
Abstract
Key message
C genome chromosome substitution lines of B. juncea constitute a key genetic resource for increased genetic diversity and hybrid performance.
Abstract
C genome chromosome substitution lines were found in the progenies of derived B. juncea (2n = 36; AABB), synthesized through hybridization between B. napus and B. carinata. These were originally recognized based on the morphology and genomic in situ hybridization. Genotyping using the Brassica Illumina 60K Infinium SNP array confirmed the presence of C genome chromosomes in a large number of derived B. juncea genotypes. Three whole chromosome substitutions and 13 major C genome fragment substitutions were identified. Fragment substitutions were primarily terminal, but intercalary substitution(s) involving chromosome C1 and C2 were identified in three genotypes. The size of substituted C genome fragments varied from 0.04 Mbp (C1) to 64.85 Mbp (C3). In terms of proportions, these ranged from 0.10 % (C1) to 100 % (C1, C3 and C7) of the substituted chromosome. SSR genotyping with B genome specific primers suggested that substituting C genome chromosome(s) are likely to have replaced B genome chromosome(s). C1 was the most common substituting chromosome while substituted B chromosome seemed random. Study of the phenotypic traits underlined the importance of the substitution lines (especially of chromosome C1) for conferring superior trait performance (main shoot length and pods on the main shoot). High heterosis was also indicated in hybrid combinations of substitution lines with natural B. juncea. These substitution genotypes constituted a valuable resource for targeted gene transfer and QTL identification.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Brassica juncea (L.) Czern and Coss. (2n = 36; AABB), also known as Indian mustard, is a premier oilseed crop of the Indian sub-continent. It is a condiment and edible oilseed crop in Russia and parts of Eastern Europe. It arose several times through hybridization between the diploid genome donor species B. rapa (2n = 20; AA) and B. nigra (2n = 16; BB), with both figuring as cytoplasm donors during independent hybridization events (Kaur et al. 2014). These two diploid species, along with B. oleracea (CC; 2n = 18), are primary paleopolyploids (Truco et al. 1996) having arisen from a common hypothetical hexaploid ancestor with 42 chromosomes (Cheng et al. 2013).
Like most other field crops, Indian mustard is facing challenges of stagnating yields and susceptibility to biotic and abiotic stresses. Narrow genetic diversity in the breeding pools is arguably a major cause of yield stagnation and failure to enhance current levels of resistance to biotic and abiotic stresses (Banga and Banga 2009). Intensive selection and hybridization efforts in the past helped to increase crop productivity by substituting superior alleles at bottleneck loci. Though this strategy facilitated the evolution of high-yielding varieties, it also tempted plant breeders to restrict hybridization to elite but closely related germplasm. In fact, most of the current mustard varieties in India have been bred through crosses between high-value, and closely related genotypes or are reselections from old cultivars. For example, a very old but widely adapted cultivar, Varuna, figures repeatedly in the pedigrees of most mustard genotypes released for commercial cultivation in India (Chauhan et al. 2011). A similar situation has been documented for the Australian B. napus breeding programme (Cowling 2007). It is plausible that majority of favorable alleles have become fixed in a small set of elite germplasm. Any further genetic enhancement for productivity and stress resistance would require genetic augmentation across gene pools. Genetically diverse germplasm resources have been exploited as sources of beneficial alleles in many crop species (Zamir 2001). Extensive and exploitable variation is available within B. juncea, related crop Brassica species and wild crucifers (Banga et al. 2003; Warwick 2011). It was possible to introgress wild alleles for tolerance to various biotic stresses into B. juncea (Garg et al. 2010; Kumar et al. 2011; Atri et al. 2012; Banuelos et al. 2013) or exploit alien cytoplasmic variation through development of a several alloplasmic CMS lines (Janeja et al. 2003; Prakash et al. 2009; Atri et al. 2016). Some of these CMS sources have been used to develop hybrids in both B. napus and B. juncea.
In spite of demonstrated breeding gains, exotic germplasm resources are largely underutilized in this crop. Negative trait associations, linkage drag, meiotic barriers, distorted allele segregation ratios and loss of alien genes in early generations of backcrossing are the primary impediments. Probability of unwanted linkages can be extremely high as rates of recombination can be very low in introgressed chromosome segments. Fixing beneficial genes while eliminating undesirable ones can be very difficult and may require many generations of planned hybridizations and specialized breeding strategies. B. napus and B. carinata are especially valuable and accessible reservoirs of novel genetic variation for B. juncea. However, potential utility of specific alien genes is compromised by uncontrolled and complex chromosome pairing in crosses involving allotetraploid species. Chromosome addition and chromosome substitution lines in allotetraploids offer the possibility of limiting introgressions to specific chromosomes. Chromosome addition lines have been developed in Brassica. For example, B. napus–Diplotaxis erucoides, B. napus–B. nigra and B. rapa–B. alboglabra have been produced (Delourme et al. 1989; Jahier et al. 1989; Chen et al. 1997). Although C-genome chromosome substitution lines have been reported in B. juncea (Banga 1988), these were never adequately investigated in terms of molecular characterization and breeding value.
In this communication we report molecular–cytogenetic characterization of C genome chromosome substitution lines in derived B. juncea forms obtained through hybridization between B. napus and B. carinata. We also demonstrate their value for trait introgression and increased heterosis in crosses with natural B. juncea.
Methods and materials
Spontaneous C genome chromosome substitution lines were identified in the progenies of derived B. juncea developed through hybridization between B. napus and B. carinata (Gupta et al. 2015). In addition four natural B. juncea genotypes namely PBR 210, RLC 1, AMH2 and CBJ 002 were used for developing F1 hybrids with chromosome substitution lines. Of these, PBR 210 and RLC 1 are commercial B. juncea varieties in India. AMH 2 and CBJ 002 are B. juncea introductions from China.
Fluorescent genomic in situ hybridization (fl-GISH)
Slide preparation
To prepare meiotic spreads, anthers with pollen mother cells at meiotic stages between prophase I and metaphase I were collected from derived B. juncea lines and fixed in Farmer’s solution (3 ethanol:1acetic acid) in the morning hours. For mitotic slide preparations, roots of 30 mm in size were treated with water saturated with α-bromonaphthalene for 4 h and then fixed in alcohol:acetic acid (3:l) for at least 2 h at 4 °C. Anthers and root tips were first washed in a citrate buffer for 30 min to remove the fixative and subsequently incubated in the enzyme mixture containing 2 % (w/v) cellulase and 20 % (v/v) pectinase in 4 mmol citrate buffer (Citric acid monohydrate and trisodium citrate dehydrate), pH 4.8 for about 1 h at 37 °C. The treated anthers and roots were agitated with a micropipette tip to meiotic and mitotic cells in the micro-centrifuge tubes. These were centrifuged for 3 min at 600–800g, followed by 45 min treatment in 150 mmol KCl for 20 min. The cells were washed thrice at 800g for 3 min in freshly prepared fixative to clear the cytoplasm. One drop of 7 µl of suspension was released on acid cleaned chilled slide from a height of 50 mm to spread the cells on the slide. Slides were air-dried in a desiccator before further use.
In situ procedure
Total genomic DNA from Brassica species was extracted using the DNeasy plant mini kit (Qiagen) according to manufacturer’s instructions to prepare probes and blocking DNA. To know whether the substitution happened between B and C genome or between A and C genome, two colour GISH was performed for simultaneous representation of all three genomes (A, B and C) in derived B. juncea lines. Purified and sheared DNA of B. nigra var. UP was labelled with fluorescein-12-dUTP and from B. oleracea L var. alboglabra Bailey was labelled with tetramethyl rhodamine-5-dUTP using a nick-translation kit to identify B and C genomes in lines. Autoclaved B. rapa DNA (200–500 bp) and an excess of unlabelled PCR product of the C genome 45S rDNA intergenic spacer was added to the hybridization solution as a blocker to prevent non-specific inter-genomic cross-hybridization between A and C genomes of Brassica. DNA of B. oleracea L var. alboglabra was used to develop a C genome probe as it could selectively hybridize with C genome chromosomes. The GISH protocol as proposed by Schwarzacher and Heslop-Harrison (2000) was followed with minor modifications to study C genome substitutions. For in situ hybridization, 40 µl of hybridization mixture containing 50 % formamide, 2× SSC, 10 % dextran sulphate, 0.025 µg salmon sperm DNA, 1.25 mM EDTA, 0.125 % SDS, 100 ng of each labelled probes and 200-fold blocking DNA was applied on slides with good chromosome spreads. After hybridization, the slides were washed at 42 °C for 2 min in 2× SSC and 5 min in high stringent solution (20 % formamide and 0.1× SSC at 42 °C), two times respectively. Then slides were washed in 0.1× SSC at 42 °C for 10 min and then allowed to cool at room temperature for 5 min. The slides were rinsed in detection buffer (4× SSC containing 0.2 % Tween-20) for 5 min. Chromosomes were counterstained by incubating the slides in 20 µl DAPI II Counterstain (Abbott Molecular, India) (pH = 7) under a plastic coverslip at room temperature for 10–30 min in the dark and fluorescence was visualized using Carl Zeiss microscope (Imager. Z 2 AX10). At least 25 cells were observed for each preparation. These were photographed using a computer assisted cooled charge-coupled device (CCD) camera, and images were merged with Image-J software.
DNA extraction, molecular characterization and data analysis
DNA was extracted from fresh leaves using the CTAB (Cetyl trimethyl ammonium bromide) method (Doyle and Doyle 1990). The Illumina Infinium B. napus SNP array was used for SNP genotyping (Mason et al. 2014a, b). Initial SNP data curating was carried out according to methods described previously (Mason et al. 2015). SNP positions were obtained on the published B. rapa and B. oleracea genome sequences (Wang et al. 2011; Parkin et al. 2014) using BLAST to align probe sequences to the reference genome using an e value cutoff of 0.000001. Single nucleotide polymorphisms were filtered through Genome Studio software to retain genome-specific SNPs, using parental genotypes as controls. All SNPs giving >20 % heterozygous calls (AB), or >10 % no calls (NC) were removed. Finally, 1458 A-genome and 1936 C-genome SNPs (a total of 3394) were retained. No B-genome specific SNPs that did not also amplify A or C genome loci were able to be identified. The chromosome substitution lines were also analyzed using 48 B genome specific SSR markers distributed at the rate of six markers per chromosome across the B genome as per the procedure described earlier (Gupta et al. 2015). Sequence information of these SSR primers was obtained from Dr. Isobel Parkin, under a materials transfer agreement with Agriculture and Agri Food Canada.
Circular genome data visualization software (CIRCOS) was used to depict C genome chromosome substitutions (Krzywinski et al. 2009). Software Graphical GenoTypes (GGT) (Van Berloo 2008) was used for graphical representation of molecular marker data to confirm the presence/absence of B genome chromosomes. Software SAS was used for statistical analyses.
Results
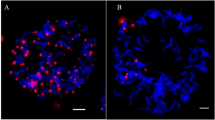
Chromosome substitution lines were initially identified in the progenies of the derived B. juncea genotypes based on distinct morphological features normally associated with the Brassica C genome. These included delayed flowering, bolting habit and leathery leaves. Meiotic analysis of selected genotypes showed a euploid chromosome number (2n = 36 with 18 II) for all these lines. Dual colour GISH allowed unambiguous identification of A, B and C genome chromosomes (Fig. 1). In nine derived genotypes namely, DJ15, DJ 16, DJ17, DJ19, DJ20, DJ21, DJ22, DJ31, and DJ 41, appearance of seven chromosome pairs strongly labeled with green colour and one chromosome pair with red colour distinctly indicated that B genome chromosome substituted a C genome chromosome each in these substitution lines (Fig. 1a). Fourteen green painted chromosomes and two red painted chromosomes in mitotic spreads confirmed disomic chromosome substitutions (Fig. 1b).

C genome chromosome substitution in derived B. juncea. a Diakinesis, all the 10 A genome chromosomes are stained blue due to staining with DAPI. Seven B genome chromosomes stained green due to labelling with B genome probe. C genome chromosome substitution for one B genome chromosome is indicated by red colour due to labelling with C genome specific probe. b Mitotic metaphase showing disomic C genome chromosome substitution (colour figure online)
Identifying C-genome chromosome substitutions with SNP genotyping
Results from SNP genotyping assays confirmed the presence of 10 intact A-genome chromosomes in all the derived B. juncea lines, with no large missing regions detected. C-genome chromosomes were absent in most derived B. juncea lines. However, in some instances C genome chromosomes revealed amplification for the entire length or part of a chromosome. These were construed to be C genome whole chromosomes or fragment substitutions (Fig. 2). Availability of physical positions for all C genome SNPs allowed us to delineate these chromosomes and chromosome regions (Figs. 2, 3; Table 1). Complete substitutions of chromosome C1 were indicated in DJ15, DJ17, DJ19, DJ21, DJ22 and DJ41. Very large fragment substitutions were also recorded in the genotypes DJ14 (34.75 Mbp), DJ20, 31 (38.79 Mbp), DJ24 (31.50 Mbp), DJ25 (33.07 Mbp), DJ70 (28.93 Mbp) and DJ5 (29.56 Mbp). Comparatively smaller fragment substitutions involving chromosome C1 were observed in DJ16 (13.86 Mbp) and DJ28 (10.85 Mbp). Almost all the fragment substitutions were terminal, but there were variations in the chromosome segments substituted. Three derived B. juncea genotypes: DJ25 (44.52 Mbp), DJ17 (41.29 Mbp) and DJ 52, 71 (26 Mbp), harboured very large chromosome fragment substitutions from chromosome C2. These covered almost 50 to 84 % of the chromosome. DJ52, DJ71 and DJ70 were unique as these carried intercalary C genome substitutions (Fig. 3). Chromosome C3 was identified as a complete chromosome substitution in DJ17 (64.85 Mbp). This genotype also harboured almost a complete (97 %) chromosome substitution for C8 (39.79 Mbp). Small terminal substitutions were also observed in DJ5 and DJ17, constituting 26.9 % of the donor C8 chromosome. Chromosomes C4, C5, C6 and C9 were not recovered in any chromosome substitution line using a minimum cut-off value of 10 % for fragment substitution. However, very small substituted chromosome fragments (less than 10 %) were recovered in many derived B. juncea genotypes involving all of the chromosomes, and ranged from 0.04 to 6.83 Mbp in size. Stable inheritance of whole chromosome substitutions was established by confirming the expected chromosome number and meiotic stability in the progenies obtained following selfing of identified chromosome substitution lines.

Circos diagramme documenting the C genome whole chromosome or chromosome fragment substitutions for eight B genome chromosomes in different genotypes of derived B. juncea. Each chromosome is indicated by different colour (colour figure online)

Chromosome cartoons showing B genome chromosomes (blue) by complete or segment substitution from C genome chromosome(s) as indicated by red colour (colour figure online)
Genotyping with B-genome specific SSR primers
A set of 48 B-genome specific SSR primers, distributed at six per chromosome were used to detect the presence/absence of B chromosomes in 14 derived B. juncea lines showing either complete or a large chromosome fragment substitution. B. rapa (AA), B. oleracea (CC), B. nigra (BB), B. carinata (BBCC), B. juncea (AABB) and B. napus (AACC) were used as controls. Forty eight markers amplified 67 alleles. Of these 62 alleles were retained to develop graphical genotypes as these were also present in all the three B genome bearing species, but were absent in B. rapa (AA), B. oleracea (CC) or B. napus (AACC). Graphical genotypes are presented in Fig. 4. These indicated absence of either whole B chromosome(s) or fragments thereof in the test genotypes. The presence of C genome chromosome in a test genotype coupled with the corresponding absence of B chromosome in the same genotype helped us to identified both substituted B genome chromosome and substituting C genome chromosome (Table 1). There seemed to be no pattern in substituted B or substituting C genome chromosome in the test genotypes. For example, C1 was the most prevalent substitution. However, it replaced different B genome chromosomes (B2, B3, B4, B5, etc.).

Graphical genotyping based on polymorphism for B- genome specific SSR markers. These indicate absence of either whole B chromosome(s) or fragments thereof in the test genotypes as indicated by blue colour (colour figure online)
Breeding values
All the chromosome substitution lines were fully fertile. The pollen grain fertility as evidenced by pollen grain stainability with acetocarmine varied from 93 to 97 %. The corresponding values for natural B. juncea were in excess of 96 %. Available agronomic data (Gupta et al. 2015) from selected genotypes was used to establish the breeding values of C-genome substitution lines carrying complete chromosome substitutions or at least 50 % fragment substitutions. The morpho-physiological traits included: chlorophyll values at flowering using SPAD meter, days to flowering, days to maturity, plant height, branch number, main shoot length, no. of pods on the main shoot, pod length and seed yield. Data regarding oil, protein and meal glucosinolates were also collected from both normal euploid derived B. juncea and corresponding substitution lines with complete chromosome and chromosome fragment (>10 %) substitutions. Data are depicted in Figs. 5 and 6 over 2 years. Based on averages over both the datasets, it was apparent that the euploid B. juncea and substitution lines differed primarily for main shoot length and number of pods on the main shoot. There was a marginal depression in the pod length in the chromosome/chromosome fragment substitution lines as compared to euploid B. juncea. For other traits, the differences were statistically non-significant,

Average performance of C genome chromosome substitution (white-DJS) in comparison to the normal euploid genotypes (grey-DJG) for key agronomic traits (colour figure online)

Average performance of C genome chromosome substitution (white-DJS) in comparison to the normal euploid genotypes (grey-DJG) for key yield components and seed quality traits (colour figure online)
In order to analyze the impact of chromosome substitutions on these two traits, we compared the performance of individual chromosome/chromosome substitution lines. As is apparent from Fig. 7, genotype DJ16 that carried almost complete chromosome substitutions for two C genome chromosomes (C7 and C8) outperformed the derived B. juncea genotypes for main shoot length, and pods on the main shoot. One substitution line (DJ20) carrying a complete chromosome C1 showed varied performance. On the basis of the average over six genotypes carrying a C1 substitution, the main shoot length and consequently the number of pods on the main shoot were higher than those observed for euploid B. juncea genotypes. The longest shoot length and highest number of pods on the main shoot were recorded for the genotype DJ19, while DJ20 showed the lowest values for these two traits.

Average performance of different chromosome substitution lines for main shoot length and pods on the main shoot under timely sown (TS) and late sown (LS) conditions
Better parent heterosis
F1 hybrids were produced between C genome chromosome substitution lines of derived B. juncea with four natural B. juncea (PBR 210, RLC 1, AMH2 and CBJ 002). These were compared against a set of corresponding derived B. juncea genotypes showing no C genome presence. The trials were conducted over 2 years. The results are summarized in Figs. 8 and 9. It was very interesting to note that all derived B. juncea lines carrying disomic substitutions for chromosome C1 showed excellent heterosis for yield (7.5–31.5 % with an average of 24.6 %) in crosses with natural B. juncea. The only exception was DJ31 (C1). The results were consistent over both the years. However, maximum heterosis was recorded between the genotypes DJ25 (C1, C2) and PBR 210 (59.3 %). In contrast to the chromosome substitution lines, the average heterosis in crosses involving euploid derived B. juncea was 5.6 %, which was significantly lower than the average heterosis (13.25 %) recorded for the crosses involving chromosome substitution lines. Against consistent heterotic performance of chromosome substitution lines in the hybrid combinations, the euploid B. juncea lines showed varied performance with heterosis ranging from −30.83 to 44 %. There were certain genotypes (e.g., DJ73 and DJ1) which produced much higher heterosis (38.42 %, 44 %) than that recorded by the combinations involving C-genome chromosome substitution lines. Superior performance of chromosome substitution lines in hybrid combinations was also reflected in crosses involving natural B. juncea cvs. RLC 1 and PBR 210 as testers. However, the average performance of hybrids involving C chromosome substitution lines and two exotic B. juncea testers, AMH2 and CBJ002 were only marginally superior to the average performance of corresponding hybrids involving euploid derived B. juncea genotypes. All the substitution lines (except for DJ31-C1) carrying C1 as a complete chromosome or chromosome fragment substitution produced heterotic hybrids, out-yielding the check by margins ranging from 7.5 to 31.5 %. In contrast, the average heterosis for euploid derived B. juncea lines was negative, and it varied from −30.83 to 44 %. This broadly confirmed the results for other two testers.

Expression of heterosis in the hybrids developed by crossing chromosome substitution lines and derived B. juncea with natural B. juncea genotypes from India: PBR 210 and RLC 1

Expression of heterosis in in the hybrids developed by crossing chromosome substitution lines and derived B. juncea with natural B. juncea genotypes from China: AMH 2 and CBJ 002
Discussion
Introgressing useful genetic variation from a related allotetraploid species into the B. juncea gene pool is difficult in spite of the extensive synteny that exists in crop Brassica species. This is primarily due to genetic limitations, such as the level of accessible genetic variation and meiotic complexities resulting from simultaneous occurrence of a large number of homologous, homoeologous and even paralogous associations. An alternative approach to promote controlled recombination among Brassica allotetraploids is to limit recombination to individual chromosomes by using chromosome substitution lines. A similar and possibly superior approach is marker-assisted chromosome segment substitution, as first demonstrated in Lycopersicon “introgression lines (ILs)”. Random chromosome substitutions occur naturally among the interspecific crosses from the crop Brassica species, owing to their origin from a common ancient polyploidization event. Cytological observations of interspecific hybrids and comparative molecular mapping suggest that synteny and colinearity are still conserved among three Brassica genomes. However, major cytostructural barriers exist to interspecific gene transfer from chromosomes belonging to the B genome.
We had previously reported an alternate approach to synthesize B. juncea through hybridization between B. napus and B. carinata (Gupta et al. 2015). An unintended outcome of this procedure was the development of chromosome and chromosome segment substitution (CS) lines, which we used for downstream quantitative evaluation. GISH is an excellent procedure for characterizing alien chromosome segments in expected introgressed lines, allowing identification of alien chromosomes and large translocations (Schwarzacher et al. 1992). Major C genome chromosome substitutions greater than 10 % were recorded for 18 derived B. juncea genotypes out of 89 genotypes assessed. These included three complete C genome chromosome substitutions, putatively for B genome chromosomes. All these were disomic chromosome and were stably inherited. This was evident from GISH and simple cytogenetic analysis of whole chromosome substitution lines and their selfed progenies. That the C genome chromosome substitutions occurred at the cost of B chromosomes was evident when results of SNP genotyping were supplemented with graphical genotypes developed on the basis of genotyping with B genome specific SSR primers. Large size of many fragment substitutions and their terminal locations seemed to suggest that chromosome fragment substitutions may have arisen due to breakages and reunion of chromosomes or due to homeologous recombination between C and B genome chromosomes. In the case of DJ52, DJ71, which carried intercalary substitutions for chromosome C2 and DJ70 which carried intercalary substitutions for chromosome C1, a large part of the C chromosome may have participated in recombination events with B chromosome fragments from two different chromosomes or two fragments of the same B chromosome. Reciprocal segment exchange between A and C genome chromosomes may be a less likely factor as GISH analysis did not support this possibility for substitution lines involving whole chromosomes or large chromosome fragments (>50 %). Allosyndesis in Brassica genomes is generally observed as A–C genome associations (mean 4.0 per cell) and less frequently as A–B genome associations (0.8 per cell) and B–C genome associations (0.3 per cell), although B–C allosyndesis may be influenced by the genome structure and allelic composition (Mason et al. 2010, 2014a, b).
In a series of complementary quantitative analyses, the chromosome substitutions were related to available data for different agronomic traits and expression of heterosis in hybrid combinations between derived B. juncea with C chromosome substitutions and natural B. juncea. An increase in main shoot length and consequently, the number of pods on the main shoot was observed following substitution of C1, C7 and C8, which seemed to confirm past reports suggesting the presence of QTL for shoot length traits on chromosomes C1 and C8 and for pod number on chromosomes C1 and C9 in B. napus (Ding et al. 2012; Zhou et al. 2014). Raman et al. (2014) also identified QTL for pod number on chromosomes C3 and C5. Shi et al. (2009) associated chromosomes C1 and C6 for trait pod number in B. napus. These are very important findings, as these opened up the concept of QTL transportability across species as all these substituted C genome chromosomes in B. juncea were obtained from B. napus. The corresponding genes were not only transferred to B. juncea, but also retained their expression and breeding value. Apparently, these chromosome substitution lines constitute important breeding assets for increasing the genetic diversity available in B. juncea.
Testing of hybrids involving chromosome substitution lines and euploid B. juncea demonstrated positive as well as negative effects of chromosome substitution on yield heterosis. With the exception of DJ31 (C1) all the derived B. juncea lines carrying disomic substitutions for chromosome C1 expressed excellent yield heterosis (7.5–31.5 % with an average of 24.6 %) in crosses with Indian genotypes of natural B. juncea. In contrast, the average performance of hybrids involving C chromosome substitution lines and Chinese B. juncea testers, AMH 2 and CBJ 002 were only marginally superior. This indicated the role of background genotypes on the expression of heterosis. In spite of this, use of CSLs in the study of heterosis may help in understanding heterosis at a single chromosome level.
Interspecific transfer of alien genes associated with key economic traits can be difficult due to restricted allosyndetic pairing in B–C genomes. Chromosome substitution allows greater chances of allosyndetic pairing between targeted traits located on a substituted chromosome, as single chromosome substitutions generally do not impose an extra biological penalty in terms of reproductive fitness of the recipient genotype. Restricting recombination to target chromosomes may also help to reduce linkage drag compared to the complex genome interactions observed in interspecific hybrids with a full chromosome complement. It may also be possible to develop mapping populations by involving chromosome substitution lines to initiate fine mapping of large as well as small effect QTLs which can later be used for map-based cloning. Studies in cotton have shown that for QTL localization does not require segregating populations and chromosome substitution lines provide statistical advantages and opportunities to associate QTLs with a specific substituted chromosome (Nadeau et al. 2000). Characterized chromosome substitution lines can also potentially be used to validate NGS based genome assemblies.
Author contribution statement
SSB developed basic genetic resources and designed the experiments. MG performed all the experiments. AM and JB carried out SNP genotyping, SNP data curating and editing of the manuscript. SSB and MG analysed the data and wrote the manuscript.
References
Atri C, Kumar B, Kumar H, Kumar S, Sharma S, Banga SS (2012) Development and characterization of Brassica juncea–fruticulosa introgression lines exhibiting resistance to mustard aphid (Lipaphis erysimi Kalt). BMC Genet 13:104
Atri C, Kaur B, Sharma S, Gandhi N, Verma H, Goyal A, Banga SS (2016) Substituting nuclear genome of Brassica juncea (L.) Czern and Coss. In: Cytoplasmic background of Brassica fruticulosa results in cytoplasmic male sterility. Euphytica. doi:10.1007/s10681-015-1628-4
Banga SS (1988) C-genome chromosome substitution lines in Brassica juncea (L.) Czern and Coss. Genetica 77:81–84
Banga SS, Banga SK (2009) Crop improvement strategies in rapeseed-mustard. In: Hegde DM (ed) Vegetable oils scenario: approaches to meet the growing demands. ISOR, Hyderabad, pp 13–35
Banga SS, Bhaskar PB, Ahuja I (2003) Synthesis of intergeneric hybrids and establishment of genomic affinity between Diplotaxis catholica and crop Brassica species. Theor Appl Genet 106:1244–1247
Banuelos GS, Dhillon SK, Banga SS (2013) Oilseed Brassicas. In: Singh BP (ed) Biofuel crops: production, physiology and genetics. Centre for Agricultural Biosciences International, Egham, pp 339–368
Chauhan JS, Singh KH, Singh VV, Kumar S (2011) Hundred years of rapeseed-mustard breeding in India: accomplishments and future strategies. Indian J Agr Sci 81:1093–1109
Chen BY, Cheng BF, Jorgensen RB, Heneen WK (1997) Production and cytogenetics of Brassica campestris-alboglabra chromosome addition lines. Theor Appl Genet 94:633–640
Cheng F, Mandakova T, Wu J, Xie Q, Lysak MA, Wang X (2013) Deciphering the diploid ancestral genome of the mesohexaploid Brassica rapa. Plant Cell 25:1541–1544
Cowling W (2007) Genetic diversity in Australian canola and implications for crop breeding for changing future environments. Field Crop Res 104:103–111
Delourme R, Eber F, Cheavre AM (1989) Intergeneric hybridization of Diplotaxis erucoides and Brassica napus I. Cytogenetic analysis of F1 and BC1 progeny. Euphytica 41:123–128
Ding G, Zhao Z, Liao Y et al (2012) Quantitative trait loci for seed yield and yield-related traits, and their responses to reduced phosphorus supply in Brassica napus. Ann Bot 109:747–759
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Garg H, Atri C, Sandhu PS, Kaur B, Renton M, Banga SK, Singh H, Singh C, Barbetti MJ, Banga SS (2010) High level of resistance to Sclerotinia sclerotiorum in introgression lines derived from hybridization between wild crucifers and the crop Brassica species B. napus and B. juncea. Field Crop Res 117:51–58
Gupta M, Gupta S, Kumar H, Kumar N, Banga SS (2015) Population structure and breeding value of a new type of Brassica juncea created by combining A and B genomes from related allotetraploids. Theor Appl Genet 128:221–234
Jahier J, Chevre A, Tanguy A, Eber F (1989) Extraction of disomic addition lines of Brassica napus–B. nigra. Genome 32:408–413
Janeja HS, Banga SK, Bhaskar PB, Banga SS (2003) Alloplasmic male sterile Brassica napus with Enarthrocarpus lyratus cytoplasm: introgression and molecular mapping of an E. lyratus chromosome segment carrying a fertility restoring gene. Genome 46(5):792–797
Kaur P, Banga S, Kumar N, Gupta S, Akhatar J, Banga SS (2014) Polyphyletic origin of Brassica juncea with B. rapa and B. nigra (Brassicaceae) participating as cytoplasm donor parents in independent hybridization events. Am J Bot 101(7):1157–1166
Krzywinski MI, Schein JE, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19(9):1639–1645
Kumar S, Atri C, Sangha MK, Banga SS (2011) Screening of wild crucifers for resistance to mustard aphid, Lipaphis erysimi (Kaltenbach) and attempt at introgression of resistance gene(s) from Brassica fruticulosa to Brassica juncea. Euphytica 179:461–470
Mason AS, Huteau V, Eber F, Coriton O, Yan G, Nelson M, Cowling WA, Chevre AM (2010) Genome structure affects the rate of autosyndesis and allosyndesis in AABC, BBAC and CCAB Brassica interspecific hybrids. Chromosome Res 18:655–666
Mason AS, Batley J, Bayer PE, Hayward A, Cowling WA, Nelson MN (2014a) High-resolution molecular karyotyping uncovers pairing between ancestrally related Brassica chromosomes. New Phytol 202(3):964–974
Mason AS, Nelson MN, Takahira J, Cowling WA, Alves GM, Chaudhuri A, Chen N, Ragu ME, Dalton-Morgan J, Coriton O, Huteau H, Eber F, Chèvre AM, Batley J (2014b) The fate of chromosomes and alleles in an allohexaploid Brassica population. Genetics 197:273–283
Mason AS, Zhang J, Tollenaere R, Vasquez Teuber P, Dalton-Morgan J, Hu L et al (2015) High-throughput genotyping for species identification and diversity assessment in germplasm collections. Mol Ecol Resour. doi:10.1111/1755-0998.12379
Nadeau JH, Singer JB, Matin A, Lander ES (2000) Analysing complex genetic traits with chromosome substitution strains. Nat Genet 24:221–225
Parkin IAP, Koh C, Tang H, Robinson SJ, Kagale S, Clarke WE, Town CD, Nixon J, Krishnakumar K et al (2014) Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol 15:R77
Prakash S, Bhat SR, Qurios CF, Kirit PB, Chopra VL (2009) Brassica and its close allies: cytogenetics and evolution. Plant Breed Rev 31:21–187
Raman H, Raman R, Kilian A, Detering F, Carling J, Coombes N, Diffey S, Kadkol G, Edwards D, McCully M, Kumar P, Parkin IAP, Batley J et al (2014) Genome-wide delineation of natural variation for pod shatter resistance in Brassica napus. PLoS ONE 9:e101673
Schwarzacher T, Heslop-Harrison JS (2000) Practical in situ hybridization. BIOS Scientific Publishers, Oxford
Schwarzacher T, Anamthawat-Jónsson K, Harrison GE, Islam AKMR, Jia JZ, King IP et al (1992) Genomic in situ hybridization to identify alien chromosomes and chromosome segments in wheat. Theor Appl Genet 84:778–786
Shi J, Li R, Qiu D, Jiang C, Long Y, Morgan C, Bancroft I, Zhao J, Meng J (2009) Unraveling the complex trait of crop yield with quantitative trait loci mapping in Brassica napus. Genetics 182:3851–3861
Truco MJ, Hu J, Sadowski J, Quiros CF (1996) Inter- and intragenomic homology of the Brassica genomes: implications for their origin and evolution. Theor Appl Genet 93:1225–1233
Van Berloo R (2008) GGT 2.0: versatile software for visualization and analysis of genetic data. J Hered 99:232–236
Wang X, Wang H, Wang J, Sun R, Wu J, Liu S, Bai Y, Mun JH, Bancroft I, Cheng F, Huang S, Li X, Hua W, Wang J, Wang X, Freeling M, Pires JC, Paterson AH, Chalhoub B, Wang B, Hayward A, Sharpe AG, Park BS, Weisshaar B, Liu B, Li B, Liu B, Tong C, Song C, Duran C et al (2011) The genome of the mesopolyploid crop species Brassica rapa. Nat Gen 43:1035–1039
Warwick SI (2011) Brassicaceae in agriculture. In: Schmidt R, Bancroft I (eds) Genetics and genomics of the Brassicaceae. Plant genetics and genomics: crops and models. Springer, New York, pp 33–65
Zamir D (2001) Improving plant breeding with exotic genetic libraries. Nat Rev Genet 2:983–989
Zhou QH, Fu DH, Mason AS, Zeng YJ, Zhao CX, Huan YJ (2014) In silico integration of quantitative trait loci for seed yield and yield-related traits in Brassica napus. Mol Breed 33:881–894
Acknowledgments
Research was carried out with the financial assistance from Department of Biotechnology, Government of India in form of Centre of Excellence and Innovation in Biotechnology “Germplasm enhancement for crop architecture and defensive traits in Brassica juncea l. czern. and coss.” Surinder Banga also acknowledges salary support from Indian Council of Agricultural Research. Jacqueline Batley is supported by an Australian Research Council Future Fellowship (FT130100604). Annaliese Mason acknowledges salary support from an Australian Research Council Discovery Early Career Researcher Award (DE120100668) and a Deutsche Forschungsgemeinschaft Emmy Noether award (MA 6473/1-1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by P. Heslop-Harrison.
Rights and permissions
About this article

Cite this article
Gupta, M., Mason, A.S., Batley, J. et al. Molecular-cytogenetic characterization of C-genome chromosome substitution lines in Brassica juncea (L.) Czern and Coss.. Theor Appl Genet 129, 1153–1166 (2016). https://doi.org/10.1007/s00122-016-2692-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-016-2692-4