
Abstract
Drought stress adversely affects soybean at various developmental stages, which collectively results in yield reduction. Unpredictable rainfall has been reported to contribute about 36% to variation of yield difference between the rain-fed and irrigated fields. Among the drought resistance mechanisms, drought avoidance in genotypes with fibrous roots was recognized to be associated with drought resistance in soybean. Plant introduction PI416937 was shown to possess fibrous roots and has been used as a parent in breeding programs to improve soybean productivity. Little information is available on relative contribution and chromosomal location of quantitative trait loci (QTL) conditioning fibrous roots in soybean. To identify the genomic locations and genetic bases of this trait, a recombinant inbred line population was derived from a cross between PI416937 and ‘Benning’. To detect associated QTLs, phenotypic data were collected and analyzed for 2 years under rain-fed field conditions. The selective genotyping approach was used to reduce the costs and work associated with conducting the QTL analysis. A total of five QTLs were identified on chromosomes Gm01 (Satt383), Gm03 (Satt339), Gm04 (Sct_191), Gm08 (Satt429), and Gm20 (Sat_299), and together explained 51% of the variation in root score. Detected QTLs were co-localized with QTLs related to root morphology, suggesting that fibrous roots QTL may be associated with other morpho-physiological traits and seed yield in soybean. Genetic dissection of the fibrous roots trait at the individual marker loci will allow for marker-assisted selection to develop soybean genotypes with enhanced levels of fibrous roots.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Higher plants have evolved multiple, interconnected strategies that enable them to survive abiotic stresses. However, these strategies are not well understood in most crops. Among these, drought is the major abiotic stress that generally reduces yield to less than half of that possible under ideal growing conditions (Boyer 1982). Drought affects soybean seed yield to some degree at almost all growth stages. When the plant suffers a water deficit, its turgor pressure approaches zero, cells begin to collapse, membranes suffer damage, proteins and enzymes are denatured and their structure is altered, and finally the cells die. Under water deficit conditions, a soybean plant reduces its metabolism to survive the stress and then resumes its activities when the stress is removed, which affects the seed yield as a final plant product (Korte et al. 1983; Dornbos and Mullen 1992). Inadequate and unpredictable rainfall has been reported to contribute about a 36% yield difference between the rain-fed and irrigated soybean fields (Specht et al. 1999).
The mechanisms of drought resistance in plants can be grouped into three categories as drought escape, drought avoidance, and drought tolerance (Carrow 1996). The escape mechanism maximizes the plant’s ability to complete its life cycle before a serious deficit occurs. This mechanism involves rapid physiological development characters such as early flowering and early maturity, and remobilization of pre-anthesis assimilates to seed. The avoidance mechanism is characterized by the ability to maintain relatively high tissue water potential, despite a shortage of soil moisture. Plants that utilize the avoidance mechanism survive by balancing the maintenance of turgor and the reduction of water loss, through increased rooting depth, an efficient root system, and increased hydraulic conductance. This results in increased water availability or reduced water loss through reduced stomatal conductance, reduced absorption of radiation by leaf rolling or folding, and reduced evaporation surface. Drought tolerance is the ability of plants to withstand water-deficit with low tissue water potential. The mechanisms of drought tolerance include maintenance of turgor through osmotic adjustment, increased plasticity in the cell, and decreased cell size and desiccation tolerance by protoplasmic resistance. Drought resistance is a complex trait, and plants may employ more than one mechanism at a time to resist drought. Resistant plants could contain a combination of different characters including morphological, physiological, and biochemical characters to reduce plant damage (Jones et al. 1981).
Secondary traits evaluated to assess drought tolerance include morphological and/or physiological traits such as yield components, leaf area, leaf wilting, flowering time, anthesis period, and root morphology (Richards 1996). A secondary trait should be genetically associated with seed yield in the target environment, highly heritable, genetically variable, inexpensive and rapid to measure, and observed at or before flowering; (Monneveux and Ribaut 2006). Canopy wilting (slow wilting) has been used as secondary trait to predict drought resistance in soybean (Charlson et al. 2009).
Several studies indicated that deeper rooting might be the underlying mechanism of drought resistance for the tolerant soybean genotypes, which is positively related to soybean yield under drought stress (Brown et al. 1985; Cortes and Sinclair 1986; Hudak and Patterson 1995). The root system is particularly important in response to drought stress since plant exposure to limited soil water occurs in the root zone first (Hoogenboom et al. 1987). Root plasticity is another trait associated with drought resistance. A deep and thick root system provided access to water deep in the soil profile in upland rice (Oryza sativa) (Fukai and Cooper 1995). On the other hand, a fibrous root system has been reported to be more efficient for water absorption in soybean because of the increase in surface area and the number of root tips (Hudak and Patterson 1995; Pantalone et al. 1996a).
Genetic diversity in soybean root traits has been reported for root elongation, root nodule formation, fibrous root, and root mass in response to various abiotic/biotic stresses such as aluminum toxicity (Bianchi-Hall et al. 1998; Villagarcia et al. 2001), flooding (Bacanamwo and Purcell 1999), soybean cyst nematode (Heterodera glycines) (Concibido et al. 1994; Miltner et al. 1991), iron deficiency (Charlson et al. 2005; Lin et al. 1997; Wang et al. 2008), manganese deficiency (Kassem et al. 2004), and phosphorus deficiency (Li et al. 2005; Zhang et al. 2009a, b). Exotic soybean accessions have been found to possess greater drought tolerance than elite cultivars and to have unique root characteristics when evaluated in drought stress environments (Hudak and Patterson 1995). In the field studies under moisture deficit field conditions PI416937 possessed a slower wilting canopy than an elite cultivar and its leaves possessed lower solute potential, higher turgor, and higher relative water content which resulted in a relatively small yield reduction when compared to the elite cultivar (King et al. 2009; Sloane et al. 1990). The fibrous root morphology of PI416937 was reported to have a positive association with seed protein concentration and was shown to be a heritable trait indicating its potential use in breeding programs for enhancing seed yield under drought conditions (Pantalone et al. 1996a).
Screening soybean for root traits in breeding populations is tedious and expensive due to difficulties of measuring root characteristics under field conditions. This has resulted in relatively limited genetic studies of soybean root morphology under field conditions. The application of QTL analysis allows the identification of chromosomal regions conditioning the phenotypic variation in quantitative traits such as root morphology and identifies the desirable alleles at these QTLs for use in marker assisted selection (MAS). Previous QTL mapping of drought related traits in soybean includes yield under drought stress conditions (Du et al. 2009b; Specht et al. 1999), slow canopy wilting (Charlson et al. 2009), water use efficiency (WUE; Mian et al. 1996, 1998; Specht et al. 2001), iron deficiency (Charlson et al. 2005; Lin et al. 1997; Wang et al. 2008), manganese deficiency (Kassem et al. 2004), phosphorus deficiency (Li et al. 2005; Zhang et al. 2009a, b), aluminum tolerance (Bianchi-Hall et al. 2000), and salt tolerance (Lee et al. 2004). Molecular markers can be also used to obtain genetic information about root traits, including location of the loci, inheritance of positive alleles, the components of genetic variance, pleiotropic effects of the loci, and epistasis. Following this strategy, high-throughput screening methods have been used in rice and maize (Price and Tomos 1997; Zhang et al. 2001; Tuberosa et al. 2002). No studies have been published on fibrous roots QTL in soybean even though the trait holds potential for yield improvement in rain-fed environments. The objectives of this study were to identify QTL(s) associated with the fibrous root trait in a soybean recombinant inbred line (RIL) population developed from a cross of ‘Benning’ (few fibrous roots) and PI416937 (many fibrous roots) and to elucidate genetic characteristics of the QTL(s) to facilitate their use in MAS.
Materials and methods
Plant materials and phenotypic evaluation
A cross between cultivar Benning (PI595645) and PI416937 was made during the summer of 1996 at the University of Georgia Plant Sciences Farm near Watkinsville, GA. PI416937 is a Japanese landrace that phenotypically appears distinctly different from the ancestors of North American soybean cultivars. When compared with elite US cultivars it has a slower wilting canopy and a more fibrous root system (Pantalone et al. 1996a, b; King et al. 2009). Benning is a Maturity Group VII cultivar developed by University of Georgia (Boerma et al. 1997). The F1 seeds were grown in the USDA/ARS winter nursery located near Isabella, Puerto Rico during the winter of 1996–1997. The F2–F5 generations were advanced by the single seed decent (SSD) method in Georgia and Puerto Rico. The F5 plants were harvested individually, and the F5-derived lines were grown as plant rows at the University of Georgia Plant Sciences Farm during the summer of 1999. At harvest, one plant per row was selected to represent each family depending on uniform maturity and non-shattering. In 2000, a total of 240 F6 plants were grown as individual plant rows. At maturity each row was individually harvested.
In 2001 the 240 F6-derived RILs were planted in hill plots thinned to six plants per hill in a randomized complete block experimental design with nine replications at the University of Georgia Plant Sciences Farm (an Appling coarse sandy loam soil) under rain-fed conditions. Prior to planting 107 kg ha−1 P2O5 was added to the soil. Nematodes, insects, and weeds were controlled using commercial chemicals as needed. At the R4 growth stage (Fehr et al. 1971), plants of each hill were tied into a bundle and the roots were removed from the soil with a mechanical peanut inverter. Roots of the six plants in each hill were visually rated according to Pantalone et al. (1996a) on scale of 1–8; where 1 is when the roots are course and contain very few fibrous roots and 8 is when the hill contains mostly fibrous roots (Fig. 1).

The examples of roots in various root score categories; where 1 = few fibrous roots and 8 = many fibrous roots
In 2009, 128 of the original 240 RILs were selected based on to their high or low fibrous roots score in 2001. The 128 RILs and parents Benning and PI416937 each repeated three times were planted in hill plots thinned to six plants per hill at the University of Georgia Plant Sciences Farm, (in a Cecil coarse sand loam soil) in a randomized complete block design experimental design with eight replications. Prior to planting 94 kg ha−1 P2O5 and 46 kg ha−1 K2O were applied to the soil. Nematodes, insects and weeds were controlled using commercial chemicals as needed. Root harvesting and scoring followed the same methods used in 2001 experiment.
Statistical analyses
The data from the 2001 and 2009 experiments were analyzed using SAS PROC GLM as randomized complete block experimental designs with replicates and RILs considered as random effects (Statistical Analysis System, SAS institute 2001). Analysis of the data across years was conducted by SAS PROC MIXED using restricted maximum likelihood (REML) method as randomized complete block experimental design with replicates, years, and RILs considered as random effects (Steel and Torrie 1980).
Heritabilities on an entry-mean basis was calculated as h 2 = (σ 2RIL /(σ 2RIL + σ 2e /r) for 2001 and h 2 = σ 2G /[(σ 2RIL + (σ 2RIL *Y/y) + (σ 2e /y)] for the combined 2001 and 2009 data (Nyquist 1991; Holland et al. 2003); where σ 2RIL equaled the genetic variance among the RILs, σ 2RIL*Y represented the variance of RILs by year interaction, σ 2e the variance of experimental error, y the number of years, and r the number of replications. Because the 2001 and 2009 data were unbalanced, the value for r was computed as the harmonic mean of the 2001 and 2009 number of replications (Holland et al. 2003).
DNA markers, map construction and QTL analysis
DNA from each of the F6-derived lines and the two parents was extracted from the first trifoliolate leaves of greenhouse-grown plants using a CTAB (Hexadecyltrimethylammonium acid) procedure modified from Keim et al. (1988). PCR reactions were based on the protocol of Diwan and Cregan (1997) with slight modifications. The reactions were performed in a dual 384-well or a 96-well GeneAmp PCR System 9700 (PE-ABI, Foster City, CA). Data were collected on 276 SSR markers, that were polymorphic between the parents. The primers were labeled with fluorescent dyes. A loading sample for each lane of a gel was prepared with loading mixture (2.0 μl of deionized formamide, 2.0 μl of loading buffer, 0.3 μl of Genescan Rox-500) and 1–2 μl of the PCR products. DNA was denatured at 95°C for 5 min, prior to separation on a 4.8% (w/v) polyacrylamide gel with 1× TAE (Tris acetate EDTA) running buffer using ABI PRISM 377 DNA Sequencer (PE-ABI, Foster City, CA). The marker fragments were analyzed with GeneScan and scored with Genotyper software (PE-ABI, Foster City, CA).
Linkage maps were constructed using Map Manager QTX (Meer et al. 2002) with a minimum logarithm of the likelihood-of-odds (LOD) score of 3.0 and a maximum recombination fraction of 0.25 as thresholds for detecting possible linkages and multipoint ordering of markers. Observed recombination frequencies were converted to map distance and expressed as centiMorgans using the Kosambi’s mapping function.
Significant associations of the markers with the fibrous root trait were tested using single-factor analysis of variance (SF-ANOVA) of SAS (SAS Institute 2001) based on RIL means for each year separately and least square means across years. QTL analysis was performed using QTL Cartographer 2.5 (Wang et al. 2007) using genotypic least square means across environments and the multiple interval mapping (MIM) procedure of Kao et al. (1999) and Zeng et al. (1999). The MIM is a stepwise model adaptation procedure combined with an initial model selection of markers. The MIM was performed to test the presence of significant QTLs and QTL effects including additive and epistatic effects by simultaneous analysis of the QTLs in multiple regression models (Zeng et al. 1999) with the following steps: (1) the initial model included representatives of significant markers that associated with the variation in fibrous roots using SF-ANOVA. To select representatives, the significant markers were sorted by chromosomes and position within each chromosome, then one marker with largest phenotypic variation from each significant marker cluster was chosen as representative of this chromosomal region, (2) the pre-selected MIM model was optimized using the “optimize QTL positions” option, then (3) the whole genome was re-scanned searching for new main effect QTL and epistatic effect between main QTL using “search for new QTL” and “QTL interaction” options respectively; (4) model was re-evaluated to fit all the significant main and epistatic QTLs in MIM model using “test existing QTLs” option, (5) the MIM model with minimum Bayesian information criterion (BIC) was chosen, where c(n) = ln(n), and search walk speed was 1 cM. After identifying the best model (BIC-M0 = 185.1993), the 5 QTL effects, their total phenotypic variation, and the proportion of the variation explained by each QTL of the model was estimated using the “summary” option.
Results
SSR linkage map of Benning × PI416937
A total of 629 SSR markers were screened for polymorphism between Benning and PI416937, and 281 markers (44.6%) were polymorphic. A linkage map was constructed with 276 informative markers (Supplemental Fig. 1). The linkage maps consisted of 20 linkage groups which corresponds to soybean’s 20 chromosomes and collectively covered 2169 cM or 94% of the 2276 cM soybean genome (based on the USDA consensus soybean linkage map; Song et al. 2004). This resulted in an average of 7.86 cM between adjacent SSR markers on the Benning × PI416937 linkage maps (Supplemental Table 1). Among the 20 linkage groups, chromosome Gm13 included the most linked markers with 20 while chromosome Gm15 had the fewest with nine linked markers. In general, the SSR marker order and distance between adjacent markers were similar to those on the soybean genetic linkage consensus map of Song et al. (2004) and SoyBase (http://soybase.org/; Supplemental Table 1).
Fibrous roots variation of the parents and RILs
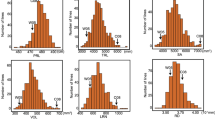
In 2001 PI416937 averaged significantly (P < 0.05) higher fibrous roots score (7.2 vs. 4.5) than Benning (Table 1). The root scores of the 240 RILs ranged from 3.4 to 7.2. The scores for the 240 RILs were normally distributed and showed transgressive segregation with some of the RILs producing less fibrous roots than Benning (Fig. 2, Table 1). None of the RILs produced more fibrous roots than PI416937. The entry-mean heritability for selection based on means of nine replications was 0.74.

Phenotypic frequencies of fibrous root score in 240 RILs of Benning × PI416937 population in 2001
A selective genotyping approach was employed to identify the RILs for inclusion in the 2009 experiment. Based on the 2001 mean root scores, 64 RILs were selected for their low root scores and 64 RILs were selected for their high root scores. In 2009 both parents averaged few fibrous roots than in 2001 (Table 1). PI416937 had an average root score of 5.6 compared to a root score of 3.6 for Benning. The range of root scores (3.4–5.6) among the 128 RILs was similar to the two parents and none of the RILs expressed transgressive segregation.
The combined analysis of variance of root score over years showed significant (P < 0.01) effects for year, genotype, and the genotype by year interaction. The 2001/2009 root score means for the 128 RILs were distributed in a bimodal fashion (data not shown). This bimodal distribution was expected based on the selective genotyping approach used in 2009. When the genotype by year mean squares was used as the error term (random-effect model), there were significant (P < 0.05) differences for the 2-year means among the 128 RILs and between the parents, Benning and PI416937. Benning averaged a root score of 3.8 compared to a score of 6.4 for PI416937. The range in root scores among the 128 RILs (3.6–6.2) was similar to the range between their parents. The RILs did not show transgressive segregation for root score. The entry-mean heritability estimate (based on selection of the 2-year means) was 0.37 (data not shown).
QTL analysis
Single-factor analysis of variance (SF-ANOVA) was employed to putatively identify QTL conditioning the fibrous roots trait for each year and across years. If a marker was significantly (P < 0.01) associated with root scores for the combined data across years, it was then tested for significance (P < 0.05) in 2001 and 2009. The rationale for using the less stringent probability level for the single-year analysis was to test the consistency of QTL detection across years. Nine SSR markers on nine soybean chromosomes identified putative QTLs associated with root scores based on 2-year means (Table 2). Of these nine putative QTLs, three (Satt429 on Gm08, Satt383 on Gm01, and Satt339 on Gm03) were identified in both the 2001 and 2009 experiments. Five putative QTLs (Sct_191 on Gm04, Sat_227 on Gm02, Satt568 on Gm12, Sat_229 on Gm20, and Satt196 on Gm09) were significant (P < 0.05) only in the 2001 environment when 240 RILs were evaluated. Satt383 and Sat_345, which are located ~11 cM apart on Gm01, are likely associated with the same fibrous roots QTL. The remaining QTL on Gm19 (Sat_071) was the only putative QTL identified based on the 2-year data and not found significant (P > 0.05) in either the 2001 or 2009 environment. These putative fibrous roots QTLs explained from 5.3 to 13.5% of the phenotypic variation in 2001/2009 root scores (Table 2). The PI416937 alleles increased fibrous rooting at all the putative QTLs with the exception of the QTL identified by Sat_299 on Gm20 (Table 3).
The SSR marker and root data were then analyzed by the MIM model of QTL-Cartographer. The MIM method utilizes multiple marker intervals simultaneously to identify QTLs. This method fits all QTLs into the model and has the capacity for the evaluation of QTL epistasis. Based on the RIL least square means across environments, the MIM model identified five QTLs at LOD ≥ 2.5; Q_root_Gm08 (flanked with Satt228 and Satt429 on Gm08), Q_root_Gm04 (flanked with Satt713 and Sct_191 on Gm04), Q_root_Gm01 (flanked with Satt179 and Satt383 on Gm01), Q_root_Gm20 (flanked with Sat_420 and Sat_299 on Gm20), and Q_root_Gm03 (flanked with Satt339 and Sat_091 on Gm03). The LOD scores of the detected QTLs ranged from 2.62 to 5.71 (Table 3, Fig. 3). The phenotypic variation for the fibrous root trait explained by each of these QTLs ranged from 7.3% for Q_root_Gm04 to 13.5% for Q_root_Gm03, and the total phenotypic variation explained by model was 51.1%. At four of these QTLs (Q_root_Gm08, Q_root_Gm04, Q_root_Gm01 and Q_root_Gm03) the PI416937 alleles increased the root scores, while at Q_root_Gm20 the Benning allele increased the root score (Table 3).

Interval mapping fibrous root score QTLs in the Benning × PI416937 RIL population based on the combined 2001/2009 data. QTL nomenclature is in the form of Q-trait-chromosome-position. Length of QTL bar indicates the LOD-1 confidence interval and QTL line is extended to LOD-2 confidence interval drops from the maximum likelihood value
In order to gain insight into the potential effect the five fibrous roots QTLs may have on other soybean traits, we conducted a comparative QTL analysis. The genomic locations of the five fibrous roots QTLs were aligned on the soybean linkage/QTL maps in Soybase (http://soybase.org/). The soybean linkage/QTL maps of chromosomes Gm01, Gm03, Gm04, Gm08, and Gm20 were aligned with the fibrous root QTL intervals by use of the SSR markers defining the genomic locations of the fibrous root QTL (Table 3). For this analysis we arbitrarily included QTLs conditioning other traits if these trait QTLs were mapped within a bin that included the fibrous root QTL interval ±5 cM beyond the two markers that defined the fibrous root QTL boundary. A total of 35 QTLs were reported for traits related to abiotic stresses, plant morphology and development, seed composition, and nitrogen accumulation (Fig. 4). In addition, there were three QTLs for seed yield near Q_root_Gm01 and Q_root_Gm03, and two QTLs for seed weight near to Q_root_Gm01 and Q_root_Gm20.

Comparative QTL analysis of the fibrous root QTLs detected in the Benning × PI416937 population with coinciding QTLs found on Soybase. These QTLs were identified by alignment of both maps with common SSR markers (http://soybase.org/)
On Gm08 near the fibrous roots QTL, Q_root_Gm08, there were two QTLs for length of reproductive period (R3_1-3 and R7_1-7), one for maturity (Pod_mat_13-1), and one for leaf water loss (LWL). Data from an unpublished study with this RIL population indicated that this region of the chromosome Gm08 did not contain QTL conditioning maturity. The LWL QTL was reported to map in this region by Du et al. (2009b) in a different population.
The fibrous root QTL Q_root_Gm04 mapped to the same region as QTLs for maturity, seed protein and oil content, plant height, seed abortion (Tischner et al. 2003), and water use efficiency. The plant height (Pl_ht 5-4), maturity (Pod_mat_6-6), water-use efficiency (WUE_2-3), and protein (Prot_4-3) QTLs were originally mapped in a ‘Young’ × PI416937 F4-derived population (Mian et al. 1996, 1998). The PI416937 alleles at these QTLs resulted in later maturity, more efficient water use, higher protein content, and taller plants than the Young alleles. The WUE_2-2 QTL was mapped in an S100 × Tokyo population and mapped to the same genomic location as the WUE_2-3 QTL.
The trait QTLs associated with the Q_root_Gm01 fibrous root QTL were NitR7_1-2 which conditions nitrogen accumulation during the R7 stage of development, oil content (Oil_13-2), plant height (Pl_ht_17-10), and resistance to race 2 of SCN nematode (SCN_race2). In the interval of the Q_root_Gm20 QTL, the Cnpy_wdth_1-3 QTL was found which conditions canopy width. A total of two drought tolerance QTLs were found outside of the interval of Q_root_Gm20, one for leaf water loss (LWL) (at 15 cM away) and wilt coefficient (wilt Coe) (22 cM away). Located near the Q_root_Gm03 fibrous root QTL were four iron efficiency uptake QTLs, a salt tolerance QTL, two protein-related QTLs (Beta_conglycinin_1-1 and Prot_21-9), and a plant height QTL.
Discussion
Drought affects soybean growth and seed yield to some degree at almost all growth stages. Balancing water loss by modifying soybean root morphology is one of the drought avoidance mechanisms. Screening plant root morphology in large breeding populations is tedious and expensive due to difficulties of measuring root characteristics in the field and the relatively low heritability of the trait. Pantalone et al. (1996b) reported an entry-mean heritability for fibrous roots score of 0.39 (selection based on means across 2 years and 2 locations). In the present study we found a similar entry-mean heritability of 0.37 based on selection of 2-year means (data not shown), but this heritability estimate must be considered somewhat biased based on non-random selection of RILs for inclusion in the 2009 experiment by selective genotyping. The QTL analysis of this population identified five fibrous root QTLs, Q_root_Gm08, Q_root_Gm04, Q_root_Gm01, Q_root_Gm20, and Q_root_Gm03. These QTLs can be employed in a MAS program and which should overcome many of the previous limitations in the selection of soybean genotypes with modified root morphology.
These five QTLs were found to explain from 7 to 15% of the phenotypic variation in fibrous roots scores. None of these QTLs is considered a major fibrous root QTL. This was unexpected considering the unique fibrous roots system of PI416937 when compared to other soybean genotypes. We are unaware of other soybean genotypes with PI416937’s level of fibrous roots or even genotypes with intermediate levels of fibrous roots. This may be due to their lack of existence or due to the limited sampling of soybean germplasm for root morphology. The identification of four fibrous root QTLs where the PI416937 alleles conditioned greater levels of fibrous roots and one QTL where Benning contained the allele for more fibrous roots provides breeders the opportunity to develop soybean lines with intermediate levels of fibrous roots and to evaluate the agronomic value of these modified rooting patterns.
To minimize the population size and maximize genetic information gained from this study, a selective genotyping (SG) method was used in 2009 to select RILs from the tails of whole population (Lander and Botstein 1989). Selective genotyping has been used to study single traits related to biotic/abiotic stresses in several plant species including stripe rust in barley (Vales et al. 2005), salt tolerance in tomato (Foolad et al. 2001), sclerotinia midstalk rot resistance in sunflower (Micic et al. 2005), phosphorus deficiency in rice (Ni et al. 1998), and drought in maize (Hao et al. 2009) and rice (Subashri et al. 2009). The present findings indicate that SG can be used in identifying QTL associated with fibrous root traits as well as other traits where population size is one of the limiting factors.
QTLs associated with soybean seed yield under well-watered conditions were reported on Gm01 (Kabelka et al. 2004; Orf et al. 1999) and Gm03 (Kabelka et al. 2004). Measuring the fibrous root trait is destructive and yield could not be conducted in the same experiment. Tuberosa et al. (2002) noticed overlap between weight of the adventitious seminal roots in hydroponics with QTLs for grain yield under both well-watered and water-stressed conditions. Co-localization of QTLs for a specific trait and yield under drought stress could lead to improved yield via improving root traits (Babu et al. 2003; Lebreton et al. 1995). Drought stress during flowering and early pod expansion resulted in decreased pod set (Desclaux and Roumet 1996), which affected the number of seeds per pod and in turn the seed weight and yield (Liu et al. 2003). Liu et al. (2004) suggested that soybean root signals control pod growth under drought conditions.
Soybean protein and oil concentrations are affected by environmental stresses (Bellaloui and Mengistu 2008; Carrera et al. 2009; Dornbos and Mullen 1992; Specht et al. 2001). Nitrogen fixation activity has also been shown to be highly sensitive to moisture deficits, where drought depressed its activity resulting in reduced N2 accumulation rate, protein content, and plant growth (King and Purcell, 2001; Streeter 2003; Sinclair et al. 2007). QTLs for oil content and root score in the same genomic regions were reported on, Gm04 (Lg-C1) (Fasoula et al. 2004; Lee et al. 1996) and Gm01 (Lg-D1a) (Hyten et al. 2004). Seed protein QTLs were located in the same genomic regions as fibrous root QTL on Gm08 (Vollmann et al. 2002), Gm04 (Brummer et al. 1997; Lee et al. 1996), and Gm03 (Kabelka et al. 2004). A QTL for β-conglycinin (β-subunit; 48 kDa) was reported on chromosome Gm03 (Panthee et al. 2004a). Samarah et al. (2006) indicated that severe drought during seed development delayed the appearance of both the basic unit of glycinin and β-subunit of β-conglycinin. It is known that environmental stresses, including drought, modifies protein expression, where some proteins are increased (expressed) and others are decreased (suppressed). A previously detected QTL for nitrogen accumulation on Gm01 at reproductive stages R7 (Panthee et al. 2004b) coincided with the genomic locations of the root QTL Q_root_Gm01 suggesting that the fibrous root system of PI416937 could increase root surface area resulting in greater nodule numbers and that could enhance the N2 fixation (Pantalone et al. 1999).
Chromosomal locations harboring fibrous root QTLs were also found to be associated with QTLs for different abiotic stresses. Mian et al. (1998) mapped a QTL for water use efficiency (WUE 2-3) to the same genomic location as Q_root_Gm04 using progeny from the Young × PI416937 population. The positive allele at QTL WUE 2-3 was also inherited from PI416937. Although not located within ±5 cM of the Q_root_Gm20 QTL interval, a leaf wilting coefficient (Wilt-Coe) QTL was reported within 22 cM of Q_root_Gm20, as well as a leaf water loss (LWL) QTL that was located within 15 cM (Fig. 4). Leaf water loss QTLs were also located near the Q_root_Gm08 and Q_root_Gm20 (Du et al. 2009a). The similar genomic location of QTLs for water use efficiency, leaf water loss, and fibrous roots is interesting given the obvious potential for each of these traits to contribute to drought tolerance.
King et al. (2009) found that PI416937 may possess more than one mechanism for drought resistance. PI416937 was initially identified based on its slow canopy wilting trait (Sloane et al. 1990). Fletcher et al. (2007) found that PI416937 may conserve soil moisture by limiting transpiration during periods of high evaporative demand by reaching its maximum transpiration rate at a vapor pressured deficit (VPD) of 2.0 kPa. PI416937 showed no additional increase in transpiration at higher VPD while two fast canopy wilting cultivars continued to increase their transpiration rates as VPD was increased above 2.0 kPa.
Four QTLs on Gm03 (Fe_effic_5-2, Fe_effic_6-2, Fe_effic_10-2, and Fe_effic_12-2) were reported to be associated with the iron deficiency, a trait that depends on the iron absorption by soybean roots (Lin et al. 1997, 2000). Salt tolerance is another trait that is likely impacted by the root morphology. A salt tolerance QTL was previously detected and mapped in the same genomic location as Q_root_Gm03 (Lee et al. 2004). Q_root_Gm01 and a seed yield QTL has been mapped to the same interval in a population of ‘BSR 101’ × LG82-8379 (Kabelka et al. 2004). The LG82-8379 breeding line was selected from a cross between two exotic accessions, PI 68508 and FC 04007B.
North American soybean cultivars have a narrow genetic base (Gizlice et al. 1994), where 80% of the alleles found in public soybean cultivars released between 1947 and 1988 were derived from 13 ancestral lines. The exotic line, PI416937 has positive alleles for important traits such as drought resistance and high seed yield, which can be introduced into elite germplasms to improve soybean productivity. The results suggest that the identified fibrous root QTLs may be associated with other morpho-physiological traits which eventually affect a soybean plant’s ability to tolerate moisture stress. Clustering of QTLs for various abiotic and nitrogen/seed protein-related traits near these five fibrous roots QTLs could be pleiotropic effects of the same gene affecting these related traits, or this chromosomal region has tightly linked genes responsible for the variation in these traits. The challenge is introgression of these favorable alleles into an elite cultivar while not replacing large regions of the recipient genome with chromosomal segments of an unadapted donor parent. The effect of combining the favorable alleles at different fibrous roots loci into one background would be a straightforward application of MAS. Backcrossing the fibrous roots alleles from PI416937 into elite cultivars would allow the effective evaluation of the fibrous root traits and its ability to produce stable seed yield under drought conditions.
References
Babu RC, Nguyen BD, Chamarerk V, Shanmugasundaram P, Chezhian P, Jeyaprakash P, Ganesh SK, Palchamy A, Sadasivam S, Sarkarung S, Wade LJ, Nguyen HT (2003) Genetic analysis of drought resistance in rice by molecular markers: association between secondary traits and field performance. Crop Sci 43:1457–1469
Bacanamwo M, Purcell LC (1999) Soybean root morphological and anatomical traits associated with acclimation to flooding. Crop Sci 39:143–149
Bellaloui N, Mengistu A (2008) Seed composition is influenced by irrigation regimes and cultivar differences in soybean. Irrig Sci 26:261–268
Bianchi-Hall CM, Carter TE Jr, Rufty TW, Arellano C, Boerma HR, Ashley DA (1998) Heritability and resource allocation of aluminum tolerance derived from soybean PI 416937. Crop Sci 38:513–522
Bianchi-Hall CM, Carter TE Jr, Bailey MA, Mian MAR, Rufty TW, Ashley DA, Boerma HR, Arellano C, Hussey RS, Parrott WA (2000) Aluminum tolerance associated with quantitative trait loci derived from soybean PI 416937 in hydroponics. Crop Sci 40:538–545
Boerma HR, Hussey RS, Phillips DV, Wood ED, Rowan GB, Finnerty SL (1997) Registration of ‘Benning’ soybean. Crop Sci 37:1982
Boyer JS (1982) Plant productivity and environment. Science 218:443–448
Brown EA, Cavines CE, Brown DA (1985) Response of selected soybean cultivars to soil moisture deficit. Agron J 77:274–278
Brummer EC, Graef GL, Orf J, Wilcox JR, Shoemaker RC (1997) Mapping QTL for seed protein and oil content in eight soybean populations. Crop Sci 37:370–378
Carrera C, Martinez MJ, Dardanelli J, Balzarini M (2009) Water deficit effect on the relationship between temperature during the seed fill period and soybean seed oil and protein concentrations. Crop Sci 49:990–998
Carrow RN (1996) Drought avoidance characteristics of diverse tall fescue cultivars. Crop Sci 36:371–377
Charlson DV, Grant D, Bailey TB, Cianzio SR, Shoemaker RC (2005) Molecular marker Satt481 is associated with iron-deficiency chlorosis resistance in a soybean breeding population. Crop Sci 45(6):2394–2399
Charlson DV, Bhatnagar S, King CA, Ray JD, Sneller CH, Carter TE, Purcell LC (2009) Polygenic inheritance of canopy wilting in soybean [Glycine max (L.) Merr.]. Theor Appl Genet 119:587–594
Concibido VC, Denny RL, Boutin SR, Hautea R, Orf JH, Young ND (1994) DNA marker analysis of loci underlying resistance to soybean cyst-nematode (Heterodera glycines Ichinohe). Crop Sci 34:240–246
Cortes PM, Sinclair TR (1986) Gas-exchange of field-grown soybean under drought. Agron J 78:454–458
Desclaux D, Roumet P (1996) Impact of drought stress on the phenology of two soybean (Glycine max L. Merr) cultivars. Field Crops Res 46:61–70
Diwan N, Cregan PB (1997) Automated sizing of fluorescent-labeled Simple Sequence Repeat (SSR) markers to assay genetic variation in soybean. Theor Appl Genet 95:723–733
Dornbos DL, Mullen RE (1992) Soybean seed protein and oil contents and fatty-acid composition adjustments by drought and temperature. J Am Oil Chem Soc 69:228–231
Du WJ, Fu SX, Yu DY (2009a) Genetic analysis for the leaf pubescence density and water status traits in soybean [Glycine max (L.) Merr.]. Plant Breed 128:259–265
Du WJ, Yu DY, Fu SX (2009b) Detection of quantitative trait loci for yield and drought tolerance traits in soybean using a recombinant inbred line population. J Integr Plant Biol 51:868–878
Fasoula VA, Harris DK, Boerma HR (2004) Validation and designation of quantitative trait loci for seed protein, seed oil, and seed weight from two soybean populations. Crop Sci 44:1218–1225
Fehr WR, Cavlness CE, Burmood LT, Penninglon IS (1971) Stage of development descriptions for soybeans, Glycine max (L.) Merrill. Crop Sci 11:929–931
Fletcher AL, Sinclair TR, Allen LH (2007) Transpiration responses to vapor pressure deficit in well watered ‘slow-wilting’ and commercial soybean. Environ Exp Bot 61:145–151
Foolad MR, Zhang LP, Lin GY (2001) Identification and validation of QTLs for salt tolerance during vegetative growth in tomato by selective genotyping. Genome 44:444–454
Fukai S, Cooper M (1995) Development of drought-resistant cultivars using physio-morphological traits in rice. Field Crops Research 40:67–86
Gizlice Z, Carter TE, Burton JW (1994) Genetic base for north-american public soybean cultivars released between 1947 and 1988. Crop Sci 34:1143–1151
Hao Z, Liu X, Li X, Xie C, Li M, Zhang D, Zhang S, Xu Y (2009) Identification of quantitative trait loci for drought tolerance at seedling stage by screening a large number of introgression lines in maize. Plant Breed 128:337–341
Holland JB, Nyquist WE, Cervantes-Martinez CT (2003) Estimating and interpreting heritability for plant breeding: an update. Plant Breed Rev 22:9–111
Hoogenboom G, Huck MG, Peterson CM (1987) Root-growth rate of soybean as affected by drought stress. Agron J 79:607–614
Hudak CM, Patterson RP (1995) Vegetative growth analysis of a drought-resistant soybean plant introduction. Crop Sci 35:464–471
Hyten DL, Pantalone VR, Sams CE, Saxton AM, Landau-Ellis D, Stefaniak TR, Schmidt ME (2004) Seed quality QTL in a prominent soybean population. Theor Appl Genet 109:552–561
Jones MM, Turner NC, Osmond CB (1981) Mechanisms of drought resistance. In: Paleg LG, Aspinall D (eds) The physiology and biochemistry of drought resistance in plants, 1st edn. Academic Press, New York, pp 15–37
Kabelka EA, Diers BW, Fehr WR, LeRoy AR, Baianu IC, You T, Neece DJ, Nelson RL (2004) Putative alleles for increased yield from soybean plant introductions. Crop Sci 44:784–791
Kao CH, Zeng ZB, Teasdale RD (1999) Multiple interval mapping for quantitative trait loci. Genetics 152:1203–1216
Kassem MA, Meksem K, Kang CH, Njiti VN, Kilo V, Wood AJ, Lightfoot DA (2004) Loci underlying resistance to manganese toxicity mapped in a soybean recombinant inbred line population of ‘Essex’ × ‘Forrest’. Plant Soil 260:197–204
Keim P, Olson TC, Shoemaker RC (1988) A rapid protocol for isolating soybean DNA. Soybean Genet Newsl 15:150–152
King CA, Purcell LC (2001) Soybean nodule size and relationship to nitrogen fixation response to water deficit. Crop Sci 41:1099–1107
King CA, Purcell LC, Brye KR (2009) Differential wilting among soybean genotypes in response to water deficit. Crop Sci 49:290–298
Korte LL, Specht JE, Williams JH, Sorensen RC (1983) Irrigation of soybean genotypes during reproductive ontogeny. II. Yield component responses. Crop Sci. 23:528–533
Lander ES, Botstein D (1989) Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121:185–199
Lebreton C, Lazic-Jancic V, Steed A, Pekic S, Quarrie SA (1995) Identification of QTL for drought responses in maize and their use in testing causal relationships between traits. J Exp Bot 46:853–865
Lee SH, Bailey MA, Mian MAR, Carter TE Jr, Ashley DA, Hussey RS, Parrott WA, Boerma HR (1996) Molecular markers associated with soybean plant height, lodging, and maturity across locations. Crop Sci 36:728–735
Lee GJ, Boerma HR, Villagarcia MR, Zhou X, Carter TE, Li Z, Gibbs MO (2004) A major QTL conditioning salt tolerance in S-100 soybean and descendent cultivars. Theor Appl Genet 109:1610–1619
Li XJ, An P, Eneji AE, Hamamura K, Lux A, Inanaga S (2005) Growth and yield responses of two soybean cultivars to defoliation and water stress. Biologia 60:467–472
Lin S, Cianzio SR, Shoemaker RC (1997) Mapping genetic loci for iron deficiency chlorosis in soybean. Mol Breed 3:219–229
Lin SF, Grant D, Cianzio S, Shoemaker R (2000) Molecular characterization of iron deficiency chlorosis in soybean. J Plant Nutr 23:1929–1939
Liu FL, Andersen MN, Jensen CR (2003) Loss of pod set caused by drought stress is associated with water status and ABA content of reproductive structures in soybean. Funct Plant Biol 30:271–280
Liu F, Andersen MN, Jensen CR (2004) Root signal controls pod growth in drought-stressed soybean during the critical, abortion-sensitive phase of pod development. Field Crops Res 85:159–166
Meer JM, Manly KF, Cudmore RHJ (2002) User manual for map manager QTX. Roswell Park Cancer Institute, Buffalo
Mian MAR, Bailey MA, Ashley DA, Wells R, Carter TE Jr, Parrott WA, Boerma HR (1996) Molecular markers associated with water use efficiency and leaf ash in soybean. Crop Sci 36:1252–1257
Mian MAR, Ashley DA, Boerma HR (1998) An additional QTL for water use efficiency in soybean. Crop Sci 38:390–393
Micic Z, Hahn V, Bauer E, Melchinger AE, Knapp SJ, Tang S, Schon CC (2005) Identification and validation of QTL for Sclerotinia midstalk rot resistance in sunflower by selective genotyping. Theor Appl Genet 111:233–242
Miltner ED, Karnok KJ, Hussey RS (1991) Root response of tolerant and intolerant soybean cultivars to soybean cyst nematode. Agron J 83:571–576
Monneveux P, Ribaut JM (2006) Secondary traits for drought tolerance improvement in cereals. In: Ribaut JM (ed) Drought adaptation in cereals. Haworth Press, USA, pp 97–144
Ni JJ, Wu P, Senadhira D, Huang N (1998) Mapping QTLs for phosphorus deficiency tolerance in rice (Oryza sativa L.). Theor Appl Genet 97:1361–1369
Nyquist WE (1991) Estimation of heritability and prediction of selection response in plant-populations. Crit Rev Plant Sci 10:235–322
Orf JH, Chase K, Adler FR, Mansur LM, Lark KG (1999) Genetics of soybean agronomic traits: II. Interactions between yield quantitative trait loci in soybean. Crop Sci 39:1652–1657
Pantalone VR, Rebetzke GJ, Burton JW, Carter TE (1996a) Phenotypic evaluation of root traits in soybean and applicability to plant breeding. Crop Sci 36:456–459
Pantalone VR, Burton JW, Carter TE (1996b) Soybean fibrous root heritability and genotypic correlations with agronomic and seed quality traits. Crop Sci 36:1120–1125
Pantalone VR, Rebetzke GJ, Burton JW, Carter TE Jr, Israel DW (1999) Soybean PI 416937 root system contributes to biomass accumulation in reciprocal grafts. Agron J 91:840–844
Panthee DR, Kwanyuen P, Sams CE, West DR, Saxton AM, Pantalone VR (2004a) Quantitative trait loci for beta-conglycinin (7S) and glycinin (11S) fractions of soybean storage protein. J Am Oil Chem Soc 81:1005–1012
Panthee DR, Pantalone VR, Sams CE, Saxton AM, West DR, Rayford WE (2004b) Genomic regions governing soybean seed nitrogen accumulation. J Am Oil Chem Soc 81:77–81
Price AH, Tomos AD (1997) Genetic dissection of root growth in rice (Oryza sativa L). 2. Mapping quantitative trait loci using molecular markers. Theor Appl Genet 95:143–152
Richards RA (1996) Defining selection criteria to improve yield under drought. Plant Growth Regul 20:157–166
Samarah NH, Mullen RE, Cianzio SR, Scott P (2006) Dehydrin-like proteins in soybean seeds in response to drought stress during seed filling. Crop Sci 46:2141–2150
Sinclair TR, Purcell LC, King CA, Sneller CH, Chen P, Vadez V (2007) Drought tolerance and yield increase of soybean resulting from improved symbiotic N2 fixation. Field Crops Res 101:68–71
Sloane RJ, Patterson RP, Carter TE (1990) Field drought tolerance of a soybean plant introduction. Crop Sci 30:118–123
Song QJ, Marek LF, Shoemaker RC, Lark KG, Concibido VC, Delannay X, Specht JE, Cregan PB (2004) A new integrated genetic linkage map of the soybean. Theor Appl Genet 109:122–128
Specht JE, Hume DJ, Kumudini SV (1999) Soybean yield potential—a genetic and physiological perspective. Crop Sci 39:1560–1570
Specht JE, Chase K, Macrander M, Graef GL, Chung J, Markwell JP, Germann M, Orf JH, Lark KG (2001) Soybean response to water: a QTL analysis of drought tolerance. Crop Sci 41:493–509
Statistical Analysis System institute (2001) SAS user’s guide: statistics. SAS Inst, Cary
Steel RGD, Torrie JH (1980) Principles and procedures of statistics: a biometrical approach, 2nd edn. McGraw-Hill, New York
Streeter JG (2003) Effects of drought on nitrogen fixation in soybean root nodules. Plant Cell Environ 26:1199–1204
Subashri M, Robin S, Vinod KK, Rajeswari S, Mohanasundaram K, Raveendran TS (2009) Trait identification and QTL validation for reproductive stage drought resistance in rice using selective genotyping of near flowering RILs. Euphytica 166:291–305
Tischner T, Allphin L, Chase K, Orf JH, Lark KG (2003) Genetics of seed abortion and reproductive traits in soybean. Crop Sci 43:464–473
Tuberosa R, Sanguineti MC, Landi P, Michela Giuliani M, Salvi S, Conti S (2002) Identification of QTLs for root characteristics in maize grown in hydroponics and analysis of their overlap with QTLs for grain yield in the field at two water regimes. Plant Mol Biol 48:697–712
Vales MI, Schon CC, Capettini F, Chen XM, Corey AE, Mather DE, Mundt CC, Richardson KL, Sandoval-Islas JS, Utz HF, Hayes PM (2005) Effect of population size on the estimation of QTL: a test using resistance to barley stripe rust. Theor Appl Genet 111:1260–1270
Villagarcia MR, Carter TE, Rufty TW, Niewoehner AS, Jennette MW, Arrellano C (2001) Genotypic rankings for aluminum tolerance of soybean roots grown in hydroponics and sand culture. Crop Sci 41:1499–1507
Vollmann J, Schausberger H, Bistrich H, Lelley T (2002) The presence or absence of the soybean Kunitz trypsin inhibitor as a quantitative trait locus for seed protein content. Plant Breed 121:272–274
Wang SC, Basten J, and Zeng Z-B (2007). Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC. http://statgen.ncsu.edu/qtlcart/WQTLCart.htm
Wang J, McLean PE, Lee R, Goos RJ, Helms T (2008) Association mapping of iron deficiency chlorosis loci in soybean (Glycine max L. Merr.) advanced breeding lines. Theor Appl Genet 116:777–787
Zeng ZB, Kao CH, Basten CJ (1999) Estimating the genetic architecture of quantitative traits. Genet Res 74:279–289
Zhang WP, Shen XY, Wu P, Hu B, Liao CY (2001) QTLs and epistasis for seminal root length under a different water supply in rice (Oryza sativa L.). Theor Appl Genet 103:118–123
Zhang D, Cheng H, Geng LY, Kan GZ, Cui SY, Meng QC, Gai JY, Yu DY (2009a) Detection of quantitative trait loci for phosphorus deficiency tolerance at soybean seedling stage. Euphytica 167:313–322
Zhang XY, Chen SY, Sun HY, Wang YM, Shao LW (2009b) Root size, distribution and soil water depletion as affected by cultivars and environmental factors. Field Crops Res 114:75–83
Acknowledgments
This research was supported by funds allocated to the Georgia Agricultural Experiment Stations and grants from the United Soybean Board.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by I. Rajcan.
Electronic supplementary material
Below is the link to the electronic supplementary material.
122_2010_1500_MOESM2_ESM.ppt
Supplementary Figure 1. Linkage map of Benning x PI416937 RIL population with 267 SSR markers distributed over 20 linkage groups.(PPT 495 kb)
Rights and permissions
About this article
Cite this article
Abdel-Haleem, H., Lee, GJ. & Boerma, R.H. Identification of QTL for increased fibrous roots in soybean. Theor Appl Genet 122, 935–946 (2011). https://doi.org/10.1007/s00122-010-1500-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-010-1500-9