
Abstract
Key message
A QTL qMrdd8 that confers resistance to MRDD was fine mapped into an interval of 347 kb; one SNP and two InDels identified in the interval were significantly associated with resistance to MRDD.
Abstract
Maize rough dwarf disease (MRDD) is highly prevalent in the summer maize-growing areas in China, and leads to significant yield losses in maize (Zea mays L.). In this study, the quantitative trait locus (QTL) qMrdd8, which confers resistance to MRDD, was fine mapped. Initially, qMrdd8 was consistently identified in the interval between the simple sequence repeat markers umc1617 and phi121 in three F2 sub-populations derived from a cross between the resistant recombinant inbred line NL203 and the susceptible line B73. Subsequently, qMrdd8 was fine mapped into an interval of 347 kb defined by the markers IDRQ2 and IDRQ20 using a recombinant-derived progeny test strategy. Based on single nucleotide polymorphism (SNP) genotypes identified using the MaizeSNP50 BeadChip, a long haplotype including qMrdd8 was identified in four resistant inbred lines. One SNP, the 2549-bp insertion/deletion polymorphism (InDel) InDel25, and the 2761-bp InDel27, which all were significantly associated with resistance to MRDD in a set of 226 maize inbred lines (P < 0.05), were detected within qMrdd8. Furthermore, two candidate genes, CG1 and CG2, were detected in the interval using RNA sequencing (RNA-Seq), and InDel25 was localized within the candidate gene CG1. In conclusion, the fine mapping of qMrdd8 will be helpful in cloning the resistance gene, and the three polymorphic markers identified in this study could be used to improve MRDD resistance via a marker-assisted selection approach.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Maize rough dwarf disease (MRDD) is a serious viral disease that leads to severe yield losses in maize (Bai et al. 2002; Dovas et al. 2004; Lenardon et al. 1998). Especially on the Yellow-Huai-Hai River Plain in China, maize is sown after the harvest of wheat which serves as the reservoir for the causal virus and its insect vector Laodelphax striatellus (Zhang et al. 2001). Maize rough dwarf virus, Mal de Rio Cuarto virus, and rice black-streaked dwarf virus (RBSDV) are the major pathogens that cause MRDD in Europe (Dovas et al. 2004), South America (Arneodo et al. 2002), and Asia (Bai et al. 2002; Fang et al. 2001), respectively. These closely related viruses affect common plant hosts such as maize and wheat, are carried by a common planthopper vector, show similar disease symptoms, and have genomic segments with highly homologous sequences (Zhang et al. 2001). These three viruses belong to the genus Fijivirus within the family Reoviridae.
In China, RBSDV can be transmitted among maize, rice, and wheat (Bai et al. 2002), and the virus particles reside in the phloem of the affected plants (Arneodo et al. 2002). It causes serious growth abnormalities, such as dwarfing, internode shortening, poor radical system development, and dark green leaves (Zhang et al. 2001). MRDD can be alleviated to some degree by using pesticides to reduce the population of its insect vector. However, the economic input costs and the environmental pollution risks of this approach are considerable. Maize sowing dates can also be delayed to avoid peak migration of the Laodelphax striatellus vector, which is also known as small brown planthopper, but that can reduce maize yield (March et al. 2002). Consequently, the use of resistant cultivars is the most economic and environmentally sound means to control this disease.
Therefore, evaluation of maize germplasm for resistance to MRDD is fundamental for identification of resistant germplasm and subsequent cloning of resistance genes, which are prerequisites for developing resistant cultivars (Ali and Yan 2012). Inbred lines commonly used in maize breeding programs in China have been screened for resistance to MRDD under natural infection (Xue et al. 2012; Yang et al. 2010). MRDD resistance has been found in the inbred lines Qi319, Shen137, SH15, 89-1, Dan3130, Jinhuang59, X178, P138, and others. Most of them were recycled from the US hybrid P78599 (Liu et al. 2015; Lu et al. 2001).
Plant breeders and pathologists have paid much attention to understanding polygenic traits, using QTL mapping to shed light on their genetic architecture and to facilitate their management. QTL mapping is an important way to detect statistically significant associations between molecular markers and QTL underlying traits of interest in segregating populations. Inclusive composite interval mapping (ICIM) is a statistical method for QTL mapping proposed by Li et al. (2007) that takes into account the absorption of additive and epistatic effects by the two flanking markers (Zhang et al. 2008). Extensive simulations in different genomes and various genetic models indicate that ICIM can increase the power of QTL detection, reduce false positive rates, and reduce bias in estimation of QTL effects compared to composite interval mapping (Li et al. 2007). ICIM has been used in a wide range of QTL studies (Cui et al. 2011; Lan et al. 2010; Lu et al. 2009; Luan et al. 2012; Wang et al. 2012a, b). For example, ICIM was used to identify QTL for cotton yield and fiber quality in a backcross inbred line population comprised of 146 lines (Yu et al. 2013).
Previous studies consistently reported that resistance to MRDD is quantitatively inherited (Bonamico et al. 2012; Di Renzo et al. 2004; Liu et al. 2014; Luan et al. 2012; Shi et al. 2012). Using the resistant inbred line BLS14 and the susceptible line Mo17, Di Renzo et al. (2004) identified two QTL for resistance to Mal de Río Cuarto disease, which were located on maize chromosome bins 1.03 and 8.03/4. Using an F2 population derived from a cross between the resistant inbred line 90110 and the susceptible inbred Ye478, Wang (2007) detected three QTL for resistance to MRDD on chromosome bins 6.02, 7.02, and 8.07. Another study using recombinant inbred lines (RIL) also derived from 90110 × Ye478 identified five QTL including one on chromosome bin 8.07 that explained up to 28.9 % of the phenotypic variance in MRDD resistance (Luan et al. 2012). A major QTL originating from the resistant inbred line X178 on chromosome bin 8.03 explained 24.6–37.3 % of the phenotypic variance for MRDD (Shi et al. 2012). A major QTL, qMrdd1, which explained 33.7 and 41.3 % of the phenotypic variance in MRDD resistance in two locations, was also mapped into an interval of 1.2 Mb on chromosome bin 8.03 (Tao et al. 2013b). However, gene(s) have not yet been reported.
After primary mapping of QTL for traits of interest, fine mapping should be conducted to facilitate efficient marker-assisted selection (MAS) and cloning of target genes. The recombinant-derived progeny test strategy is an efficient and powerful method for QTL fine mapping (Yang et al. 2012). This method can accurately locate QTL by testing associations between molecular markers and phenotypic variance in progenies derived from recombinants. If target genes act in a dominant manner, recombinants could be backcrossed to susceptible inbred lines or to wild type to produce progenies for mapping. If target genes act in a recessive manner, recombinants should be self-pollinated to produce progenies. The recombinant-derived progeny test has been used in several studies to fine map QTL underlying traits of interest (Hurni et al. 2015; Nair et al. 2015; Tao et al. 2013a; Zhang 2012). For example, this method was used to fine map the QTL qHSR1 and cloned the target gene ZmWAK, which confers quantitative resistance to maize head smut (Zuo et al. 2015).
Improving the resistance of maize cultivars to MRDD is an important objective for maize breeding. Fine mapping of major QTL for MRDD resistance and subsequent MAS of resistant genotypes will facilitate the development of resistant maize cultivars. The objective of this study was to fine map the QTL qMrdd8, which confers resistance to MRDD in X178 (Shi et al. 2012), and to identify other markers tightly linked to qMrdd8 to use for subsequent MAS of MRDD resistance.
Materials and methods
Plant materials
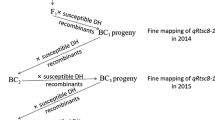
Based on the previous study (Shi et al. 2012), the resistant RIL NL203 derived from a cross between X178 and B73 was crossed with the susceptible inbred line B73 to produce F2 sub-populations. Three F2 sub-populations from three F1 ears, F2-1, F2-2, and F2-5, consisting of 241, 253, and 312 individual plants, respectively, were used to map the QTL for MRDD resistance. Recombinant plants identified in the sub-population F2-5 were used to fine map the QTL qMrdd8 (Fig. 1).

Experimental flow chart for fine mapping of the QTL qMrdd8 conferring resistance to maize rough dwarf disease. “recombinants” indicates selection of recombinant plants
Evaluation of sub-populations for resistance to MRDD
In 2012, the sub-populations F2-1 and F2-2 were evaluated for resistance to MRDD under artificial inoculation following the method established by Di et al. (2005) at Nanjing in Jiangsu Province (32.06°N, 118.78°E). The sub-population F2-5 was evaluated for resistance to MRDD under natural infection during the summer at Yancheng in Jiangsu Province (33.39°N, 120.14°E). Together with the resistant control NL203 and the susceptible control B73, the sub-population F2-5 was planted in 21 rows 0.6 m wide and 4.5 m long on 5 May 2012 during the period that MRDD virus was transmitted by Laodelphax striatellus. At growth stage R6, resistance to MRDD was scored using a disease rating scale based on the severity of MRDD symptoms with scores from 0 to 4 (Miao et al. 2005; Shi et al. 2012), in which a score of 0 indicated the most highly resistant phenotype and 4 represented the most seriously susceptible phenotype.
The inbred lines X178 (resistant) and B73 (susceptible) were also evaluated for resistance to MRDD under artificial inoculation. Leaves from ten individual plants from each line were harvested 72 h after inoculation and stored at −80 °C. RNA was extracted from these leaves following the protocol of TRIZOL reagent established by Invitrogen life technologies Co. Ltd. and subjected to high-throughput sequencing using the Illumina 2000 platform by BerryGenomics Co. Ltd (Beijing, China) to identify candidate genes within qMrdd8.
Genotyping using SSRs
Genomic DNA for genotyping was extracted from the young upper leaves of plants using a CTAB procedure (Murray and Thompson 1980). The quality and quantity of DNA were evaluated by running samples on 1.0 % agarose gels and by determining absorbance ratios using a spectrophotometer (Nanodrop 2000, Thermo Scientific, US). SSR and InDel primer sequences were obtained from MaizeGDB (http://www.maizegdb.org/), and the primers were synthesized at Beijing AuGCT Bio-technology Co. Ltd. PCR amplicons were separated by electrophoresis on 8 % polyacrylamide gels and visualized by silver staining.
Construction of linkage map and QTL identification
Linkage map construction was carried out using QTL IciMapping version 3.2 (http://www.isbreeding.net). Markers with LOD scores of at least 3.0 were grouped together. Grouped markers were then ordered using the nnTwoOpt algorithm. After ordering, marker positions were rippled for fine-tuning using sums of adjacent recombination frequencies. The linkage map and phenotype information were then combined to identify QTL for resistance to MRDD using QTL IciMapping software version 3.2 (Li et al. 2007).
Development of InDel markers
Within the physical interval identified in initial QTL mapping using the three F2 sub-populations, 26 InDel markers were designed to fine map the QTL based on the known InDels between the inbred lines B73 and Mo17 that have been deposited in MaizeGDB (http://www.maizegdb.org/). These InDels were designated with the prefix ‘ID’. Another 47 InDel markers were designed based on the known InDels between the inbred lines Qi319 and Ye478 that were identified by ~30× genome resequencing (Zhou et al. 2016). This set of InDels was designated by the prefix ‘IDRQ’. These markers were all screened for polymorphisms between the parental lines NL203 and B73.
Fine mapping strategy of qMrdd8
Fine mapping of qMrdd8 was carried out using a recombinant-derived progeny test strategy (Fig. 1) (Yang et al. 2012). The 11 plants in the F2-5 sub-population that were heterozygous at qMrdd8 were self-pollinated in the summer of 2012. A total of 335 progenies were planted in a winter nursery in Hainan in the winter of 2012 to be screened for recombinants at qMrdd8. F3 recombinants were then backcrossed to B73 to produce BC1F1 progenies. In the summer of 2013, 1537 progenies were genotyped and new recombinants within the mapped QTL region were backcrossed to B73 to produce BC2F1 progenies. In the winter of 2013, heterozygous recombinant BC2F1 progenies were self-pollinated to produce BC2F2 segregating populations with a segregation ratio equivalent to that of an F2 population with respect to the mapped QTL region. These segregating populations were then used to fine map qMrdd8.
In the summer of 2014, 50–80 randomly selected kernels from the BC2F2 segregating populations were planted to evaluate for resistance to MRDD under artificial inoculation at Nanjing in Jiangsu Province. Another 60–120 kernels from the BC2F2 segregating populations were planted at Jining in Shandong Province to evaluate for resistance to MRDD under natural infection. Individuals from the BC2F2 segregating populations were then classified into one of the three possible marker genotypes in the qMrdd8 region: homozygous NL203/NL203, homozygous B73/B73, or heterozygous NL203/B73. Disease scores among the three genotypic classes were compared using one-way ANOVA in SAS version 9.2 (SAS, Inc., Cary, NC, US). A significant (P < 0.05) difference in disease scores among the three genotypic classes indicated that a MRDD resistance gene was located within a heterozygous region. Disease severity index (DSI) was calculated for all genotypic classes of the recombinant-derived progenies and was used as the basis for phenotypic comparison. DSI was defined as \( {\text{DSI}} = \sum \left( {{\text{disease score}} \times {\text{number of plants with each score}}} \right)/\left( {{\text{maximum disease score}} \times {\text{total number of plants of one genotype}}} \right) .\). Because of the low incidence of MRDD under natural infection in 2014, the proportions of the three genotypic classes with disease scores of 1, 2, 3, or 4 were tested against a segregation ratio of 1:2:1 using a Chi-square test in SAS version 9.2. In 2015, the difference in disease scores between a homozygous recombinant-derived family and B73 was measured using a Student’s t-test in SAS version 9.2 under artificial inoculation, and under natural infection, the difference was measured using a one-way ANOVA followed by Duncan’s multiple comparisons (P = 0.05) in SAS version 9.2.
Haplotype and association analysis
A panel of 226 Chinese maize inbred lines was evaluated for resistance to MRDD under natural infection in Shandong and Jiangsu provinces (Liu et al. 2014) and was genotyped using the MaizeSNP50 BeadChip. Haplotypes for the qMrdd8 region were analyzed and the annotated gene GRMZM2G435373 that lies within the qMrdd8 region was sequenced in this panel. A mixed linear model was used to analyze correlations between polymorphisms and resistance to MRDD using the program TASSEL standalone version 3.1 (Bradbury et al. 2007; Liu et al. 2014). The structure parameter and the kinship parameter were calculated as described in Liu et al. (2014).
Results
Phenotype of the three F2 sub-populations
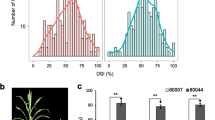
The F2-1 and F2-2 sub-populations were evaluated for resistance to MRDD under artificial inoculation. The average DSI of NL203 and B73 were 0.09 and 0.89, respectively. In the two sub-populations, there were more plants with disease scores of 0 or 4 than with scores of 1, 2, or 3. The distributions of disease scores were similar between the two sub-populations, and the Pearson correlation (r = 0.94) between the distribution of the disease scores of F2-1 and that of F2-2 was statistically significant at P = 0.01. The F2-5 sub-population was evaluated for resistance to MRDD under natural infection. The DSI of NL203 and B73 were 0.08 and 0.75, respectively. The plants with disease scores of 1, 2, or 3 were more frequent than the plants with disease scores of 0 or 4. The distribution of the disease scores differed from that of the F2-1 and F2-2 sub-populations, and the Pearson correlations between the disease score distributions of these sub-populations were not statistically significant at P = 0.05.
Construction of linkage maps
A total of 157 markers including 124 SSRs and 33 InDels on chromosome 8 were screened using the genomic DNA of NL203 and B73, and 18 of these markers (11 %) were polymorphic. The F2-1 and F2-2 sub-populations were genotyped using ten polymorphic markers covering qMrdd8. The lengths of the F2-1 and F2-2 linkage maps were 35.28 and 38.36 cM, respectively (Fig. 2). The maximum intervals between adjacent markers in these two sub-populations were 12.00 and 11.84 cM, respectively. The order of the markers in these two sub-populations was consistent with that of IBM 2008 neighbor map (http://www.maizegdb.org). The F2-5 sub-population was genotyped using 18 polymorphic markers to produce a linkage map 153.81 cM in length. The maximum interval between adjacent markers was 25.53 cM (Fig. 2). The order of these 18 markers was consistent with that in IBM 2008 neighbor map except for the SSR bnlg2037 (http://www.maizegdb.org).

LOD scores for qMrdd8 in three F2 sub-populations derived from inbred lines NL203 and B73
Identification of QTL for resistance to MRDD
By combining the disease scores and the linkage map for the F2-1 sub-population to perform ICIM using the QTL IciMapping software, one QTL with a LOD value of 5.10 flanked by the SSR markers umc1617 and phi121 was identified that explained 11 % of the phenotypic variance for MRDD resistance (Fig. 2; Table 1). In the F2-2 sub-population, one QTL with a LOD value of 6.18 that was also flanked by the SSR markers umc1617 and phi121 was detected and explained 11 % of the phenotypic variance for MRDD resistance (Fig. 2; Table 1). In the F2-5 sub-population, a QTL was identified with a LOD value of 18.80 that was flanked by the SSR markers umc1735 and phi121 (Fig. 2) and explained 25.71 % of the phenotypic variance for MRDD resistance (Table 1). The QTL flanked by the SSR markers umc1617 and phi121 was consistently identified in these three F2 sub-populations, and was designated qMrdd8 (Fig. 2).
Fine mapping of qMrdd8
A total of 11 InDel markers were polymorphic between NL203 and B73 (Table 2). To fine map qMrdd8, the flanking markers umc1617 and phi121 were first used to identify recombinants from among 3899 BC2F1 plants in Hainan province during the winter of 2013, and the genotypes of the recombinants were subsequently resolved using 11 polymorphic InDel markers. Five categories of recombinants were detected and self-pollinated to produce BC2F2 families for fine mapping (Figs. 1, 3).

Fine mapping of qMrdd8 in 2014 and 2015. The years 2014 and 2015 indicate the fine mapping results obtained in 2014 and 2015, respectively. Genetic composition of each recombinant category is shown in different colors. Black, gray, and white rectangles indicate homozygous NL203 genotype, heterozygous NL203/B73 genotype, and homozygous B73 genotype, respectively. Statistically significant differences in disease scores at P = 0.05 among three genotypic classes within one recombant-derived family indicates that the resistance QTL is located in a heterozygous region while no significant difference indicates that the resistance QTL lies in a homozygous region. And their corresponding parental recombinants are deduced as segregant (S) or non-segregant (NS), respectively. In 2015 under artificial inoculation, a statistically significant difference (P = 0.05) in disease scores between a homozygous NL203 recombinant-derived family and B73 indicates that the resistance QTL is located in homozygous NL203 region. And under natural infection, statistically significance (SG) or insignificance (NSG) (P = 0.05) indicates that a homozygous NL203 recombinant-derived family did or did not differ significantly from the susceptible control B73 in disease scores. Finally, qMrdd8 was located between markers IDRQ2 and IDRQ20 with a 347-kb region. DP deduced phenotype, No. P number of progenies, Na not available, B73 homozygous B73 genotype, Het heterozygous genotype NL203/B73, NL203 homozygous NL203 genotype
In 2014, the five categories of recombinant-derived BC2F2 families were evaluated for resistance to MRDD using artificial inoculation and natural infection. Three genotypic classes within each BC2F2 family derived from individual heterozygous recombinant plants were identified: homozygous NL203, heterozygous (NL203/B73), and homozygous B73. DSI was calculated for these three genotypic classes within each family under artificial inoculation. The number of the plants with a disease score of 1, 2, 3, or 4 was also recorded for these three genotypic classes within each family under natural infection. Recombinant categories 3, 4, and 6 were considered susceptible to MRDD because there was no statistically significant difference among the three genotypic classes (Fig. 3). Recombinant category 5 was considered resistant to MRDD because there was a significant difference in MRDD resistance among the three genotypic classes. Although there did not seem to be a significant difference in MRDD resistance among the three genotypic classes in recombinant category 2 according to one-way ANOVA (P > 0.20), a small P value of Chi-square (P = 0.0464) and the significant difference in disease scores between the homozygous NL203 and B73 genotypic classes (Student’s t-test, P < 0.05) suggested that recombinant category 2 was resistant to MRDD. Finally, the QTL was located between the markers IDRQ2 and ID16 (Fig. 3).
Individual plants from BC2F2 families were self-pollinated to generate BC2F3 families in the summer of 2014, and recombinants were screened from 7605 BC2F3 plants at Hainan Province in the winter of 2014. According to the interval of qMrdd8 mapped in the summer of 2014, nine categories of recombinant-derived families, including four categories of homozygous recombinant-derived families, were used to fine map qMrdd8 in the summer of 2015 (Fig. 3). In 2015, InDel25 and InDel27, 2549 and 2761 bp in length, respectively, were found to be polymorphic within the interval between the markers IDRQ2 and ID16. These two InDels were used to develop the polymorphic markers designated IDP25K and IDP27K, respectively. Nine categories of recombinants were genotyped using these two markers. Heterozygous recombinant-derived families included three genotypic classes (homozygous NL203, heterozygous (NL203/B73), and homozygous B73) and were evaluated for MRDD resistance. Seeds of each of the four categories of the homozygous recombinant-derived families (recombinant categories 1-1, 1-2, 1-5, and 1-9) were evaluated for MRDD resistance under natural infection and also mixed with B73 seeds in a ratio of 1:1 (Fig. 3), and these four mixtures were subsequently evaluated for MRDD resistance under artificial inoculation. After the evaluation under artificial inoculation and natural infection, recombinant category 1-1 was resistant to MRDD because of the statistically significant difference from B73 in disease scores (Fig. 3). This implied that the resistance QTL was located within the homozygous NL203 region of recombinant category 1-1. The significant difference in MRDD scores among the three genotypic classes within the families derived from recombinant categories 1–3, 1–4, 1–6, 1–7, and 1–8 indicated that the resistance QTL was located in the heterozygous regions of these recombinant categories (Fig. 3). Recombinant categories 1-2, 1-5, and 1-9 were susceptible to MRDD because of insignificant difference in MRDD scores between B73 and the families derived from these three recombinant categories and the high DSI of the three families. Thus, qMrdd8 was mapped into the 347-kb interval between the markers IDRQ2 and IDRQ20 (Table 2; Fig. 3).
Haplotype and polymorphisms within qMrdd8 associated with MRDD resistance
Based on the SNP genotypes of these 226 maize inbred lines analyzed using the MaizeSNP50 BeadChip, the haplotype, which was detected in five resistant inbred lines by Liu et al. (2014) and was 81.57 Mb in length, was identified in other four inbred lines including Jinghuang96B, Liao68, 141, and Shen137. The DSI of these four inbred lines were lower than the average DSI of all 226 inbred lines, and were significantly different from that of the susceptible control Ye478 based on ANOVA followed by Duncan’s multiple comparisons (P < 0.05).
The gene GRMZM2G435373 was located within the qMrdd8 interval, and was annotated as a GDP dissociation inhibitor related to tobacco mosaic virus infection (Kramer et al. 2011). This gene was resequenced in all 226 maize inbred lines, and one polymorphic SNP marker, SNP7, was detected on chromosome 8 at 103,446,448 according to B73 RefGen_v3 (www.maizegdb.org). This SNP consisted of a T:C transition with nine inbred lines carrying the T variant (Table S1). The DSI of the nine inbred lines ranged from 0 to 0.41 with an average of 0.18; all were significantly different from that of the susceptible control Ye478 (P < 0.05). Further, InDel25 and InDel27 were detected on chromosome 8 at positions 103,537,713 and 103,634,975, respectively, according to B73 RefGen_v3. They were in the same phase with SNP7 across all 226 maize inbred lines analyzed here (Table S1). Association mapping indicated that SNP7, InDel25, and InDel27 within the 81.57-Mb haplotype were all significantly associated with MRDD resistance (P < 0.05).
Identification of candidate genes associated with MRDD resistance
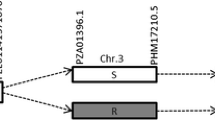
According to maize B73 genome annotation available at MaizeGDB (http://www.maizegdb.org), one annotated gene GRMZM2G435373 was detected in the qMrdd8 interval between the markers IDRQ2 and IDRQ20. However, this gene might not be the resistance gene underlying qMrdd8 because there was only one synonymous mutation in its mRNA and its expression did not significantly differ between X178 and B73. Two candidate genes, CG1 (Fig. 4a) and CG2 (Fig. 4b), were identified within the qMrdd8 interval using RNA-Seq analysis. InDel25 was located within CG1 according to its physical position. CG2, which is about 60 kb from InDel27, might only be expressed in X178 (Fig. 4b).

Candidate genes identified by RNA-Seq. RNA-Seq reads are shown in the Integrative Genomics Viewer. “a” indicates CG1, and “b” indicates CG2
Discussion
Evaluation of MRDD resistance under natural infection and artificial inoculation
Accurate evaluation of resistance to MRDD is crucial for fine mapping of associated QTL. Both natural infection and artificial inoculation were used to evaluate the MRDD resistance of individual plants in this study. Natural infection has commonly been used to evaluate maize germplasm for resistance to MRDD (Xue et al. 2012; Yang et al. 2010) and to dissect the genetic architecture of MRDD resistance (Hao et al. 2015; Liu et al. 2014; Tao et al. 2013b). The main advantage of natural infection is the large-scale evaluation of plants with replications in one or multiple environments. However, variance in maize plant age, the number and peak migration times of the planthopper vector, and field conditions all could result in the fluctuation in the MRDD resistance phenotype (Tao et al. 2013b). Artificial inoculation is another method used to evaluate maize plants for resistance to MRDD that provides relatively stable and uniform infection conditions but generally allows fewer plants to be inoculated (Di et al. 2005; Wang et al. 2006). In the present study, the distributions of the disease-resistance phenotypes in the F2-1 and F2-2 sub-populations were significantly correlated, which indicated that relatively stable and uniform infection conditions were obtained by artificial inoculation. Disease-resistance phenotypes in the F2-5 sub-population were distributed nearly normally. The correlation between the distributions of the disease-resistance phenotypes in the F2-1 and F2-5 sub-populations was not significant, which could have resulted from the variance in the dynamics, density, or other properties of the planthopper vector. The association of qMrdd8 with MRDD resistance was identified consistently under both natural infection and artificial inoculation. This implied that the resistance evaluation and disease severity scoring systems used here were valid.
Potential of tightly linked markers for MAS
Previous studies have reported that resistance to MRDD is quantitatively inherited (Bonamico et al. 2012; Di Renzo et al. 2004; Liu et al. 2014; Luan et al. 2012; Shi et al. 2012). Polygenic traits, such as yield, flowering time, and resistance to disease, are difficult to manage for geneticists, plant breeders, and pathologists. However, molecular markers now allow the genetic architecture underlying quantitative traits to be explored using linkage mapping and association mapping. The establishment of associations between molecular markers and QTL underlying quantitative traits then facilitates the management of these traits. A main goal of QTL mapping is to identify markers significantly associated with traits of interest to use for MAS in plant breeding programs. The application of MAS is especially meaningful in breeding for resistance to MRDD, because it will allow more rapid development of resistant cultivars by pyramiding disease resistance genes from different germplasm sources.
In the present study, the QTL qMrdd8 that confers resistance to MRDD was fine mapped into a 347-kb interval within the 1.2-Mb interval previously identified by Tao et al. (2013b). SNP7, InDel25 and InDel27 in the interval were all significantly associated with resistance to MRDD. Because these molecular markers were tightly linked within 5 cM of the QTL, they can be efficiently used for MAS of MRDD resistance (Michelmore 1995). All of these three markers were polymorphic between the resistant haplotype and all susceptible inbred lines in the present study (Table S1). Thus, it should be possible to use these three markers to transfer this MRDD resistance gene into many other susceptible lines that have other desirable traits. According to the association analysis performed in this study, elite inbred lines such as Chang7-2, Dan340, Mo17, and Shen5003, which played an important role in Chinese maize breeding (Liu et al. 2015), were susceptible to MRDD because of their high DSI. The three markers can be employed to transfer the resistance gene underlying qMrdd8 from one of the nine resistant lines into elite breeding populations to improve their MRDD resistance using MAS. The knowledge of the linkage of these markers to MRDD resistance will be particularly valuable because the evaluation of resistance to MRDD is time consuming and difficult under natural field conditions.
The genetic architecture of resistance to MRDD
Several studies have shown that the control of resistance to MRDD is polygenic (Bonamico et al. 2012; Di Renzo et al. 2004; Hao et al. 2015; Liu et al. 2014; Luan et al. 2012; Shi et al. 2012; Tao et al. 2013b). These studies have identified many QTL conferring resistance to MRDD through linkage mapping or genome-wide association mapping. However, only a few of these QTL were consistently associated with MRDD across these studies. For example, a QTL on chromosome bin 8.03 identified by Di Renzo et al. (2004), Shi et al. (2012), Tao et al. (2013b), and Liu et al. (2014) was not detected in the studies conducted by Hao et al. (2015) and Luan et al. (2012). This incongruity might have been due to differences in scoring systems, distinct genetic backgrounds of different mapping populations, the genetic architecture underlying resistance to MRDD, or genotype-by-environment interactions. For example, Luan et al. (2012) evaluated resistance to MRDD using four indices reflecting symptoms such as shortening of the upper internode, waxy enations, and tassel phenotype. However, our results indicated that the genetic architecture underlying resistance to MRDD is an important factor that might have resulted in the inconsistent detection of QTL in previous studies. One long haplotype associated with MRDD resistance was identified in nine maize inbred lines that were all recycled from the hybrid P78599 and differed significantly in DSI from the susceptible control Ye478 (Liu et al. 2014). This haplotype may play an important role in improving MRDD tolerance of susceptible lines. The cloning of the resistance gene underlying qMrdd8 located in this haplotype can be conducted to reveal the mechanism for resistance to MRDD.
Author contribution statement
C. Liu and J. Weng performed the experiments and wrote the paper. D. Zhang, Z. Hao, H. Yong, C. Xie, M. Li, S. Zhang, and X. Li performed the experiments and revised the paper. J. Weng and X. Li designed the experiments.
References
Ali F, Yan JB (2012) Disease resistance in maize and the role of molecular breeding in defending against global threat. J Integr Plant Biol 54:134–151
Arneodo JD, Guzman FA, Conci LR, Laguna IG, Truol GA (2002) Transmission features of Mal de Rio Cuarto virus in wheat by its planthopper vector delphacodes kuscheli. Ann Appl Biol 141:195–200
Bai FW, Yan J, Qu ZC, Zhang HW, Xu J, Ye MM, Shen D (2002) Phylogenetic analysis reveals that a dwarfing disease on different cereal crop in china is due to rice black streaked dwarf virus (RBSDV). Virus Genes 25:201–206
Bonamico NC, Di Renzo MA, IbaÑEz MA, Borghi ML, Díaz DG, Salerno JC, Balzarini MG (2012) QTL analysis of resistance to Mal de Río Cuarto disease in maize using recombinant inbred lines. J Agric Sci 150:619–629
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Cui F, Li J, Ding A, Zhao C, Wang L, Wang X, Li S, Bao Y, Li X, Feng D, Kong L, Wang H (2011) Conditional QTL mapping for plant height with respect to the length of the spike and internode in two mapping populations of wheat. Theor Appl Genet 122:1517–1536
Di Renzo MA, Bonamico NC, Díaz DG, IbaÑEz MA, Faricelli ME, Balzarini MG, Salerno JC (2004) Microsatellite markers linked to QTL for resistance to Mal de Río Cuarto disease in Zea mays L. J Agric Sci 142:289–295
Di DP, Miao HQ, Lu YG, Tian LZ (2005) Study on the method of inoculation and identification for the resistance of maize to maize rough dwarf virus. J Agric Univ Hebei 28:76–78 (in Chinese)
Dovas CI, Eythymiou K, Katis NI (2004) First report of maize rough dwarf virus (MRDV) on maize crops in Greece. Plant Pathol 53:238
Fang S, Yu J, Feng J, Han C, Li D, Liu Y (2001) Identification of rice black-streaked dwarf fijivirus in maize with rough dwarf disease in China. Arch Virol 146:167–170
Hao D, Cheng Y, Chen G, Lu H, Shi M, Zhang Z, Huang X, Mao Y, Xue L (2015) Identification of significant single nucleotide polymorphisms for resistance to maize rough dwarf disease in elite maize (Zea mays L.) inbred lines. Euphytica 203:109–120
Hurni S, Scheuermann D, Krattinger SG, Kessel B, Wicker T, Herren G, Fitze MN, Breen J, Presterl T, Ouzunova M, Keller B (2015) The maize disease resistance gene Htn1 against northern corn leaf blight encodes a wall-associated receptor-like kinase. Proc Natl Acad Sci USA 112:8780–8785
Kramer SR, Goregaoker SP, Culver JN (2011) Association of the Tobacco mosaic virus 126 kDa replication protein with a GDI protein affects host susceptibility. Virology 414:110–118
Lan C, Liang S, Zhou X, Zhou G, Lu Q, Xia X, He Z (2010) Identification of genomic regions controlling adult-plant stripe rust resistance in Chinese landrace Pingyuan 50 through bulked segregant analysis. Phytopathology 100:313–318
Lenardon SL, March GJ, Nome SF, Ornaghi JA (1998) Recent outbreak of “Mal de Rio Cuarto” virus on corn in Argentina. Plant Dis 82:448
Li H, Ye G, Wang J (2007) A modified algorithm for the improvement of composite interval mapping. Genetics 175:361–374
Liu CL, Weng JF, Zhang DG, Zhang XC, Yang XY, Shi LY, Meng QC, Yuan JH, Guo XP, Hao ZF, Xie CX, Li MS, Ci XK, Bai L, Li XH, Zhang SH (2014) Genome-wide association study of resistance to rough dwarf disease in maize. Eur J Plant Pathol 139:205–216
Liu C, Hao Z, Zhang D, Xie C, Li M, Zhang X, Yong H, Zhang S, Weng J, Li X (2015) Genetic properties of 240 maize inbred lines and identity-by-descent segments revealed by high-density SNP markers. Mol Breed 35:146
Lu YG, Di DP, Miao HQ, Tian LZ (2001) Identification and analysis on resistance of introduced foreign and domestic maize inbreds to MRDV. J Hebei Agric Sci 5:22–24 (in Chinese)
Lu Y, Lan C, Liang S, Zhou X, Liu D, Zhou G, Lu Q, Jing J, Wang M, Xia X, He Z (2009) QTL mapping for adult-plant resistance to stripe rust in Italian common wheat cultivars Libellula and Strampelli. Theor Appl Genet 119:1349–1359
Luan J, Wang F, Li Y, Zhang B, Zhang J (2012) Mapping quantitative trait loci conferring resistance to rice black-streaked virus in maize (Zea mays L.). Theor Appl Genet 125:781–791
March GJ, Ornaghi JA, Beviacqua JE, Giuggia J, Rago A, Lenardon SL (2002) Systemic insecticides for control of delphacodes kuscheli and the Mal de Río Cuarto virus on maize. Int J Pest Manag 48:127–132
Miao HQ, Lu YG, Tian LZ, Di DP (2005) A preliminary report on the criterion for evaluating the disease resistance of maize to the disease caused by maize rough dwarf fijivirus. J Northwest Sci Technol Univ Agric For 33:1 (in Chinese)
Michelmore R (1995) Molecular approaches to manipulation of disease resistance genes. Annu Rev Phytopathol 15:393–427
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucl Acids Res 8:4321–4325
Nair SK, Babu R, Magorokosho C, Mahuku G, Semagn K, Beyene Y, Das B, Makumbi D, Lava Kumar P, Olsen M, Boddupalli PM (2015) Fine mapping of Msv1, a major QTL for resistance to Maize Streak Virus leads to development of production markers for breeding pipelines. Theor Appl Genet 128:1839–1854
Shi LY, Hao ZF, Weng JF, Xie CX, Liu CL, Zhang DG, Li MS, Bai L, Li XH, Zhang SH (2012) Identification of a major quantitative trait locus for resistance to maize rough dwarf virus in a Chinese maize inbred line X178 using a linkage map based on 514 gene-derived single nucleotide polymorphisms. Mol Breed 30:615–625
Tao Y, Jiang L, Liu Q, Zhang Y, Zhang R, Ingvardsen CR, Frei UK, Wang B, Lai J, Lubberstedt T, Xu M (2013a) Combined linkage and association mapping reveals candidates for Scmv1, a major locus involved in resistance to sugarcane mosaic virus (SCMV) in maize. BMC Plant Biol 13:162
Tao YF, Liu QC, Wang HH, Zhang YJ, Huang XY, Wang BB, Lai JS, Ye JR, Liu BS, Xu ML (2013b) Identification and fine-mapping of a QTL, qMrdd1, that confers recessive resistance to maize rough dwarf disease. BMC Plant Biol 13:145
Wang F (2007) Molecular mapping of three loci conferring resistance to maize (Zea mays L.) rough dwarf disease. Doctoral Dissertation: Shandong University (in Chinese)
Wang F, Qin GZ, Sui ZH, Wang ZH, Wang ZY, Yu JL, Zhang JR (2006) Improved method for assaying maize plant resistance to maize rough dwarf disease by artificial inoculation and real-time RT-PCR. Eur J Plant Pathol 116:289–300
Wang Z, Chen Z, Cheng J, Lai Y, Wang J, Bao Y, Huang J, Zhang H (2012a) QTL analysis of Na+ and K+ concentrations in roots and shoots under different levels of NaCl stress in rice (Oryza sativa L.). PLoS One 7:e51202
Wang Z, Cheng J, Chen Z, Huang J, Bao Y, Wang J, Zhang H (2012b) Identification of QTLs with main, epistatic and QTL x environment interaction effects for salt tolerance in rice seedlings under different salinity conditions. Theor Appl Genet 125:807–815
Xue L, Zhang D, Xu L, Jin MM, Peng CJ, Xu CW (2012) Mining and analyzing genetic diversity for maize rough dwarf disease resistant gerplasms and its application in maize breeding. Acta Agron Sin 37:2123–2129 (in Chinese)
Yang XF, Wen GB, Yang Y (2010) Resistance identification and analysis of different maize germplasms to maize rough dwarf virus. J Maize Sci 18:144–146 (in Chinese)
Yang Q, Zhang DF, Xu ML (2012) A sequential quantitative trait locus fine-mapping strategy using recombinant-derived progeny. J Integr Plant Biol 54:228–237
Yu J, Zhang K, Li S, Yu S, Zhai H, Wu M, Li X, Fan S, Song M, Yang D, Li Y, Zhang J (2013) Mapping quantitative trait loci for lint yield and fiber quality across environments in a Gossypium hirsutum × Gossypium barbadense backcross inbred line population. Theor Appl Genet 126:275–287
Zhang YJ (2012) Mapping of QTL for resistance to maize rough dwarf disease using near-isogenic lines and their application in molecular marker-assisted selection. Master Dissertation: Shandong Agricultural University (in Chinese)
Zhang H, Chen J, Lei J, Adams MJ (2001) Sequence analysis shows that a dwarfing disease on rice, wheat and maize in China is caused by Rice black-streaked dwarf virus. Eur J Plant Pathol 107:563–567
Zhang L, Li H, Li Z, Wang J (2008) Interactions between markers can be caused by the dominance effect of quantitative trait loci. Genetics 180:1177–1190
Zhang D, Liu Y, Guo Y, Yang Q, Ye J, Chen S, Xu M (2012) Fine-mapping of qRfg2, a QTL for resistance to Gibberella stalk rot in maize. Theor Appl Genet 124:585–596
Zhou Z, Zhang C, Zhou Y, Hao Z, Wang Z, Zeng X, Di H, Li M, Zhang D, Yong H, Zhang S, Weng J, Li X (2016) Genetic dissection of maize plant architecture with an ultra-high density bin map based on recombinant inbred lines. BMC Genom 17:178
Zuo W, Chao Q, Zhang N, Ye J, Tan G, Li B, Xing Y, Zhang B, Liu H, Fengler KA, Zhao J, Zhao X, Chen Y, Lai J, Yan J, Xu M (2015) A maize wall-associated kinase confers quantitative resistance to head smut. Nat Genet 47:151–157
Acknowledgments
We thank Jianhua Yuan, Qingchang Meng, Yanping Chen, and Xiaoming Kong for their assistance in evaluation of resistance to MRDD. This research was jointly funded by the Pilot Project of Breeding of the Seven Major Crops (2016YFD0101803), the International Science & Technology Cooperation Program of China (2014DFG1690), the National Hi-Tech Research Program and Development Program of China (2012AA101104), and the Chinese Academy of Agriculture Science (CAAS) Innovation Project.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
We claim that the experiments described herein comply with the ethical standards in China.
Conflict of interest
A patent regarding the polymorphisms SNP7, InDel25, and InDel27 and tightly linked markers has been applied for at the State Intellectual Property Office of the People’s Republic of China.
Additional information
Communicated by T. Lubberstedt.
C. Liu and J. Hua contributed equally to this article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, C., Hua, J., Liu, C. et al. Fine mapping of a quantitative trait locus conferring resistance to maize rough dwarf disease. Theor Appl Genet 129, 2333–2342 (2016). https://doi.org/10.1007/s00122-016-2770-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-016-2770-7