
Abstract
Key message
Angular leaf spot is a devastating bacterial disease of strawberry. Resistance from two wild accessions is highly heritable and controlled by a major locus on linkage group 6D.
Abstract
Angular leaf spot caused by Xanthomonas fragariae is the only major bacterial disease of cultivated strawberry (Fragaria ×ananassa). While this disease may cause reductions of up to 8 % of marketable yield in Florida winter annual production, no resistant cultivars have been commercialized. Wild accessions US4808 and US4809 were previously identified as resistant to the four genetic clades of X. fragariae, and introgression of the trait into commercial quality perennial-type germplasm was initiated. Previous reports indicated high heritability for the trait but proposed both single-locus and multi-locus inheritance models. The objective of this study was to determine the mode of inheritance of resistance, to identify causal loci, and to begin introgression of resistance into Florida-adapted germplasm. Resistance was observed in two years of field trials with inoculated plants that assayed four full-sib families descended from US4808 to US4809. Resistance segregated 1:1 in all families indicating control by a dominant allele at a single locus. Using a selective genotyping approach with the IStraw90 Axiom® SNP array and pedigree-based QTL detection, a single major-effect QTL was identified in two full-sib families, one descended from each resistant accession. High-resolution melt curve analysis validated the presence of the QTL in separate populations. The QTL was delimited to the 33.1–33.6 Mbp (F. vesca vesca v1.1 reference) and 34.8–35.3 Mbp (F. vesca bracteata v2.0 reference) regions of linkage group 6D for both resistance sources and was designated FaRXf1. Characterization of this locus will facilitate marker-assisted selection toward the development of resistant cultivars.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Angular leaf spot (ALS) caused by Xanthomonas fragariae (Kennedy and King 1962a, b; Maas 1984) is the only major bacterial disease of cultivated strawberry (Fragaria ×ananassa Duchesne). First described in Minnesota in 1960 (Kennedy and King 1960), ALS has since been formally reported in most major strawberry producing regions worldwide. Symptoms include water-soaked foliar lesions that are transparent when viewed with transmitted light and that produce bacterial ooze at high relative humidity (Kennedy and King 1960, 1962a). Lesions often coalesce and become necrotic, causing a tattered leaf appearance and a reduction in total yield of up to 7.4 % by weight during the Florida winter growing season (Kennedy and King 1962b; Roberts et al. 1997). Similarly, marketable yields may be reduced by up to 8.6 % due to presence of necrotic lesions on fruit calyxes, although the pathogen does not directly affect the fruit itself (Epstein 1966; Roberts et al. 1997).
In production areas with temperate climate and perennial growing systems, the primary source of inoculum is likely overwintered leaf litter (Kennedy and King 1962b). Conversely, in annual production systems, inoculum most likely is derived from asymptomatic transplants with latent infection that become symptomatic after establishment in the fruiting field (Roberts et al. 1996). Pathogen spread is facilitated by rain events or the use of overhead irrigation, the latter of which is used intensively in winter annual production systems for transplant establishment and to protect flowers during frosts. Epidemic ALS appears to be linked to high relative humidity and temperatures between 18 to 25 °C (Hildebrand et al. 2005), suggesting that conditions on the days following frost events in Florida are ideal for symptom development.
Chemical control with copper formulations or acibenzolar-s-methyl is marginally effective but may result in a similar amount of yield loss to that caused by ALS, especially in years with low disease pressure (Mertely et al. 2010). As such, commercial fruit growers rarely attempt chemical control of ALS. Due to the lack of viable control options, the European and Mediterranean Plant Protection Organization has placed the pathogen on its A2 list of pests recommended for regulation (CABI/EPPO 1997).
Thus far, no ALS-resistant cultivated strawberry (2n = 8× = 56) has been commercialized. In a screen of diverse Fragaria spp. germplasm, Maas et al. (2000) and (2002) identified two octoploid accessions as resistant to bacterial isolates representing the four proposed genetic clades of X. fragariae (Pooler et al. 1996; Roberts et al. 1998; Hildebrand et al. 2005). The first, known as US4808, is a F. virginiana accession collected in the wild in Minnesota in 1986 (Maas et al. 2002). The second, US4809, is the offspring of the cross F. virginiana accession SG-26 (PI414129) × F. ×ananassa ‘Earliglow’; SG-26 was collected on a family farm in Georgia where it had been grown since 1900 (Maas et al. 2002).
Reports of initial introgression efforts described 3–18 % resistant progeny in resistant × susceptible crosses when either of the two resistant accessions was used as the trait donor, suggesting that resistance was regulated by at least three unlinked genetic loci (Hartung et al. 2003; Lewers et al. 2003; Jamieson et al. 2013). However, a later report described approximately 50 % resistant progeny in resistant × susceptible crosses and 75 % resistant progeny in resistant × resistant crosses, suggesting that resistance was conferred by a dominant allele at a single locus (Jamieson et al. 2014). The reason for this inconsistency in segregation ratios is unknown but may relate to the introgression process. Broad sense heritability (H 2) estimates were at least 0.69 for both resistant accessions for resistance against all four known X. fragariae genetic clades, indicating that phenotypic selection was effective for trait introgression (Pooler et al. 1996; Roberts et al. 1998; Lewers et al. 2003). This was confirmed by rapid progress in introgression efforts made to date in perennial production systems (Jamieson et al. 2013, 2014).
The objective of this study was to elucidate the genetic architecture of resistance to ALS derived from accessions US4808 and US4809. To this end, a complex multifamily population derived from the two accessions was assayed for resistance in two years of field trials, followed by QTL identification using pedigree-based QTL analysis and QTL validation using high-resolution melting analysis.
Materials and methods
Germplasm
Four full-sib families were phenotyped in two years of field trials with inoculated plants. Crosses were made in the greenhouse in January–March of 2013 and 2014 for field trials planted in October of 2013 and 2014, respectively. In the 2013–2014 season, the cross ‘Strawberry Festival’ × K12-10 generated family 13.77 (n = 137) whereas the cross ‘Strawberry Festival’ × K08-17 generated family 13.78 (n = 134; Fig. 1). Parents K08-17 and K12-10 were resistant selections arising from the resistance introgression efforts of Jamieson et al. (2014) in Nova Scotia, whereas ‘Strawberry Festival’ is a susceptible University of Florida cultivar grown widely in winter and spring annual production systems.

Pedigree of full-sib families generated in this study. a Families 13.78 and 14.100; b families 13.77 and 14.101. Female parents are to the left and male parents to the right in each cross. Gray boxes indicate resistant individuals or families segregating for resistance
After phenotyping and observation during the 2013–2014 field trial, one resistant individual from family 13.77 (identified as 13.77−5) was crossed to Sensation™ ‘Florida127’, a recent University of Florida release (Whitaker et al. 2015), to generate family 14.101 (n = 287), and one resistant progeny from family 13.78 (13.78−57) was crossed to advanced selection FL 11.28–34 to generate family 14.100 (n = 125; Fig. 1). Families 14.100 and 14.101 were phenotyped in 2014–2015 field trials with inoculated plants as a validation population (Fig. 1). Resistant progeny from 14.100 to 14.101 with fruit quality and horticultural traits approaching ideotypic levels were selected for further evaluation.
Field trials
Seedlings were planted in a high-elevation summer nursery in Center, Colorado, in June of each year for runner propagation (clonal replicates). Bare root daughter plants were planted in October of both years into black plastic mulch on two-row raised beds on 38 cm centers at the University of Florida Gulf Coast Research and Education Center research farm in Wimauma, Florida. Overhead sprinklers were used for plant establishment for 10 d after planting. Soil fumigation, pest and pathogen management, fertilization, and irrigation were conducted according to industry standards.
Field trials were organized into a randomized complete block design whereby each block contained two neighboring raised beds. One clonal replication per progeny individual was included per block (n progeny = 4) with one replication per progeny genotype planted to every second bed. One replication per control genotype was included per bed (n control = 8), for two replications of each control genotype per block. Susceptible controls included ‘Strawberry Festival’ and WinterStar™ ‘FL 05-107’ in the 2013–2014 season, and all of the previous in addition to ‘Earliglow’, Sensation™ ‘Florida127’, and advanced breeding selection FL 11.28–34 in the 2014–2015 season. ‘Treasure’ was included as a moderately susceptible control (Seijo et al. 2012) in both years. Resistant controls included resistance donors K08-17 and K12-10 in 2013–2014 and both donors plus resistant selections 13.77−5 and 13.78−57 in 2014–2015.
Pathogen isolation
As is often the case, many plants arrived from nurseries in 2013 with ALS symptoms. To isolate X. fragariae for use as inoculum, affected leaf tissue from six unique progeny of families 13.77 and 13.78 was disinfested in 62 mM sodium hypochlorite (NaClO) for 1 min followed by a triple rinse in sterile diH2O. Lesions were excised, macerated in 1 mL sterile diH2O, vortexed, and allowed to rest for 15 min to encourage streaming of bacteria into suspension. A 1:10 dilution was plated and later subcultured on Wilbrink’s media and incubated at 28 °C until single colonies were isolated (Koike 1965). Colonies were confirmed as X. fragariae using colony PCR with primers Xf9 and Xf11 (Roberts et al. 1996) and confirmed as pathogenic in preliminary greenhouse assays (data not shown).
Inoculation and rating
Equal numbers of plates from each of the six X. fragariae isolates corresponding to the progeny from which they were isolated were grown for 4 d and diluted to 106 CFU mL−1 (OD600 = 0.025) in tap water as one combined inoculum. Inoculum was applied during late afternoon to foliage and crowns with a hand pump sprayer until runoff (approximately 58 mL per plant) on 11 and 26 December 2013, and 2 and 18 February 2015, during the two respective fruiting field seasons. Overhead irrigation was applied during the day preceding inoculation to wound leaf tissue and increase entry points for the pathogen. To spread inoculum and increase relative humidity in the plant microenvironment (e.g., Hildebrand et al. 2005), overhead irrigation was then applied for 1 min immediately after inoculation, during daylight hours for the 7 d following inoculation, and for 2–6 h/d from days 8 to 14 after inoculation based on weather conditions. Individual plants were scored for percent diseased leaf area on a 0-100 % scale in 5 % increments (Roberts et al. 1997) on 15 January and 17 February, 2014, and 26 February and 17 March, 2015.
Statistical analysis
A clonal mean score was calculated as the sum of all replicate scores for an individual or control genotype divided by the number of scores collected in a given year. Broad-sense heritability was calculated as
where σ 2 g = genetic variance, σ 2 r = replication/block variance, and σ 2 e = error variance. Calculation of heritability included progeny phenotypes only whereas control genotypes were included in field trials to allow detection of location effects. Progeny were rated as resistant or susceptible based on their position in the frequency distribution for each full-sib family, such that progeny with clonal mean scores less than the antimode of the observed bimodal phenotypic distribution were considered resistant and those with scores greater than the antimode were considered susceptible (e.g. Welz et al. 1998). A Chi-square (χ 2) analysis to test goodness of fit to a 1:1 trait segregation ratio indicative of single-locus control was conducted using the chisq.test function of R software (R Core Team 2013) using these resistance and susceptibility categorizations.
DNA extraction, SNP genotyping, and QTL analysis
Unexpanded trifoliate leaves were harvested on ice from the field and stored at −80 °C until use. Approximately 60 mg leaf tissue per sample was ground to a fine powder in a PowerGen™ homogenizer (Fisher Scientific, Pittsburgh, Pennsylvania) with two 2 mm diameter stainless steel beads. Samples for SNP array genotyping were processed with the E-Z 96® Plant DNA kit according to the manufacturer’s instructions (Omega Bio-Tek, Norcross, Georgia) and quantified with the Quant-iT™ PicoGreen® Assay (Life Technologies, Thermo Fisher Scientific, Waltham, MA).
Selective genotyping of progeny in the lower and upper tails of phenotypic frequency distributions was identified as the ideal genotyping method to identify major effect QTL while reducing genotyping costs (Darvasi and Soller 1992; Lebowitz et al. 1987). Given the hypothesis that one or a few loci controlled most of the variation for resistance in the two lineages used in this study, and to allow inclusion into one 96-well plate, 37 highly resistant and 37 highly susceptible individuals from families 13.77 and 13.78, along with replications of immediate parents ‘Strawberry Festival’, K08-17 and K12-10, were genotyped with the IStraw90 Axiom® SNP array developed for allo-octoploid strawberry (Bassil and Davis et al. 2015).
A whole-genome linkage map of 3,622 SNP markers showing unique segregation patterns and spanning 24.9 Morgan was developed in JOINMAP 4.1 software (Van Ooijen 2006, 2011) using 75 progeny from the population ‘Holiday’ × ‘Korona’. The map was adapted from the poly-high resolution SNP marker map of Bassil and Davis et al. (2015) by adding no-minor-homozygote and off-target variant markers according to the procedures and quality control measurements detailed for LG6D by the same authors and Van de Weg and Koehorst-van Putten (version 20150310, unpublished). The final map contained 12,966 SNP markers and spanned 21.0 Morgan.
From this initial map, markers were added and removed as to achieve maximum polymorphism in University of Florida germplasm with the minimum number of markers (Verma, unpublished data). Default maximum likelihood method and population type “CP” settings were used in the construction of this second map, which was the map used for QTL analysis. In order to identify and exclude low performance markers, fraction values of observed (oDR) and expected (eDR) double recombinant singletons were obtained from FlexQTL™ software and markers with oDR minus eDR >0.05 were excluded from the map (Clark et al. 2014). Next, linkage maps were further curated using graphical genotyping following the procedure of Bassil and Davis et al. (2015) for linkage groups that showed a QTL. Spurious singleton calls identified by JoinMap’s genotype probability module that could not be resolved by manual re-ordering of markers were excluded, and graphical genotyping-derived marker orders were used.
Using this genetic map, a Bayesian multiple QTL model was fitted using FlexQTL™ software (Bink et al. 2002, 2008, 2014; Guan et al. 2015). Whole-genome QTL analysis was then repeated three times with varying chain lengths and seed numbers to create independence between each iteration and allowing for a maximum of four QTL and using a prior number of one QTL, using clonal mean percent disease leaf area phenotypic data collected in the field. Each of the three iterations converged (effective chain samples, or ECS, ≥100 for each of the parameters mean, variance of the error, number of QTL, and the variance for the number of QTL) as recommended by Bink et al. (2014). Further analysis specific to a single linkage group was then conducted for the detected major QTL. Full FlexQTL™ parameters are provided in Online Resource 1.
Haplotype analysis
SNP markers in the predicted QTL region that were not included in the whole-genome linkage map were added and ordered based on F. vesca ssp. vesca v1.1 physical map locations (Bassil and Davis et al. 2015; Shulaev et al. 2011), conservative alignment of SNP probe sequences to the F. vesca ssp. bracteata reference (Tennessen et al. 2014), relative positions derived from the linkage map used in this study and that of Bassil and Davis et al. (2015), and manual reorientation (when clear marker order patterns were observed based on recombination patterns). Haplotypes spanning 4.45 Mbp were then constructed based on marker segregation and parental diplotypes (0 = AA, 1 = AB, and 2 = BB where, for example, 1 is the diplotype and AB is a heterozygous SNP marker). Analysis of variance of haplotype effect on disease score was conducted [R function aov()] and Tukey’s HSD test [function HSD.test() from R package ‘agricolae’] was used to detect significant differences between haplotype means (de Mendiburu 2010; R Core Team 2013).
Recombination events during paternal gamete formation were manually identified in non-parental/recombinant haplotypes. To accomplish this, a resistance-associated haplotype was identified on one portion of a progeny haplotype while a susceptibility-associated haplotype was identified on the other portion of that same progeny haplotype, with a clear and apparent recombination break points between the two sections. Only haplotypes comprised of at least three neighboring non-parental marker calls were considered recombinant. A QTL region was thus delimited by aligning haplotypes of multiple progeny such that upstream and downstream recombination events were apparent.
QTL validation
In order to validate the QTL predicted in families 13.77 and 13.78, all progeny of families 14.100 and 14.101 were genotyped by high-resolution melting (HRM) analysis of SNPs identified within and nearby the QTL region. To identify SNPs, paired-end reads from Bassil and Davis et al. (2015) were sorted and aligned to the F. vesca ssp. bracteata reference (Tennessen et al. 2014) using SAMtools version 1.2 (Li et al. 2009) and GMAP/GSNAP (Wu and Watanabe 2005; Wu and Nacu 2010) in the identified QTL interval. Novel single-nucleotide variants were called in Geneious version 8.1.3 (http://www.geneious.com; Kearse et al. 2012). SNPs identified from the SNP array probes of Bassil and Davis et al. (2015) were also targeted. Primer pairs targeting these variants were constructed to yield 60–120 bp PCR products using Primer3 (Rozen and Skaletsky 2000; Untergasser et al. 2012) in Geneious software.
Genomic DNA was extracted using a CTAB/chloroform method derived from Haymes (1996) that is detailed in Online Resource 2. HRM analysis was conducted using a LightCycler 480 (Roche Diagnostics, Indianapolis, Indiana) according to the manufacturer’s protocol (Online Resource 2). PCR and HRM were conducted in one step beginning with 5 min at 95 °C, 45 cycles of 10 s at 95°, 10 s annealing, and 10 s extension at 72°, followed by melt curve analysis from 65 to 95 °C. Touchdown PCR annealing temperatures began at 65 °C and decreased to 55 °C in steps of 0.5 °C per cycle. All progeny of families 14.100 and 14.101 were genotyped using HRM6D_33.083 and HRM6D_33.110 (Online Resource 2). Marker call concordance with phenotype was determined as the proportion of progeny with resistant or susceptible phenotype having a melt curve profile similar to that of the dominant HRM profile curve of each phenotypic group. A χ 2 goodness of fit analysis was then conducted to determine independence between HRM curve profile and resistance phenotype.
Results
Field results, heritability, and trait segregation
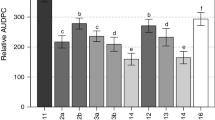
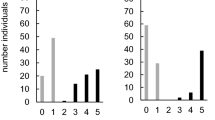
Two full-sib families descended from each of the two resistant accessions were phenotyped during two years of field trials (Fig. 1). A strong bimodal phenotypic distribution was observed in all four full-sib families (Fig. 2). The threshold, based on antimodes, for resistance or susceptibility was 12 % in 2013–2014 and 7 % in 2014–2015, as overall disease scores were higher in the first year. Chi square analysis for rejecting the hypothesis of 1:1 segregation was not significant at the P < 0.05 level in any family (Table 1), suggesting that the hypothesized 1:1 segregation ratio is appropriate. Broad-sense heritability estimates were at least 0.70 in all families (Table 1) and no field/location effects were observed. Clonal mean scores from a subset of progeny in the first year field trial were verified in inoculated growth chamber experiments with multiple isolates (Online Resource 3).

Frequency distributions of clonal mean scores for progeny of four full-sib families segregating for resistance to angular leaf spot caused by Xanthomonas fragariae. R and S arrows indicate scores of resistant and susceptible parents of each family, respectively; a family 13.77, b family 13.78, c family 14.100, and d family 14.101. The dotted line represents the antimode of each distribution, which was used to divide individuals into resistant and susceptible classes
All resistant controls showed mean scores of 0.0–3.4 % diseased leaf area in both years whereas susceptible controls showed mean scores of 12.3-18.0 % (Fig. 2). No field location effects were apparent in either year. Moderately susceptible control ‘Treasure’ showed scores of 14.0 % in 2013–2014 and 6.8 % in 2014–2015, confirming its status as moderately susceptible (Seijo et al. 2012).
QTL identification
Analysis with FlexQTL™ software repeatedly predicted the existence of a single large-effect QTL (Fig. 3). Bayes factors, which estimate statistical significance of posterior probability distributions, and which are analogous to traditional LOD scores, were >33 for the presence of a QTL on LG 6D in all genome-wide QTL analysis runs, which is a value considered “decisive” (> 10) evidence for QTL presence on that linkage group (Bink et al. 2008, 2014). For repeated analyses of LG 6D alone, Bayes factors for a QTL between 82 and 89 cM were >24, again considered “decisive” by a large margin (Bink et al. 2008, 2014).

Representative QTL prediction output of FlexQTL™ software for resistance to angular leaf spot caused by Xanthomonas fragariae for linkage group 6D. Each trace plot point represents a predicted QTL model for a subsample of 1000 out of the 40,000 Markov chain Monte Carlo iterations. The figure was generated using VisualFlexQTL™ (Bink et al. 2002, 2008, 2014)
Haplotype analysis
Orientation of SNP diplotypes based on physical location and manual ordering allowed conversion to SNP haplotypes and observation of informative recombination events. Five unique haplotypes, including two parental and three recombinant haplotypes, were observed in each of families 13.77 and 13.78 (Tables 2, 3; Fig. 4). The common susceptible parent ‘Strawberry Festival’ was homozygous at all marker loci in the region, thus indicating that genetic variation at the locus was exclusively due to donor (paternal) parent effects. The effect of haplotype on disease score was highly significant with P < 10−16 (Tables 2, 3). The three observed recombinant haplotypes in each family delimited the region of interest to 33.08–33.60 Mbp in family 13.77 and 32.74–33.66 Mbp in family 13.78 based on the F. vesca ssp. vesca v1.1 chromosome six assembly (Shulaev et al. 2011). This region was equivalent to 34.81–35.33 and 34.47–35.39 Mbp for families 13.77 and 13.78, respectively, on the F. vesca ssp. bracteata chromosome six assembly (Tennessen et al. 2014).

Delimiting a QTL for resistance to angular leaf spot caused by Xanthomonas fragariae in allo-octoploid strawberry. SNP diplotypes were organized into SNP diplotype blocks that were analogous to SNP haplotypes according to diploid reference physical location (Shulaev et al. 2011; Tennessen et al. 2014), linkage mapping on linkage group 6D in this study and Bassil and Davis et al. (2015), or observed recombination patterns. Homozygous (AA) diplotypes are represented by moderately shaded, heterozygous (AB) by lightly shaded, and homozygous (BB) by darkly shaded cells. Missing marker calls are indicated by unshaded cells. Outlined diplotype blocks indicate putative recombination sites. All diplotype blocks correspond to haplotype order, count, and nomenclature assigned in Table 3
QTL validation
Of the novel and previously identified SNPs tested in HRM assays, two markers clearly segregated with resistance to ALS in families 14.100 and 14.101 (Table 4; Online Resource 2). The first HRM marker, HRM6D_33.083, mapped to 33.083 Mbp in F. vesca vesca v1.1 LG6 and was identified by Bassil and Davis et al. (2015) as SNP array probe AX-89898194. The second HRM marker, HRM6D_33.110, mapped to a novel variant at 33.110 Mbp in F. vesca vesca v1.1 chromosome 6. In family 14.100, 100 progeny were classified, based on melt curve profile similarity, into groups associated with resistance or susceptibility for both HRM markers. In family 14.101, 259 (HRM6D_33.083) and 228 (HRM6D_33.110) progeny were grouped based on melt curve profile similarities (Table 4). Two melt curve profiles were identified in three of the four marker/family combinations, whereas family 14.100/marker HRM6D_33.110 displayed five melt curve profiles, including two for resistant and three for susceptible progeny (Online Resource 2). Concordance between phenotypes and melt curve profiles varied between 89 and 96 %, and χ 2 analyses were not significant (P > 0.05; Table 4).
Discussion
Initial reports of segregation for resistance to ALS in octoploid strawberry suggested that resistance was controlled by two or three unlinked loci (Hartung et al. 2003; Lewers et al. 2003; Jamieson et al. 2013), whereas a later report (Jamieson et al. 2014) indicated that resistance was controlled by a dominant allele at a single locus. A possible explanation for this discrepancy may be the loss of susceptibility alleles at unknown loci in later stages of backcrossing of the resistance trait from these lines into elite octoploid strawberry germplasm (Fig. 1). Given the observed 1:1 segregation and high broad sense heritability in the present study, rejection of the single-locus control of resistance hypothesis is unwarranted (Table 1). As all QTL analyses and haplotype effect analyses indicated very strong support for presence of a large-effect QTL at the distal end of LG 6D (Fig. 3), we conclude that at least one dominant allele at the novel locus, which we name FaRXf1 (for Fragaria ×ananassa Resistance to Xanthomonas fragariae 1), imparts resistance in progeny derived from US4808 to US4809. Annotation of the FaRXf1 region in the F. vesca genome reveals the presence of several resistance-associated genes including nucleotide-binding leucine-rich repeat (NB-LRRs), zinc finger and serine/threonine-protein kinase genes (http://rosaceae.org/species/fragaria_vesca/genome_v2.0.a1; Shulaev et al. 2011; Tennessen et al. 2014), which may provide guidance for future fine-mapping and functional characterization of the locus.
Allelic diversity at the FaRXf1 locus is unknown. The two resistant parents K08-17 and K12-10 showed different haplotypes for their resistant homologue due to two neighboring SNP markers within the resistance locus (Fig. 4), suggesting that sequence differences between the US4808 and US4809 resistance sources are present within the locus. While insufficient to conclude that multiple functional alleles are present in the assayed germplasm, this indicates that further work may uncover multiple resistance alleles, genes, or resistance mechanisms. The possibility of allelic diversity is further supported by differences in time and place of collection between US4808 (1986 as a wild accession in Minnesota) and the resistant parent of US4809 (1900 as an heirloom variety in Georgia; Maas et al. 2002). However, it is possible that the mutations responsible for different resistance-associated haplotypes occurred more recently than mutations causing resistance. In this case, sequence variation in the region would be expected even if the two sources share the same ancestral resistance mechanism. Regardless, as progeny derived from both sources were shown to be resistant to representatives of the four known clades of X. fragariae (Hildebrand et al. 2005; Maas et al. 2002), there is currently no evidence to indicate isolate-specific resistance for either source.
Field trials in this study were conducted under winter annual growing conditions typical of commercial production in the region. As resistant progeny in the field trials reported here descended from resistant individuals that were assayed in greenhouse and growth chamber trials (Hildebrand et al. 2005; Jamieson et al. 2013, 2014) and against a panel of genetically diverse isolates (Hildebrand et al. 2005; Maas et al. 2002), the resistance may be suited for deployment in growing conditions where X. fragariae genetic diversity is still unknown. However, the durability of resistance cannot yet be determined, given that the resistance has not been deployed on a large scale in nursery or fruit production fields. Until then it will be difficult to predict the rate at which pathogen effectors recognized by putative resistance gene products (e.g., possible NB-LRRs) may respond to increased selection pressure.
Phenotypic scores were higher in 2013–2014 than in 2014–2015 for all controls and in susceptible progeny, as inoculation occurred more than a month earlier in the first year than in the second. Regardless, antimode values and frequency distributions were consistent as were inoculation and rating techniques between families within each year. Subsequent HRM analysis of novel and published SNPs both flanking and within the FaRXf1 region in families 14.100 and 14.101 indicated that resistance segregated with marker alleles with up to 96 % predictive ability depending on the marker and family, validating QTL presence and location while supporting the use of the antimode-dependent resistance categorization technique (Table 4; Online Resource 2). Interestingly, analysis of the HRM6D_33.110 marker revealed five unique melt curve profiles that segregated with the resistance trait (Online Resource 2). The number of unique profiles was potentially due to amplification in a subgenome homeologous to LG 6D that was also segregating, possibly indicating variation that was conserved in at least one of the polyploidization events predating allo-octoploidy.
Development of the IStraw90 Axiom® SNP array incorporated unique properties of the allo-octoploid F. ×ananassa genome to allow highly informative, high-throughput SNP genotyping (Bassil and Davis et al. 2015). Many probes included in the array contained secondary SNPs, indels, or other features restricted to single sub-genomes such that only genomic DNA fragments that included these secondary features would bind to the probes. Inclusion of these secondary features approximated the diploidization or reduced effective ploidy level found within a homeologous group and allowed for genetic analysis similar to that for a diploid species. Knowledge of probes that exploit secondary features may also guide further development of subgenome-specific molecular assays for the otherwise troublesome allo-octoploid genome.
Experience gained during the course of the work presented here confirms that the IStraw90 Axiom® SNP array is effective and efficient when used for high-throughput genetics and genomics research in allo-octoploid strawberry and that, when coupled with pedigree-based analysis using FlexQTL™, QTL detection and analysis may be easily and rapidly accomplished. Interestingly, FlexQTL™ analyses occasionally indicated the presence of two QTL peaks within the single predicted QTL region when both full-sib families and their parents were analyzed concurrently. This was possibly due to differing marker heterozygosity between the two families as subsequent single-family analysis indicated a single QTL peak in the region in each family. As a single QTL was later delimited using physical locations of SNP marker data in both families (Fig. 4), it may benefit future investigators to identify and analyze all SNP markers within a region and not rely solely on mapped markers for downstream applications. Indeed, use of all available markers reduced the delimited QTL region by 0.52 Mbp in family 13.78 compared to the QTL delimited by markers included in the linkage map alone. Similarly, the use of selective genotyping with regards to the prediction algorithms of FlexQTL™ is supported and should be further investigated with decreased sample sizes and traits with lower heritability.
To date, no strawberry cultivar has been reported as resistant to ALS caused by X. fragariae. Six resistant progeny from families 14.100 and 14.101 were selected for observation in expanded field trials due to resistance to X. fragariae along with large fruit size, preferable flavor, and desirable plant architecture under Florida production conditions. Exploitation of the novel FaRXf1 locus by development of high-throughput DNA markers is expected to enhance future breeding efforts and hasten the commercialization of ALS-resistant strawberry cultivars.
Author contribution statement
JR performed phenotyping, QTL analysis, and prepared the initial manuscript; SV assisted with QTL analysis and linkage mapping; NP provided pathogen isolates and guidance for inoculation and disease screening; AJ made crosses and provided germplasm; EvdW and MB provided software and guidance for QTL analysis as well as linkage mapping resources; NB facilitated DNA quantitation and genotyping submission; SL guided HRM analysis; VW initiated the research and provided project direction. All authors have reviewed and edited the manuscript.
References
Bassil NV, Davis TM, Zhang H, Ficklin S, Mittmann M, Webster T, Mahoney L, Wood D, Alperin ES, Rosyara UR, Koehorst-vanc Putten H, Monfort A, Sargent DJ, Amaya I, Denoyes B, Bianco L, van Dijk T, Pirani A, Iezzoni A, Main D, Peace C, Yang Y, Whitaker V, Verma S, Bellon L, Brew F, Herrera R, van de Weg WE (2015) Development and preliminary evaluation of a 90 K Axiom® SNP array for the allo-octoploid cultivated strawberry Fragaria ×ananassa. BMC Genom 16:155
Bink MCAM, Uimari P, Sillanpaa J, Janss LLG, Jansen RC (2002) Multiple QTL mapping in related plant populations via a pedigree-analysis approach. Theor Appl Genet 104:751–762
Bink MCAM, Boer MP, ter Braak CJF, Jansen J, Voorrips RE, van de Weg WE (2008) Bayesian analysis of complex traits in pedigreed plant populations. Euphytica 161:85–96
Bink MCAM, Jansen J, Madduri M, Voorrips RE, Durel CE, Kouassi AB, Laurens F, Mathis F, Gessler C, Gobbin D, Rezzonico F, Patocchi A, Kellerhals M, Boudichevskaia A, Dunemann F, Peil A, Nowicka A, Lata B, Stankiewicz-Kosyl M, Jeziorek K, Pitera E, Soska A, Tomala K, Evans KM, Fernandez-Fernandez F, Guerra W, Korbin M, Keller S, Lewandowski M, Plocharski W (2014) Baysian QTL analyses using pedigreed families of an outcrossing species, with application to fruit firmness in apple. Theor Appl Genet 127:1073–1090
CABI/EPPO (1997) Quarantine pests for Europe. Smith IM, McNamara DG, Scott PR, Holderness M (eds), 2nd edn. CABI International, Wallingford
Clark MD, Schmitz CA, Rosyara UR, Luby JJ, Bradeen JM (2014) A consensus ‘Honeycrisp’ apple (Malus × domestica) genetic linkage map from three full-sib progeny populations. Tree Genet Genomes 10:627–639
Darvasi A, Soller M (1992) Selective genotyping for determination of linkage between a marker locus and a quantitative trait locus. Theor Appl Genet 85:353–359
de Mendiburu F (2010) Agricolae: statistical procedures for agricultural research using R (Online) http://tarwi.lamolina.edu.pe/~fmendiburu/. Accessed date 16 Oct 2015
Epstein AH (1966) Angular leaf spot of strawberry. Plant Dis Report 50:167
Guan Y, Peace C, Rudell Dm Verma S, Even K (2015) QTLs detected for individual sugars and soluble solids content in apple. Mol Breed 35:135
Hartung JS, Gouin CC, Lewers KS, Maas JL, Hokanson S (2003) Identification of sources of resistance to bacterial angular leafspot disease of strawberry. Acta Hortic 626:155–159
Haymes KM (1996) Mini-prep method suitable for a plant breeding program. Plant Mol Biol Report 14:280–284
Hildebrand PD, Braun PG, Renderos WE, Jamieson AR, McRae KB, Binns MR (2005) A quantitative method for inoculating strawberry leaves with Xanthomonas fragariae, factors affecting infection, and cultivar reactions. Can J Plant Pathol 27:16–24
Jamieson AR, Hildebrand PD, Renderos WE (2013) Breeding strawberry plants resistant to angular leaf spot disease. Int J Fruit Sci 13:28–35
Jamieson AR, Hildebrand PD, Renderos WE, Fillmore SAE (2014) Resistance to angular leafspot disease of strawberry: influence of seedling age. Acta Hortic 1049:187–192
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Mentjies P, Drummond A (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Kennedy BW, King TH (1960) Angular leafspot, a new disease of strawberry. Phytopathology 50:641–642
Kennedy BW, King TH (1962a) Angular leaf spot of strawberry caused by Xanthomonas fragariae sp. nov. Phytopathology 52:873–875
Kennedy BW, King TH (1962b) Studies on epidemiology of bacterial angular leafspot on strawberry. Plant Dis Report 46:360–363
Koike H (1965) The aluminum-cap method for testing sugarcane varieties against leaf scald disease. Phytopathology 55:317–319
Lebowitz RJ, Soller M, Beckmann JS (1987) Trait-based analyses for the detection of linkage between marker loci and quantitative trait loci in crosses between inbred lines. Theor Appl Genet 73:556–562
Lewers KS, Maas JL, Hokanson SC, Gouin C, Hartung JS (2003) Inheritance of resistance in strawberry to bacterial angular leafspot disease caused by Xanthomonas fragariae. J Amer Soc Hortic Sci 128:209–212
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Maas JL (1984) Compendium of strawberry diseases. American Phytopathological Society, St. Paul
Maas JL, Gouin-Behe C, Hartung JS, Hokanson SC (2000) Sources of resistance for two differentially pathogenic strains of Xanthomonas fragariae in Fragaria genotypes. HortScience 35:128–131
Maas JL, Gouin CC, Hokanson SC, Hartung JS (2002) Strawberry parent clones US 4808 and US 4809 resistant to bacterial angular leafspot disease caused by Xanthomonas fragariae. HortScience 37:716–717
Mertely J, Seijo T, Martin R, Moore DN, Peres NA (2010) Evaluation of products for angular leaf spot control in annual strawberry, 2009–10. Plant Dis Manag Rep 4:SMF052
R Core Team (2013) R: a language and environment for statistical computer. R Foundation for Statistical Computing, Vienna. http://www.R-project.org. Accessed 15 Mar 2015
Pooler MR, Ritchie DF, Hartung JS (1996) Genetic relationships among strains of Xanthomonas fragariae based on random amplified polymorphic DNA PCR, repetitive extragenic palindromic PCR, and enterobacterial repetitive intergenic consensus PCR data and generation of multiplexed PCR primers useful for identification of this phytopathogen. Appl Environ Microbiol 62:3121–3127
Roberts PD, Jones JB, Chandler CK, Stall RE, Berger RE (1996) Survival of Xanthomonas fragariae on strawberry in summer nurseries in Florida detected by specific primers and nested polymerase chain reaction. Plant Dis 80:1283–1288
Roberts PD, Berger RD, Jones JB, Chandler CK, Stall RE (1997) Disease progress, yield loss, and control of Xanthomonas fragariae on strawberry plants. Plant Dis 81:917–921
Roberts PD, Hodge NC, Bouzar H, Jones JB, Stall RE, Berger RD, Chase AR (1998) Relatedness of strains of Xanthomonas fragariae by restriction fragment length polymorphism, DNA-DNA reassociation, and fatty acid analyses. Appl Environ Microbiol 64:3961–3965
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386
Seijo T, Mertely J, Whitaker VM, Peres NA (2012) Evaluation of strawberry cultivars and advanced selections for resistance to anthracnose and Botrytis fruit rots and angular leaf spot, 2011–12. Plant Dis Manag Rep 6:SMF034
Shulaev V, Sargent DJ, Crowhurst RN, Mockler TC, Folkerts O, Delcher AL, Jaiswal P, Mockaitis K, Liston A, Mane SP, Burns P, Davis TM, Slovin JP, Bassil N, Hellens RP, Evans C, Harkins T, Kodira C, Desany B, Crasta OR, Jensen RV, Allan AC, Michael TP, Setubal JC, Celton JM, Rees DJ, Williams KP, Holt SH, Ruiz Rojas JJ, Chatterjee M, Liu B, Silva H, Meisel L, Adato A, Filichkin SA, Troggio M, Viola R, Ashman TL, Wang H, Dharmawardhana P, Elser J, Raja R, Priest HD, Bryant DW Jr, Fox SE, Givan SA, Wilhelm LJ, Naithani S, Christoffels A, Salama DY, Carter J, Lopez Girona E, Zdepski A, Wang W, Kerstetter RA, Schwab W, Korban SS, Davik J, Monfort A, Denoyes-Rothan B, Arus P, Mittler R, Flinn B, Aharoni A, Bennetzen JL, Salzberg SL, Dickerman AW, Velasco R, Borodovsky M, Veilleux RE, Folta KM (2011) The genome of woodland strawberry (Fragaria vesca). Nat Genet 43:109–116
Tennessen JA, Govindarajulu R, Ashman T-L, Liston A (2014) Evolutionary origins and dynamics of octoploid strawberry subgenomes revealed by dense targeted capture linkage maps. Genome Biol Evol 6:3295–3313
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG (2012) Primer3—new capabilities and interfaces. Nucl Acids Res 40:e115
Van Ooijen JW (2006) JoinMap 4, software for the calculation of genetic linkage maps in experimental populations. Wageningen, Kyazma BV
Van Ooijen JW (2011) Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet Res 93:343–349
Welz HG, Schechert A, Pernet A, Pixley KV, Geiger HH (1998) A gene for resistance to the maize streak virus in the African CIMMYT maize inbred line CML202. Mol Breed 4:147–154
Whitaker VM, Chandler CK, Peres N, do Nascimento Nunes MC, Plotto A, Sims CA, Plotto A, Sims CA (2015) Sensation™ ‘Florida127’ strawberry. HortScience 50:1088–1091
Wu TD, Nacu S (2010) Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26:873–881
Wu TD, Watanabe CK (2005) GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21:1859–1875
Acknowledgments
The authors acknowledge Tomas Hasing, Dr. James Mertely, and Teresa Seijo for their assistance and guidance. This work was funded in part by the National Institute of Food and Agriculture, U.S. Department of Agriculture Specialty Crop Research Initiative project “RosBREED: Combining disease resistance with horticultural quality in new rosaceous cultivars” under award No 2014-51181-22378.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by C. A. Hackett.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Roach, J.A., Verma, S., Peres, N.A. et al. FaRXf1: a locus conferring resistance to angular leaf spot caused by Xanthomonas fragariae in octoploid strawberry. Theor Appl Genet 129, 1191–1201 (2016). https://doi.org/10.1007/s00122-016-2695-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-016-2695-1