation database (https://www.arabidopsis.org/) (Table 2). Based on the re-sequencing data of the parental lines, we identified 229 exonic SNPs scattered in 14 genes and seven exonic InDels in six genes (Fig. 3c). Among the exonic SNPs, 71 non-synonymous SNPs within ten candidate genes resulted in amino acid variations. Of the seven InDels, two were predicted to cause a putative frameshift mutation in BnaA03g37870D and BnaA03g37940D, respectively.
To further fine map cqSW.A03-2, we conducted a regional association analysis of TSW in a panel with 505 rapeseed lines. A general linear model was used to identify the candidate gene for TSW regulation. Notably, one locus was found to exceed the threshold value and was therefore identified to be significantly associated with TSW (Fig. 3d). Furthermore, the significantly associated SNP, SA03_18863834, fell into the region of a histidine kinase encoding gene (BnaA03g37960D). BnaA03g37960D encodes a histidine kinase homologous to arabidopsis histidine kinase 2 (AHK2), which was shown to be involved in seed size regulation in Arabidopsis (Riefler et al. 2006). Comparison of BnaA03g37960D sequence between ZY50 and 7-5 revealed multiple nucleotide substitutions in the coding sequence (Fig. 4a). Among them, eight nucleotide substitutions resulted in eight amino acid substitutions. Interestingly, the amino acid substitution (from G to W) at position 788 was located in a highly conserved motif G2, as illustrated in a previous study (Kuderova et al. 2015) (Fig. S5).

Sequence variations and expression pattern of BnaA03g37960D. aBnaA03g37960D structure and mutation sites between ZY50 and 7-5. Mutation sites on exons are illustrated. b Comparisons of BnaA03g37960D transcript levels between NIL (ZY50) and NIL (7-5) in ovary and seed development stages. DAP days after pollination. Expression levels are expressed as the relative copies of BnACTIN7 in B. napus. Data are shown as mean ± SE. *P < 0.05; **P < 0.01. ns not significant
Expression analysis of the candidate gene
To investigate the expression profiles of the identified genes in the QTL region, we analyzed the relative transcript levels of the 18 genes in two NILs, NIL (ZY50) and NIL (7-5), which represented high- and low-TSW lines, respectively (Fig. 5). The transcript levels of the identified genes at two stages during ovary and seed development (1–2 mm ovary and 2 DAP silique) were quantified using qRT-PCR. The results showed that the average Cq values of five genes (BnaA03g37810D, BnaA03g37840D, BnaA03g37880D, BnaA03g37900D and BnaA03g37930D) were greater than 35 at both stages. Therefore, these five genes were regarded as having no expression. Among the other 13 genes, four and five genes showed different expression patterns at 1–2 mm ovary and 2-DAP stage, respectively. BnaA03g37800D, BnaA03g37820D and BnaA03g37890D were specifically expressed in NIL (7-5) at both developmental stages. Notably, BnaA03g37960D showed a higher transcript level in NIL (ZY50) than in NIL (7-5) at both stages. Furthermore, BnaA03g37960D showed greater fold differences in expression levels between the two NILs than other genes at both stages. The remaining eight genes showed no significant difference in expression, among which BnaA03g37830D showed no expression at 2-DAP stage.

Expression pattern analysis of the 13 predicted genes in NIL population. a Expression pattern of candidate genes at 1–2 mm ovary stage. b Expression pattern of candidate genes at 2-DAP stage. Data are shown as mean ± SE. *P < 0.05; **P < 0.01. ns not significant
To confirm the expression pattern of BnaA03g37960D, we analyzed its expression levels at other developmental stages of ovary and seed. The results suggested that BnaA03g37960D had relatively high expression levels at the early stages of ovary and seed development (Fig. 4b). Furthermore, BnaA03g37960D had significantly more abundant transcripts during the early stages of ovary and seed development in NIL (ZY50) than in NIL (7-5), and the transcript level of BnaA03g37960D in their hybrid, NIL (Het), was intermediate between that of NIL (ZY50) and NIL (7-5), which is consistent with the partial-dominance effect of cqSW.A03-2 on TSW. The above results suggested that BnaA03g37960D is probably the candidate gene underlying cqSW.A03-2 locus.
Candidate gene association study in natural population
To validate the function of BnaA03g37960D in TSW regulation, we performed candidate gene association analysis in natural population. A total of 153 polymorphism (SNPs) were identified in the region where the candidate gene was located, including 2839 bp upstream and 5563 bp downstream of the transcription starting site, respectively (Fig. 6a). Association analysis revealed that two significant SNPs, SA03_18861910 and SA03_18862302, were located in the second exon of BnaA03g37960D (Fig. 6b, c). Student’s t test analysis of the natural population grouped by the two significant SNPs (designated as A-C, A-G, G-C and G-G) showed significant differences in TSW (Fig. 6d). Nevertheless, there was no polymorphism in ZY50 and 7-5 at the SA03_18861910 and SA03_18862302 sites, that is, the parental lines all harbored G–C allele, suggesting that the two SNPs did not segregate in the DH population and NIL population. The results suggested that the two significantly associated SNPs may merely be in LD with the causative variation in the natural population.

Candidate gene association analysis of BnaA03g37960D in natural population. a Association between TSW and candidate gene polymorphisms. The X axis indicates the distance between the markers and the predicted transcription start site (TSS) site. The Y axis indicates the − log base 10 P value. The polymorphism with MAF < 0.05 was excluded. The two significant SNPs are shown in red. b The gene structure of BnaA03g37960D. The gray boxes indicate untranslated regions, while the black boxes indicate exons. c The inverted triangle indicates the pairwise R2 value between polymorphisms within BnaA03g37960D. d TSW of groups defined by the two significant SNPs in natural population (color figure online)
Discussion
QTL mapping has been proved to be an efficient way to reveal the genetic mechanisms of complex agronomic traits. In this study, a DH population was developed to identify the genetic regions controlling TSW. The QTL analysis resulted in the identification of six consensus QTLs controlling TSW in B. napus. To ascertain the accuracy and robustness of the identified QTLs, we compared the genomic regions underlying the six consensus QTLs with those of QTLs identified in previous studies. The consensus QTL cqSW.A03-1 likely corresponds to qSW012 and DHqSW06 identified in previous studies (Shi et al. 2009, 2011). Two QTLs on A07 linkage group, cqSW.A07-1 and cqSW.A07-2, were congruent with previously identified TSWA7a and TSWA7b, respectively (Fan et al. 2010). In addition, cqSW.A07-1 was also repeatedly identified as sw7.1 (Quijada et al. 2006) and sw7.3 (Udall et al. 2006).
Genes underlying two pleiotropic QTLs on A09 chromosome have been isolated using map-based cloning strategies (Liu et al. 2015; Shi et al. 2019). However, none of these two QTLs was detected on chromosome A09 in the present study. Collinearity analysis between the linkage map and physical map of B. napus revealed that A09 was a short linkage group, which could explain the lack of TSW QTLs on chromosome A09 (Fig. S3). This result may be caused by the poor polymorphism of fertile DH lines for linkage map construction, which were selected based on the segregation of fertility caused by a fertility restorer gene (Rfp) located on A09 chromosome (Liu et al. 2012, 2016). This hypothesis was verified by linkage analysis and QTL detection on A09 chromosome in DH population consisting of 98 fertile lines and 90 sterile lines (Fig. S6). Combined with phenotypic data collected from four experiments in 2012–2013 growing season, we identified two closely linked QTLs, cqSW.A09-1 and cqSW.A09-2 (Table S6), which may be identical to the major pleiotropic QTLs for seed weight and silique length on A09 linkage group identified in previous studies (Yang et al. 2012; Li et al. 2014b).
The strategy of combining linkage mapping with association analysis has been proposed to promote the identification of the causal genes for quantitative traits. Here, we fine-mapped a major QTL and identified a candidate gene by association analysis, which was proved to be involved in seed weight regulation in Arabidopsis (Riefler et al. 2006). Nevertheless, the variation of the significant SNPs was not detected in the parental lines (Fig. 6a), suggesting that the significant SNPs were in LD with the causative sites in natural population, as reported in a previous study (Wang et al. 2018). Moreover, the effect of cqSW.A03-2 is relatively small, which could only explain 8.46–13.70% of the phenotypic variation in DH population. In NIL population, cqSW.A03-2 increased the TSW by 0.26–0.29 g across different environments in NIL (ZY50) compared with that of NIL (7-5) (Fig. S4). In contrast, association analysis revealed that different groups defined by two significant SNPs within the candidate gene showed considerable differences in TSW (Fig. 6d). These results suggested that cqSW.A03-2 is of great potential in the improvement in TSW in B. napus.
The integration of a series of components such as seed size and seed filling process determines the final seed weight (Brinton and Uauy 2019). As a determinant factor of seed weight, seed size is maternally controlled in both dicot plants and monocot plants (Li et al. 2019b). In rapeseed, the seed weight is mainly controlled by maternal genotype (Li et al. 2015). Recently, larger pod wall photosynthetic area due to longer silique length was proved to be a major contributor to final seed weight by maternally supplying carbon from the source to the sink. Moreover, morphological and cytological analyses have revealed the primary role of seed size in determining seed weight (Li et al. 2019a), which is consistent with the high correlation between seed weight and seed size in this study (Fig. S7). This regulation model provides novel insights to the regulatory mechanism of seed weight in rapeseed (Li et al. 2019a). To test the molecular function of cqSW.A03-2, we also measured the silique length and seed number per silique when determining the TSW in NIL population. Interestingly, silique length and seed number per silique showed no significant difference between large-seed and small-seed NILs, contrasting to the significant differences observed for seed size parameters (Figure S8). Therefore, we hypothesize that seed weight is controlled by multiple mechanisms in B. napus, such as cell proliferation, cell expansion, seed filling and alteration of photoassimilate supply by silique length.
Cytokinin signaling is implicated in the regulation of plant growth and development, stress tolerance and crop yield (Muller and Sheen 2007; Kuderova et al. 2015). Cytokinin signal transduction is mediated by a multistep two-component system, in which hybrid His-kinases (HK) receptors perceive cytokinin molecules. In Arabidopsis, three Arabidopsis His-kinases (AHKs) were shown to play redundant or specific roles in the regulation of root and shoot growth, leaf aging and seed size (Higuchi et al. 2004; Nishimura et al. 2004; Mahonen et al. 2006; Riefler et al. 2006). In the present study, we mapped cqSW.A03-2 to a 61.6-kb region, in which 18 genes were predicted. Expression analysis and association mapping suggested that BnaA03g37960D, which encodes a His-kinase receptor, may be the candidate for cqSW.A03-2 locus. And analysis of the amino acid sequences revealed variations on conserved domain between the two parental lines. Further functional analysis of cqSW.A03-2 will shed more light on the regulatory mechanism of seed weight in rapeseed.
Abbreviations
- AHK:
-
Arabidopsis histidine kinase
- BLAST:
-
Basic local alignment search tool
- CDS:
-
Coding sequences
- CIM:
-
Composite interval mapping
- cM:
-
Centimorgan
- DAP:
-
Days after pollination
- DH:
-
Double haploid
- GLM:
-
Generalized linear model
- GWAS:
-
Genome-wide association study
- InDel:
-
Insertion and deletion
- LD:
-
Linkage disequilibrium
- LOD:
-
Logarithm of the odds
- MAS:
-
Marker-assisted selection
- MBS:
-
Marker-assisted background selection
- MLM:
-
Mixed linear model
- NIL:
-
Near-isogenic line
- PCA:
-
Principal component analysis
- qRT-PCR:
-
Quantitative reverse transcription-PCR
- QTL:
-
Quantitative trait locus
- SEL:
-
Seed length
- SES:
-
Seed size
- SEW:
-
Seed width
- SL:
-
Silique length
- SN:
-
Seed number per silique
- SNP:
-
Single nucleotide polymorphism
- SSR:
-
Simple sequence repeat
- TSW:
-
Thousand-seed weight
References
Arcade A, Labourdette A, Falque M, Mangin B, Chardon F, Charcosset A, Joets J (2004) BioMercator: integrating genetic maps and QTL towards discovery of candidate genes. Bioinformatics 20:2324–2326
Basunanda P, Radoev M, Ecke W, Friedt W, Becker HC, Snowdon RJ (2010) Comparative mapping of quantitative trait loci involved in heterosis for seedling and yield traits in oilseed rape (Brassica napus L.). Theor Appl Genet 120:271–281
Bazakos C, Hanemian M, Trontin C, Jimenez-Gomez JM, Loudet O (2017) New strategies and tools in quantitative genetics: how to go from the phenotype to the genotype. Annu Rev Plant Biol 68:435–455
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Brinton J, Uauy C (2019) A reductionist approach to dissecting grain weight and yield in wheat. J Integr Plant Biol 61:337–358
Cai D, Xiao Y, Yang W, Ye W, Wang B, Younas M, Wu J, Liu K (2014) Association mapping of six yieldrelated traits in rapeseed (Brassica napus L.). Theor Appl Genet 127:85–96
Cai G, Yang Q, Yi B, Fan C, Zhang C, Edwards D, Batley J, Zhou Y (2015) A bi-filtering method for processing single nucleotide polymorphism array data improves the quality of genetic map and accuracy of quantitative trait locus mapping in doubled haploid populations of polyploid Brassica napus. BMC Genom 16:409
Chalhoub B, Denoeud F, Liu S et al (2014) Early allopolyploid evolution in the post-neolithic Brassica napus oilseed genome. Science 345:950–953
Chen W, Zhang Y, Liu X, Chen B, Tu J, Tingdong F (2007) Detection of QTL for six yield-related traits in oilseed rape (Brassica napus) using DH and immortalized F2 populations. Theor Appl Genet 115:849–858
Cheng X, Xu J, Xia S, Gu J, Yang Y, Fu J, Qian X, Zhang S, Wu J, Liu K (2009) Development and genetic mapping of microsatellite markers from genome survey sequences in Brassica napus. Theor Appl Genet 118:1121–1131
Cheng F, Liu S, Wu J, Fang L, Sun S, Liu B, Li P, Hua W, Wang X (2011) BRAD, the genetics and genomics database for Brassica plants. BMC Plant Biol 11:136
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Clarke WE, Higgins EE, Plieske J, Wieseke R, Sidebottom C, Khedikar Y, Batley J, Edwards D, Meng J, Li R, Lawley CT, Pauquet J, Laga B, Cheung W, Iniguez-Luy F, Dyrszka E, Rae S, Stich B, Snowdon RJ, Sharpe AG, Ganal MW, Parkin IA (2016) A high-density SNP genotyping array for Brassica napus and its ancestral diploid species based on optimised selection of single-locus markers in the allotetraploid genome. Theor Appl Genet 129:1887–1899
Cowling WA, Balazs E (2010) Prospects and challenges for genome-wide association and genomic selection in oilseed Brassica species. Genome 53:1024–1028
Dhaka N, Rout K, Yadava SK, Sodhi YS, Gupta V, Pental D, Pradhan AK (2017) Genetic dissection of seed weight by QTL analysis and detection of allelic variation in Indian and east European gene pool lines of Brassica juncea. Theor Appl Genet 130:293–307
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Elsoda M, Malosetti M, Zwaan BJ, Koornneef M, Aarts MGM (2014) Genotype × environment interaction QTL mapping in plants: lessons from Arabidopsis. Trends Plant Sci 19:390–398
Fan C, Cai G, Qin J, Li Q, Yang M, Wu J, Fu T, Liu K, Zhou Y (2010) Mapping of quantitative trait loci and development of allele-specific markers for seed weight in Brassica napus. Theor Appl Genet 121:1289–1301
Fattahi F, Fakheri BA, Solouki M, Mollers C, Rezaizad A (2018) Mapping QTL controlling agronomic traits in a doubled haploid population of winter oilseed rape (Brassica napus L.). J Genet 97:1389–1406
Fu Y, Wei D, Dong H, He Y, Cui Y, Mei J, Wan H, Li J, Snowdon R, Friedt W, Li X, Qian W (2015) Comparative quantitative trait loci for silique length and seed weight in Brassica napus. Sci Rep 5:14407
Goffinet B, Gerber S (2000) Quantitative trait loci: a meta-analysis. Genetics 155:463–473
Gupta PK, Kulwal PL, Jaiswal V (2014) Association mapping in crop plants: opportunities and challenges. Adv Genet 85:109–147
Higuchi M, Pischke MS, Mahonen AP, Miyawaki K, Hashimoto Y, Seki M, Kobayashi M, Shinozaki K, Kato T, Tabata S, Helariutta Y, Sussman MR, Kakimoto T (2004) In planta functions of the Arabidopsis cytokinin receptor family. Proc Natl Acad Sci USA 101:8821–8826
Kearse M, Moir R, Wilson A et al (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Kosambi DD (1943) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Kuderova A, Gallova L, Kuricova K, Nejedla E, Curdova A, Micenkova L, Plihal O, Smajs D, Spichal L, Hejatko J (2015) Identification of AHK2- and AHK3-like cytokinin receptors in Brassica napus reveals two subfamilies of AHK2 orthologues. J Exp Bot 66:339–353
Kumar J, Gupta DS, Gupta S, Dubey S, Gupta P, Kumar S (2017) Quantitative trait loci from identification to exploitation for crop improvement. Plant Cell Rep 36:1187–1213
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Li F, Chen B, Xu K, Wu J, Song W, Bancroft I, Harper AL, Trick M, Liu S, Gao G, Wang N, Yan G, Qiao J, Li J, Li H, Xiao X, Zhang T, Wu X (2014a) Genome-wide association study dissects the genetic architecture of seed weight and seed quality in rapeseed (Brassica napus L.). DNA Res 21:355–367
Li N, Shi J, Wang X, Liu G, Wang H (2014b) A combined linkage and regional association mapping validation and fine mapping of two major pleiotropic QTLs for seed weight and silique length in rapeseed (Brassica napus L.). BMC Plant Biol 14:114
Li N, Peng W, Shi J, Wang X, Liu G, Wang H (2015) The natural variation of seed weight is mainly controlled by maternal genotype in rapeseed (Brassica napus L.). PLoS ONE 10:e0125360
Li N, Song D, Peng W, Zhan J, Shi J, Wang X, Liu G, Wang H (2019a) Maternal control of seed weight in rapeseed (Brassica napus L.): the causal link between the size of pod (mother, source) and seed (offspring, sink). Plant Biotechnol J 17:736–749
Li N, Xu R, Li Y (2019b) Molecular networks of seed size control in plants. Annu Rev Plant Biol 70:435–463
Liu Z, Liu P, Long F, Hong D, He Q, Yang G (2012) Fine mapping and candidate gene analysis of the nuclear restorer gene Rfp for pol CMS in rapeseed (Brassica napus L.). Theor Appl Genet 125:773–779
Liu J, Hua W, Hu Z, Yang H, Zhang L, Li R, Deng L, Sun X, Wang X, Wang H (2015) Natural variation in ARF18 gene simultaneously affects seed weight and silique length in polyploid rapeseed. Proc Natl Acad Sci USA 112:E5123–E5132
Liu Z, Yang Z, Wang X, Li K, An H, Liu J, Yang G, Fu T, Yi B, Hong D (2016) A mitochondria-targeted PPR protein restores pol cytoplasmic male sterility by reducing orf224 transcript levels in oilseed rape. Mol Plant 9:1082–1084
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−△△C(T)) method. Methods 25:402–408
Lu K, Peng L, Zhang C, Lu J, Yang B, Xiao Z, Liang Y, Xu X, Qu C, Zhang K, Liu L, Zhu Q, Fu M, Yuan X, Li J (2017) Genome-wide association and transcriptome analyses reveal candidate genes underlying yield-determining traits in Brassica napus. Front Plant Sci 8:206
Luo Z, Wang M, Long Y, Huang Y, Shi L, Zhang C, Liu X, Fitt BDL, Xiang J, Mason AS, Snowdon RJ, Liu P, Meng J, Zou J (2017) Incorporating pleiotropic quantitative trait loci in dissection of complex traits: seed yield in rapeseed as an example. Theor Appl Genet 130:1569–1585
Mackay TF, Stone EA, Ayroles JF (2009) The genetics of quantitative traits: challenges and prospects. Nat Rev Genet 10:565–577
Mahonen AP, Bishopp A, Higuchi M, Nieminen KM, Kinoshita K, Tormakangas K, Ikeda Y, Oka A, Kakimoto T, Helariutta Y (2006) Cytokinin signaling and its inhibitor AHP6 regulate cell fate during vascular development. Science 311:94–98
McKenna A, Hanna M, Banks E et al (2010) The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303
Merk HL, Yarnes SC, Deynze AV, Tong N, Menda N, Mueller LA, Mutschler MA, Loewen SA, Myers JR, Francis DM (2012) Trait diversity and potential for selection indices based on variation among regionally adapted processing tomato germplasm. J Am Soc Hortic Sci 137:427–437
Muller B, Sheen J (2007) Arabidopsis cytokinin signaling pathway. Sci STKE 2007:cm5
Nishimura C, Ohashi Y, Sato S, Kato T, Tabata S, Ueguchi C (2004) Histidine kinase homologs that act as cytokinin receptors possess overlapping functions in the regulation of shoot and root growth in Arabidopsis. Plant Cell 16:1365–1377
Quijada PA, Udall JA, Lambert B, Osborn TC (2006) Quantitative trait analysis of seed yield and other complex traits in hybrid spring rapeseed (Brassica napus L.): 1. Identification of genomic regions from winter germplasm. Theor Appl Genet 113:549–561
Raboanatahiry N, Chao H, Guo L, Gan J, Xiang J, Yan M, Zhang L, Yu L, Li M (2017) Synteny analysis of genes and distribution of loci controlling oil content and fatty acid profile based on QTL alignment map in Brassica napus. BMC Genom 18:776
Raboanatahiry N, Chao H, Dalin H, Pu S, Yan W, Yu L, Wang B, Li M (2018) QTL alignment for seed yield and yield related traits in Brassica napus. Front Plant Sci 9:1127
Radoev M, Becker HC, Ecke W (2008) Genetic analysis of heterosis for yield and yield components in rapeseed (Brassica napus L.) by quantitative trait locus mapping. Genetics 179:1547–1558
Raman H, Raman R, Kilian A, Detering F, Long Y, Edwards D, Parkin IA, Sharpe AG, Nelson MN, Larkan N, Zou J, Meng J, Aslam MN, Batley J, Cowling WA, Lydiate D (2013) A consensus map of rapeseed (Brassica napus L.) based on diversity array technology markers: applications in genetic dissection of qualitative and quantitative traits. BMC Genom 14:277
Riefler M, Novak O, Strnad M, Schmulling T (2006) Arabidopsis cytokinin receptor mutants reveal functions in shoot growth, leaf senescence, seed size, germination, root development, and cytokinin metabolism. Plant Cell 18:40–54
Shi J, Li R, Qiu D, Jiang C, Long Y, Morgan C, Bancroft I, Zhao J, Meng J (2009) Unraveling the complex trait of crop yield with quantitative trait loci mapping in Brassica napus. Genetics 182:851–861
Shi J, Li R, Zou J, Long Y, Meng J (2011) A dynamic and complex network regulates the heterosis of yield-correlated traits in rapeseed (Brassica napus L.). PLoS ONE 6:e21645
Shi J, Zhan J, Yang Y, Ye J, Huang S, Li R, Wang X, Liu G, Wang H (2015) Linkage and regional association analysis reveal two new tightly-linked major-QTLs for pod number and seed number per pod in rapeseed (Brassica napus L.). Sci Rep 5:14481
Shi L, Song J, Guo C, Wang B, Guan Z, Yang P, Chen X, Zhang Q, King GJ, Wang J, Liu K (2019) A CACTA-like transposable element in the upstream region of BnaA9.CYP78A9 acts as an enhancer to increase silique length and seed weight in rapeseed. Plant J 98:524–539
Sun LJ, Wang XD, Yu KJ, Li WJ, Peng Q, Chen F, Zhang W, Fu SX, Xiong DQ, Chu P, Guan RZ, Zhang JF (2018) Mapping of QTLs controlling seed weight and seed-shape traits in Brassica napus L. using a high-density SNP map. Euphytica 214:228
Udall JA, Quijada PA, Lambert B, Osborn TC (2006) Quantitative trait analysis of seed yield and other complex traits in hybrid spring rapeseed (Brassica napus L.): 2. Identification of alleles from unadapted germplasm. Theor Appl Genet 113:597–609
Wang X, Wang H, Wang J et al (2011) The genome of the mesopolyploid crop species Brassica rapa. Nat Genet 43:1035–1039
Wang X, Wang H, Long Y, Li D, Yin Y, Tian J, Chen L, Liu L, Zhao W, Zhao Y, Yu L, Li M (2013) Identification of QTLs associated with oil content in a high-oil Brassica napus cultivar and construction of a high-density consensus map for QTLs comparison in B. napus. PLoS ONE 8:e80569
Wang H, Xu S, Fan Y et al (2018) Beyond pathways: genetic dissection of tocopherol content in maize kernels by combining linkage and association analyses. Plant Biotechnol J 16:1464–1475
Weir B (2008) The biology of Brassica napus L. (canola). Department of Health and Aging Office of the Gene Technology Regulator, Australian Government, Canberra
Wu Y, Bhat PR, Close TJ, Lonardi S (2008) Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph. PLoS Genet 4:e1000212
Wurschum T (2012) Mapping QTL for agronomic traits in breeding populations. Theor Appl Genet 125:201–210
Xiao Y, Tong H, Yang X, Xu S, Pan Q, Qiao F, Raihan MS, Luo Y, Liu H, Zhang X, Yang N, Wang X, Deng M, Jin M, Zhao L, Luo X, Zhou Y, Li X, Liu J, Zhan W, Liu N, Wang H, Chen G, Cai Y, Xu G, Wang W, Zheng D, Yan J (2016) Genome-wide dissection of the maize ear genetic architecture using multiple populations. New Phytol 210:1095–1106
Yang P, Shu C, Chen L, Xu J, Wu J, Liu K (2012) Identification of a major QTL for silique length and seed weight in oilseed rape (Brassica napus L.). Theor Appl Genet 125:285–296
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Zhang L, Yang G, Liu P, Hong D, Li S, He Q (2011) Genetic and correlation analysis of silique-traits in Brassica napus L. by quantitative trait locus mapping. Theor Appl Genet 122:21–31
Zhao W, Wang X, Wang H, Tian J, Li B, Chen L, Chao H, Long Y, Xiang J, Gan J, Liang W, Li M (2016) Genome-wide identification of QTL for seed yield and yield-related traits and construction of a high-density consensus map for QTL comparison in Brassica napus. Front Plant Sci 7:17
Zhou QH, Fu DH, Mason AS, Zeng YJ, Zhao CX, Huang YJ (2014) In silico integration of quantitative trait loci for seed yield and yield-related traits in Brassica napus. Mol Breed 33:881–894
Zhou Q, Zhou C, Zheng W, Mason AS, Fan S, Wu C, Fu D, Huang Y (2017) Genome-wide SNP markers based on SLAF-Seq uncover breeding traces in rapeseed (Brassica napus L.). Front Plant Sci 8:648
Zhu M, Zhao S (2007) Candidate gene identification approach: progress and challenges. Int J Biol Sci 3:420–427
Zhu L, Zhang D, Fu T, Shen J, Wen J (2011) Analysis of yield and disease resistance traits of new winter rapeseed varieties over the past twenty years in China. Agric Sci Technol 12:842–846
Acknowledgements
We thank Dr. Liang Guo from Huazhong Agricultural University for providing phenotypic and genotypic data collected from natural population (data unpublished). This research was supported by Grants from the National 973 Project (2015CB150202).
Author information
Authors and Affiliations
Contributions
HW carried out most of the experiments, including QTL analysis, fine mapping, gene cloning and sequence analysis; MY participated phenotypic and genotypic analyses of DH population; MX participated in fine mapping; PW participated in data analysis; DH and GY designed and supervised the project. HW wrote the original draft. YL, QX, LW, DH and GY were involved in reviewing and editing of the manuscript. All authors read and contributed to the revision of manuscript.
Corresponding authors
Additional information
Communicated by Albrecht E. Melchinger.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
122_2020_3553_MOESM9_ESM.pdf
Table S1. Primer sequences of markers used for various purposes in the study. Table S2. Descriptive statistics of TSW in parents and DH population. Table S3. Variance components for TSW in the DH population. Table S4. Summary statistics of the linkage groups. Table S5. List of all detected significant QTLs for TSW. Table S6. List of QTLs for TSW detected on A09 linkage group (PDF 163 kb)
Rights and permissions
About this article

Cite this article
Wang, H., Yan, M., Xiong, M. et al. Genetic dissection of thousand-seed weight and fine mapping of cqSW.A03-2 via linkage and association analysis in rapeseed (Brassica napus L.). Theor Appl Genet 133, 1321–1335 (2020). https://doi.org/10.1007/s00122-020-03553-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-020-03553-9