
Abstract
Understanding the host response to Uromyces sp., the causal agent of rust in many crop species, is crucial in elucidating the specific biology of rust resistance. In an attempt to unravel the Medicago truncatula–U. striatus interaction, we performed a global analysis of transcription factor (TF) expression in resistant and susceptible accessions of the model plant M. truncatula during infection with U. striatus. For this purpose, an established qPCR platform was applied, consisting of specific primer pairs for more than 1,000 predicted TF genes. A total of 107 putative TF genes out of the 1,084 studied were differentially expressed. Thirteen of the TFs that were differentially expressed between resistant and susceptible genotypes are known to be relevant in cellular defense. These data suggest that resistance could be mediated both by genes that are constitutively expressed and by genes, which are activated/repressed when plants are inoculated. These defense related TFs sequences were amplified in chickpea DNA with the aim of determining the location of these genes on the genetic map of this crop and identifying possible DNA regions involved in resistance mechanisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Legumes are valuable agricultural and commercial crops that serve as important nutrient sources for both humans and animals. Genetic analysis in cultivated legumes is difficult because of their large genome size, abundant repetitive DNA, complex polyploidy and/or difficulties in the generation of transgenic plants. In order to overcome these limitations, Medicago truncatula (Gaertn) (Barrel medic) is used as a model legume for studying various aspects of legume genomics and biology (Cook 1999; Ané et al. 2008; Rispail et al. 2009). M. truncatula has a small diploid genome, autogamous nature, short generation time, prolific seed production, and close phylogenetic relationship with many crop legumes, such as alfalfa, pea, lentil, chickpea, and faba bean. These facts increase the value of M. truncatula as a resource for understanding the molecular basis of traits of interest in both grain and forage legumes.
Alfalfa rust, incited by Uromyces striatus (J. Schröt), is an important disease with worldwide distribution, not only being particularly damaging in alfalfa (Medicago sativa L.) (Koepper 1942) but also in annual medics (Skinner and Stuteville 1995). Many other rust species of the genus Uromyces are major constraints for grain and forage legumes (Rubiales et al. 2002). Thus, pea rust (U. pisi Pers.) is a major problem in areas that are characterized by warm humid conditions. Faba bean rust (U. viciae-fabae Pers.) causes moderate to substantial yield losses (of up to 70%), in the Middle East and North Africa, particularly when the infection starts early in the season (Rashid and Bernier 1991). Common bean rust (U. appendiculatus Pers.) occurs worldwide but is most prevalent in humid tropical and subtropical areas. Indeed, yield losses can reach 100% and are directly related to earliness and the severity of infection (Statler and McVey 1987). Chickpea rust (U. ciceris-arietini (Grogon) Jacz. & Boyer) is only a severe problem locally in certain regions but is present in almost every region of the world where chickpea is grown (Sillero et al. 2006).
Breeding for rust resistance is the most feasible means of control. Different resistance mechanisms may be operative at different phases of the infection process. In particular, non-hypersensitivity resistance typically manifests as penetration failure before the formation of the first haustorium (Heath 1981). Prehaustorial resistance has been reported in M. truncatula (Rubiales and Moral 2004; Kemen et al., 2005) and this seems to be an effective and durable resistance (Niks and Rubiales 2002). However, the nature of the signaling systems involved in non-hypersensitive response is yet unknown.
Transcriptional control mediated by the activity of transcription factors (TFs) plays an important role in defense gene expression as well as in the regulation of cross talk between different signalling pathways (Lorenzo et al. 2003; Anderson et al. 2004). It may be valuable to elucidate the specific biology of disease resistance in order to identify which TFs are expressed. However, many TFs are transcribed at very low levels. Hence, the identification of transcriptionally regulated TF genes requires sensitive tools. Transcripts quantification by qPCR is more sensitive than DNA array hybridization methods (Czechowski et al. 2004) especially for the quantification of low-abundance transcripts (Horak and Snyder 2002). In an attempt to overcome the problems associated with array-based methods of expression analysis and to provide more comprehensive data on TF expression patterns in M. tuncatula, Kakar et al. (2008) developed a genome guide qPCR-based resource for quantitative measurement of transcripts of 1,084 Medicago TF genes. Specifically, the study of TF regulation by high throughput qPCR should enable us to gain insight into the TFs that may be involved in defense responses and disease development during the M. truncatula–U. striatus interaction.
In this study, we report the screening of 1,084 different TF genes from the M. truncatula TF qPCR platform with the aim of providing an overview of the transcription factor-encoding genes expressed in response to U. striatus infection. Moreover, taking advantage of the synteny between Medicago and Cicer (Cannon et al. 2006), we studied the markers derived from M. truncatula TF genes to chickpea genetic maps in order to examine the relationship of these TFs with genomic regions controlling resistance in chickpea.
Materials and methods
Plant growth and inoculation conditions
Two M. truncatula genotypes were selected according to their reported reaction to rust fungi: A17 (susceptible) and F11.008 (resistant, no hypersensitive response) (Kemen et al., 2005). Seeds were kindly provided by J. M. Prosperi (INRA, Montpellier, France). Twenty plants per line were grown in a growth chamber under controlled conditions (20°C and 12 h day/night). Ten of these plants were inoculated and 10 plants were used as non-inoculated controls. Seedlings were inoculated after the third trifoliate leaf was completely expanded. Urediospores of U. striatus monosporic isolate JE-01 were deriving from a rust population collected on alfalfa in 2001 at Jerez, Spain. Spores were stored in liquid nitrogen, multiplied on alfalfa cv. Baraca seedlings and diluted in pure talcum (1:10), resulting in a spore deposition of 280 spores/cm2 before use. For M. truncatula inoculation, plant leaves were fixed to the soil at horizontal position. Subsequently, the leave adaxial site was inoculated by dispensing 1 mg of freshly harvested urediospores per plant using a spore settling tower. Plants were first incubated in complete darkness for 24 h at 20°C and 100% relative humidity (RH) and then transferred to a growth chamber at 20°C temperature under a 12/12 day/night photoperiod. This experiment was repeated three times, each repetition is considered as one biological replicate.
Sample collection and RNA extraction
Leaves were harvested at 24 h after inoculation. This harvesting time point was chosen because resistance responses occur at the first stage of the infection, the beginning of spore penetration (Rubiales and Moral 2004). Collected samples were immediately frozen in liquid nitrogen and stored at −80°C until RNA extraction.
Total RNAs were extracted from a pool of leaves from both inoculated and non-inoculated plants using TRIZOL reagent (Invitrogen, Carlsbad, CA, USA). The RNA was stored at −80°C until use. RNA quantity and quality was determined spectrophotometrically. About 150 μg total RNA was treated with Turbo DNaseI (Ambion) to eliminate contaminations by genomic DNA. RNA integrity was checked on 1% (w/v) denaturating agarose gels prior to and after DNaseI treatment. Absence of genomic DNA contamination was confirmed by PCR using primers specific for a M. truncatula ubiquitin gene intron sequence (Kakar et al. 2008).
cDNA synthesis
Reverse transcriptions were performed using SuperScript™ III reverse transcriptase (Invitrogen GmbH). The efficiency of cDNA synthesis was assessed by real-time PCR amplification of reference genes encoding MtUbi, and MtGapdh 5′ and 3′ (Kakar et al. 2008). Using the Cq (quantification cycle, Bustin et al. 2009) value of the MtUbi gene, we adjusted all cDNAs to equal concentrations.
Primer design and real-time quantitative PCR (qPCR) assays
A primer collection representing M. truncatula TFs was generated using available genomic databases (Kakar et al. 2008). A total of 1,084 putative TFs gene-specific primers were distributed in three 384-well plates. In order to normalize the data the four reference genes MtUbi, MtGapdh, MtEf1 and MtPdf2 [UBIQUITIN, GADPH (Glyceraldehyde 3 phosphate dehydrogenase), EF1α (Elongation Factor 1 alpha) and PDF2 (Protein Phosphatase 2A Subunit A3)] were repeated twice as technical repetitions.
Polymerase chain reaction was performed as described in Kakar et al. (2008) in an optical 384-well plate with ABI PRISM® 7900 HT sequence detection System (Applied Biosystems, Foster City, CA, USA), using SYBR® Green to monitor dsDNA synthesis.
Data were analyzed using the SDS program 2.3 software (Applied Biosystems) as described in Kakar et al. (2008). Amplification reactions with efficiencies lower than 1.6 were eliminated from the analysis. In addition, we discarded reactions with more than one melting curve peak (approximately 27.9% of the total reactions (3640/13008). Efficiency values were taken into account in all subsequent calculations, including the calculation of transcript level ratios.
Prior to qPCR normalization, the four reference genes were analyzed concerning their expression levels in the analyzed cDNA samples. Reference genes with most stable expression levels reference were selected using Repeated Pair-Wise Correlation Analysis using Bestkeeper©-version 1 software (Pfaffl et al. 2004). Expression values were estimated by ECq of each individual plot. Ratios against the geometric mean of the constitutive control were used to normalize gene expression between different biological conditions using the formula: (E (Cq hk)hk )/(E (Cq gene)gene ). The ratio of the experimental control condition (non-inoculated plants) was set up to 1 as a reference to determine relative induction/repression factors.
Statistical analyses and clustering of TFs
Factorial analysis of variance (ANOVA) was performed to analyze gene profiles for each TF following the model:
where y ijk is the expression value of ith genotype for the jth treatment in the kth repetition, μ the general mean effect, G i the genotype effect, T j the treatment effect, R k the repetition effect, GT ij the interaction genotype-treatment effect and e ijk the effect of experimental error.
TFs showing significant differences between genotypes, treatment, or for the interaction genotype-treatment, were clustered using a hierarchical cluster analysis. A data matrix with the expression ratio inoculate/non-inoculate for each genotype for the TFs showing differential expression was used to calculate a Euclidean distance matrix. UPGM method was employed to generate a dendrogram using Cluster v3.0 (Eisen et al. 1998) and visualized using TreeView v1.0.5 software.
Searching orthologous TF genes between M. truncatula and Cicer arietinum
A C. arietinum (chickpea) RIL population derived from an interspecific cross Cicer arietinum (ILC72) × C. reticulatum (Cr5-9) (Cobos et al. 2006) was used for genomic DNA extraction. PCR reactions were carried out in 10 μl reactions containing 20–40 ng of plant genomic DNA, buffer (50 mM KCl, 10 mM Tris–HCl, 0.1% Triton X-100), 2.5 mM MgCl2, 250 μM of dNTPs, 0.6 μM of primer and 1 unit of Taq DNA polymerase (Promega). Genomic DNA of M. truncatula (A17) was used as control. After denaturing the DNA during 5 min at 94°C the reaction mixture was subjected to 40 cycles of the following temperature profile: 94°C for 30 s, 59°C for 30 s and 72°C for 30 s followed by a final extension at 72°C for 5 min. Amplification products were analyzed in 12% acrylamide gels and stained with ethidium bromide.
Results
M. truncatula phenotype after infection with U. striatus
Leaves for qPCR profiling were harvested at 2 dpi. Rust symptoms were not yet visible at the time the leaves were sampled for nucleic acid extraction. Infection was later verified on the remaining leaves of inoculated M. truncatula plants (Fig. 1). The susceptible line displayed well formed pustules, whereas the resistant line had no symptoms.

Macroscopic comparison between the accessions used in the study. a Susceptible line (A17), b Resistant line (F11.008)
Analysis of TF expression profile
We used the qPCR platform to identify differentially expressed TF genes in two M. truncatula lines with different levels of tolerance to U. striatus, during infection with this pathogen. RNA was extracted from inoculated and non-inoculated plants from both M. truncatula genotypes, and cDNA for qPCR was synthezised. Four reference genes (MtUbi, MtGapdh, MtPdf2, MtEf1) were assessed. MtEf1 showed the worst correlation factor compared to the other three reference genes (r = 0.31–0.36), hence it was excluded from the analysis. We then have normalized data to the geometric mean of the relative transcript levels of the remaining three reference genes (MtUbi, MtGapdh, MtPdf2).
It was found that 28.3% (307/1084) of the TF-encoding transcripts could not be amplified. Most (83.7%, 670/777) of the amplified transcripts did not show significant differences between genotypes (susceptible and resistant), treatment (inoculated and non-inoculated), or differences due to genotype-treatment interaction, suggesting that they are constitutively expressed. However, it was found that 16.3% of amplified transcripts (107/777) were differentially expressed between inoculated and non-inoculated or between resistant and susceptible genotypes. About half (59/107) of the differentially expressed TF-encoding transcripts displayed differences only between treatments (inoculated/non-inoculated). A few of them (9/107) presented significant differences only between genotypes while the remaining genes (39/107) showed a genotype–treatment interaction, that is, the genotype response depended on the treatment.
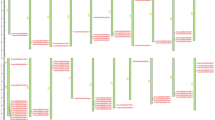
Cluster analysis of these differentially expressed TF-encoding transcripts divided these genes into two groups with different expression profiles (Fig. 2a). In the first (Cluster I), the gene expression in the resistant line was activated and in the second (Cluster II) it was repressed. We further divided these two groups into subgroups; cluster I was separated in six sub-clusters and cluster II in three (Fig. 2b).

a Heat-maps of TF expression profiles for each cluster. Down-regulation is indicated in green, up-regulation in red, normalized expression values close to the mean are in black and no detectable expression is in grey. b Averaged profiles of mRNA abundance in inoculated accessions. The Log2 of the normalized expression values and the ratio between Inoculated/non-inoculated genotypes was calculated for classification into clusters. For each cluster the median of these values in each line is represent in graphic form (±SD), sowing that each cluster has a characteristic change in each line. The number of TFs present in each cluster is indicated. The blue column is the susceptible line (A17) and the red one is the resistant line (F11.008)
Subclusters I.1, I.2 and I.3 contained 75 TFs upregulated in both susceptible and resistant inoculated genotypes. This transcription activation was stronger in the resistant line, showing a higher transcription level in subcluster I.1 (Fig. 2b). Twenty-four of these TFs were Zn-Finger family members. In addition, these clusters included four sequences with similarities to WRKY proteins, three similar to pathogenesis-related transcription factors and ethylene response factor (PR-ERF) and seven related to Myb DNA-binding proteins. The remaining 37 TFs in these clusters belonged to many different families (including 4 non-apical meristem, 2 bZip, 2 Ankyrin, 2 MADS box, 1 Helix-Turn-Helix, 1 Jumonji, or cyclin-like). Sub-clusters I.4 and I.5 included TFs-encoding transcripts upregulated in the resistant line and downregulated in the susceptible one. They consisted of 17 TFs, six of them were Zn-Finger family members, a PR-ERF, a homeodomain-like (ATPase, related with cold tolerance), a ZPT2-12, a Helix-turn-Helix TF and other sequences codifying unknown proteins. Cluster I.6 contains five TFs, two Zn-Fingers family members, two Myb-like TFs and sequences with similarities with unknown proteins. These TFs transcripts were upregulated in both genotypes with higher transcription levels in the susceptible genotype.
Within Cluster II, three subgroups were noted. A total of 13 TF-encoding transcripts were identified in clusters II.1, II.2 and II.3, most of them belonging to the Zn-Finger and MYB families. Cluster II.1 contained only one TF, a sequence similar to C2H2 Zn-finger type that was down regulated in both genotypes. Clusters II.2 and II.3 included genes down regulated in F11.008 and upregulated in A17.
We had focused our study on TFs that showed significant differences between genotypes or that displayed genotype-treatment interaction, because these TF genes were likely to be related to the plant defense responses (Table 1). Expression levels of these TFs differed between genotypes. We detected five PR-ERF (Rauscher et al. 1999). Three of them had higher transcription levels in the resistant line F11.008 (936.m00021, 1102.m00008, BF644125), one was repressed in the susceptible genotype (A17) and activated in the resistant line (1082.m00015), while the last was activated in A17 genotype and repressed in F11.008 (AL374794). Moreover, one WRKY family member transcription factor (1287.m00032) (Eulgem and Somssich 2007) exhibited a three-fold difference in transcript abundance in the resistant line (Table 1). Another TF involved in plant defense response was ZPT2-12 (1245.m00006) (Sugano et al. 2003) which was two-fold repressed in the resistant line. Finally, six MYB proteins were studied. Two (AL369166, 731.m00111) out of the six MYB TFs were present in cluster I.2 (Fig. 2); they were upregulated in both genotypes, but the transcription level was higher in the susceptible line. Two of these genes (TC96646, 1063.m00010) were included in cluster II.3 and one (TC101530) in cluster II.2 (Fig. 2); these were activated in the resistant and repressed in the susceptible line. The last MYB TF family member (1070.m00005) was located in the I.6 cluster, and showed a higher transcription level in the resistant genotype (Fig. 2).
Identifying novel defense response markers in chickpea
With the aim of identifying new markers associated with defense response in the chickpea genomic maps, we used primers for the M. truncatula TFs listed in Table 1. All of them produced amplification products using chickpea DNA, except primers for 1287.m00032 and 1102.m00008. Two of them (TC101530 y 936.m00021) showed two bands following amplification of the chickpea genome, which could be a gene duplication (Fig. 3). This needs to be studied in more detail. Only two of these markers were polymorphic in the chickpea RIL population (1070.m00005, TC101530). Linkage analysis revealed that both markers were located on linkage group 4 (LG4) of the chickpea map (Fig. 4).

Amplification products obtained from chickpea using Medicago truncatula TFs primers. The gel showed the amplification of five primers in two chickpea genotypes: ILC72 (lanes 2, 5, 8, 11 and 14) and Cr5-10 (lanes 3, 6, 9, 12 and 15) and M. truncatula “A17” (lanes 1, 4, 7, 10 and 13) as positive control. Numbers on the left are the molecular weight marker (φ×174 digested with HaeIII) (Lane M). Lanes correspond with the following primers: lanes 1–3 primers for 936.m00021; lanes 4–6 primers for 1070.m00005; lanes 7–9 primers for TC101530; lanes 10–13 primers for AL374794 and lanes 14–15 primer for AL369166

Linkage group 4 from the interspecific cross Cicer arietinum (ILC72) × Cicer reticulatum (Cr5-10). Marker names are shown on the right and the estimated map distances are shown on the left. Recombinant fractions were converted to centiMorgans using Kosambi mapping function. TFs genes positions are in boldface. The QTLAR1 and QTLAR2 regions are indicated as shaded areas
Discussion
This study represents the first high throughput real-time quantitative RT-PCR profiling of legume transcription factors during a defense process. This large-scale TF profiling approach is more sensitive and robust for monitoring changes in TF expression than the use of microarrays (Czechowski et al. 2004; Kakar et al. 2008), but requires larger amounts of RNA. Another difference between the microarray and large-scale quantitative real-time PCR approaches is that the genes targeted by the qPCR resource used in this study were selected from the International Medicago Genome Annotation Group (IMGAG) genes. Specifically, these are sequences genes which have been annotated (including potential annotation errors), while the probes contained in the microarray were mostly designed from ESTs and TC sequences, corresponding to highly expressed genes (Czechowski et al. 2004). In addition, oligo-based microarray hybridization is more prone to cross-hybridization than PCR-derived techniques.
F11.008 showed prehaustorial resistance against U. striatus infection. Prehaustorial resistance is typical of non-host interactions (Heath 2000) but is also present in so-called Partial Resistance (Rubiales and Niks 1995) and is regarded as a relatively durable type of resistance (Niks and Rubiales 2002). Most studies suggest that host compatibility requires the fungus to be able to avoid prehaustorial defenses within the substomatal cavity of the host leaf, breach the mesophyll plant cell wall to form the first haustorium and develop a biotrophic interaction with the living invaded cell to support further fungal growth (Schulze-Lefert and Panstruga 2003; Glazebrook 2005; Spanu 2006). In the resistant line employed in this study, fungal development stopped immediately before formation of the haustorial mother cells without necrosis of cells in contact with the fungus (Castillejo et al. 2010). Studying non-host resistance is the key to deciphering complex plant defense mechanisms against pathogens. This kind of resistance seems to be based on multiple defense mechanisms, as previously shown in Arabidopsis mutants inoculated with the nonpathogenic fungus U. vignae (Mellersh and Heath 2003) as well as in Hordeum vulgare cultivars inoculated with P. triticina (Neu et al. 2003).
Most research conducted into the interactions of Uromyces sp. has been focused on the impact of the disease in the field. We still know relatively little about the cellular and molecular aspects of these interactions. Understanding the transcriptional regulation of defense-associated genes is important for the improvement of disease resistance mechanisms in plants (Dangl and Jones 2001). In this study, a total of 1,084 M. truncatula putative genes were analyzed, 107 (16%) were differentially expressed. Fifty-nine out of the 107 TFs showed significant differences between treatments in both resistant and susceptible genotypes. These changes should, therefore, be regarded as general responses to inoculation. A minor subset (8.4%, 9/107) of the genes showed significant differences in expression between resistant and susceptible genotypes, suggesting that they are constitutive genes which may be related to resistance response. Four of these 9 TFs (TC102139, TC101530, AL374794, and AL369166) are implicated in known defense system pathways (Table 1). They are members of the PR-ERF and Myb TF families and have been shown to bind to promoter elements of defense-related genes, regulating their expression (Eulgem 2005). There were 10 TFs (BF644125, 1102.m00008, 936.m00021, AL374794, 1082.m00015, 1287.m00032, 1063.m00010, 1304.m00003, 1070.m00005, 731.m00111) that showed significant differences for the interaction between genotype and treatment (inoculation/non-inoculation). It is plausible to suggest that the expression of these TFs was a specific response to the infection with U. striatus. They are members of the PR-ERF, Myb and WRKY transcription factors families and consequently are related to stress response (Eulgem 2005).
The PR-ERF transcription factor family is unique to plants. They can bind to a GCC box, which is found in several PR (Pathogenesis-Related) gene promoters and confers ethylene responsiveness. The RNA levels of specific ERF genes are regulated by cold, drought, pathogen infection, wounding or treatment with ethylene, salicylic acid (SA) or jasmonic acid (JA) (Singh et al. 2002). Most of the ERF proteins whose transcriptional properties have been studied are activators of transcription, although some of them can also repress transcription (Fujimoto et al. 2000). Our results showed that in general after inoculation PR-ERF TFs pattern had higher transcription levels in the resistant genotype than in the susceptible one (Table 1). However, one of them (1082.m00015) was repressed in the resistant and activated in the susceptible genotype.
Other TFs families that showed patterns which differed between the genotypes were Myb family members (1070.m00005, 1063.m00010, 731.m00111, AL369166, TC96646, TC101530). These proteins constitute a diverse class of DNA-binding proteins of particular importance in transcriptional regulation in plants. The range of involvement of MYB transcription factors in controlling different aspects of plant gene expression is not fully understood. It is clear that a significant number of these MYB transcription factors are involved in the detailed regulation of secondary metabolism (particularly phenylpropanoid metabolism). However, some studies show that these proteins play an important role in hypersensitive response and the development of systemic acquired resistance (SAR) (Yang and Klessig 1996).
Finally, WRKY transcription factors are also unique to plants. Some WRKY members show enhanced expression and/or DNA-binding activity following induction by a range of pathogens, defense signals, and wounding (Eulgem et al. 2000). Transcriptome analyses have revealed that, in particular, putative binding sites of WRKY factors (W box) and related sequence motifs are ubiquitously conserved in the upstream regions of genes up regulated during SAR, R-mediated resistance and basal defense. Hence, WRKY factors appear to play a major role in transcriptional reprogramming during a variety of immune responses (Eulgem 2005). In this study, the WRKY TFs were activated during the M. truncatula–U. striatus interaction and differentially expressed in resistant and susceptible genotypes.
Microsynteny analysis comparing M. truncatula genome sequences revealed colinearity at different synteny blocks (Cannon et al. 2006). This microsynteny could be used to localize genes in genetic maps of closely related species. Amplification analysis of the tested primers showed high transferability between both M. truncatula and C. arietinum; 11 out of 13 primers tested produced amplification bands in chickpea. Our preliminary results from linkage analysis showed that the TFs 1070.m00005 and TC120209 were located on LG4. In this linkage group, two Quantitative Traits Loci (QTLs) for ascochyta blight resistance have been reported (Tekeoglu et al. 2002; Iruela et al. 2006). Our results showed that TF 1070.m00005 was on the QTLAR1 genomic region and TC101530 was on QTLAR2 genomic region (Fig. 4). These TFs could be candidates for future experiments to elucidate the genes that control this agronomically important trait.
In conclusion, we have identified a collection of TF genes encoding known or putative transcription factors that are differentially expressed during the interaction between M. truncatula and U. striatus. However, the roles they play in the interaction need to be clarified in order to completely understand the pathways involved in the plant’s defense against this pathogen. Further functional studies, such as RNAi analysis, are required to understand the role of these TF networks. Stress perception and signaling pathways are critical components of the adaptive response of plants that allows them to survive under environmental constraints. General-stress responses may arise from the activities of various TFs interacting among distinct biotic stress response pathways (Dangl and Jones 2001). Identification of these TFs is a crucial to understanding transcriptional control of plant defense and designing new strategies to create resistant cultivars in other legume crops. This knowledge is important for the production of plants transformed with resistance genes and in the development of diagnostic markers to differentiate resistant and susceptible genotypes.
References
Anderson JP, Badruzsaufari E, Schenk PM, Manners JM, Desmond OJ, Ehlert C, Maclean DJ, Ebert PR, Kazan K (2004) Antagonistic interaction between abscisic acid and jasmonate-ethylene signaling pathways modulates defense gene expression and disease resistance in Arabidopsis. Plant Cell 16:3460–3479
Ané JM, Zhu H, Frugoli J (2008). Recent advances in Medicago truncatula genomics. Int J Plant Genom. doi:10.1155/2008/256597
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622
Cannon SB, Sterck L, Rombauts S, Sato S, Cheung F, Gouzy J, Wang X, Mudge J, Vasdewani J, Schiex T, Spannagl M, Monaghan E, Nicholson C, Humphray SJ, Schoof H, Mayer KF, Rogers J, Quétier F, Oldroyd GE, Debellé F, Cook DR, Retzel EF, Roe BA, Town CD, Tabata S, Van de Peer Y, Young ND (2006) Legume genome evolution viewed through the Medicago truncatula and Lotus japonicas genomes. Proc Natl Acad Sci USA 103:14959–14964
Castillejo MA, Susín R, Madrid E, Fernández-Aparicio M, Jorrín JV, Rubiales D (2010) 2-DE based proteomic analysis of the Medicago truncatula–rust (Uromyces striatus) interaction. Ann Appl Biol. doi:10.1111/j.1744-7348.2010.00422.x
Cobos MJ, Rubio J, Strange RN, Moreno MT, Gill J, Millan T (2006) A new QTL for Ascochyta blight resistance in an RIL population derived from an interspecific cross in chickpea. Euphytica 149:105–111
Cook DR (1999) Medicago truncatula: a model in the making! Curr Opin Plant Biol 2:301–304
Czechowski T, Bari RP, Stitt M, Scheible WR, Udvardi MK (2004) Realtime RT-PCR profiling of over 1400 Arabidopsis transcription factors: unprecedented sensitivity reveals novel root- and shoot-specific genes. Plant J 38:366–379
Dangl JL, Jones JDG (2001) Plant pathogens and integrated defence responses to infection. Nature 411:826–833
Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA 95:14863–14868
Eulgem T (2005) Regulation of the Arabidopsis defense transcriptome. Trends Plant Sci 10:71–78
Eulgem T, Somssich IE (2007) Networks of WRKY transcription factors in defense signaling. Curr Opin Plant Biol 10:366–371
Eulgem T, Rushton PJ, Robatzek S, Somssich IE (2000) The WRKY super family of plant transcription factors. Trends Plant Sci 5:199–206
Fujimoto SY, Ohta M, Usui A, Shinshi H, Ohme-Takagi M (2000) Arabidopsis ethylene-responsive element binding factors act as transcriptional activators or repressors of GCC box-mediated gene expression. Plant Cell 12:393–404
Glazebrook J (2005) Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu Rev Phytopathol 43:205–227
Heath MC (1981) Resistance of plants to rust infection. Phytopathology 71:971–974
Heath MC (2000) Nonhost resistance and nonspecific plant defenses. Curr Opin Plant Biol 3:315–319
Horak CE, Snyder M (2002) Global analysis of gene expression in yeast. Funct Integr Genomics 2:171–180
Iruela M, Rubio J, Barro F, Cubero JI, Millan T, Gil J (2006) Detection of two quantitative trait loci for resistance to ascochyta blight in an intra-specific cross of chickpea (Cicer arietinum L.): development of SCAR markers associated with resistance. Theor Appl Genet 112:278–287
Kakar K, Wandrey M, Czechowski T, Gaertner T, Scheible WR, Stitt M, Torres-Jerez I, Xiao Y, Redman JC, Wu HC, Cheung F, Town CD, Udvardi MK (2008) A community resource for highthroughput quantitative RT-PCR analysis of transcription factor gene expression in Medicago truncatula. Plant Meth 4:18
Kemen E, Hahn M, Mendgen K, Struck C (2005) Different resistance mechanisms of Medicago truncatula ecotypes against the rust fungus Uromyces striatus. Phytopathology 95:153–157
Koepper JM (1942) Relative resistance of alfalfa species and varieties to rust caused by Uromyces striatus. Phytopathology 32:1048–1057
Lorenzo O, Piqueras R, Sanchez-Serrano JJ, Solano R (2003) ETHYLENE RESPONSE FACTOR1 integrates signals from ethylene and jasmonate pathways in plant defense. Plant Cell 15:165–178
Mellersh DG, Heath MC (2003) An investigation into the involvement of defense signaling pathways in components of the nonhost resistance of Arabidopsis thaliana to rust fungi also reveals a model system for studying rust fungal compatibility. Mol Plant Microbe Interact 16:398–404
Neu C, Keller B, Feuillet C (2003) Cytological and molecular analysis of the Hordeum vulgare–Puccinia triticina nonhost interaction. Mol Plant-Microbe Interact 16:626–633
Niks RE, Rubiales D (2002) Detection of potentially durable resistance mechanisms in plants to specialised fungal pathogens. Euphytica 124:201–216
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP (2004) Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol Lett 26:509–515
Rashid KY, Bernier CC (1991) The effect of rust on yield of faba bean cultivars and slow-rusting populations. Can J Plant Sci 71:967–972
Rauscher M, Adam AL, Wirtz S, Guggenheim R, Mendgen K, Deising HB (1999) PR-1 protein inhibits the differentiation of rust infection hyphae in leaves of acquired resistant broad bean. Plant J 19:625–633
Rispail N, Kaló P, Kiss GB, Ellis THN, Gallardo K, Thompson RD, Prats E, Larrainzar E, Ladrera R, González EM, Arrese-Igor C, Ferguson BJ, Gresshoff PM, Rubiales D (2009) Model legumes to contribute to Faba bean breeding. Field Crops Res. doi:10.1016/j.fcr.2009.03.014
Rubiales D, Moral A (2004) Prehaustorial resistance against alfalfa rust (Uromyces striatus) in Medicago truncatula. Eur J Plant Pathol 110:239–243
Rubiales D, Niks RE (1995) Characterization of Lr34, a major gene conferring nonhypersensitive resistance to wheat leaf rust. Plant Dis 79:1208–1212
Rubiales D, Emeran AA, Sillero JC (2002) Rusts on legumes in Europe and North Africa. Grain Legumes 37:8–9
Schulze-Lefert P, Panstruga R (2003) Establishment of biotrophy by parasitic fungi and reprogramming the host cells for disease resistance. Annu Rev Phytopathol 41:641–667
Sillero JC, Fondevilla S, Davidson J, Vaz Patto MC, Warkentin TD, Thomas J, Rubiales D (2006) Screening techniques and sources of resistance to rusts and mildews in grain legumes. Euphytica 147:255–272
Singh KB, Foley RC, Oñate-Sánchez L (2002) Transcription factors in plant defense and stress responses. Curr Opin Plant Biol 5:430–436
Skinner DZ, Stuteville DL (1995) Host range expansion of the alfalfa rust pathogen. Plant Dis 79:456–460
Spanu PD (2006) Why do some fungi give up their freedom and become obligate dependants on their host? New Phytol 171:447–450
Statler GD, McVey MA (1987) Partial resistance to Uromyces appendiculatus in dry edible beans. Phytopathology 77:1101–1103
Sugano S, Kaminaka H, Rybka Z, Catala R, Salinas J, Matsui K, Ohme-Takagi M, Takatsuji H (2003) Stress-responsive zinc finger gene ZPT2–3 plays a role in drought tolerance in petunia. Plant Journal 36(6):830–841
Tekeoglu M, RajeshPN PN, Muehlbauer FJ (2002) Integration of sequence tagged microsatellite sites to the chickpea genetic map. Theor Appl Genet 105:847–854
Yang Y, Klessig DF (1996) Isolation and characterization of a tobacco mosaic virus-inducible myb oncogene homolog from tobacco. Proc Nat Acad Sci USA 93:14972–14977
Acknowledgments
Financial support by FP6-2002-FOOD-1-506223 European Union Integrated Project and by Spanish projects AGL2005-07497-CO2-01/AGR and AGL2008-01239 is acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by H. T. Nguyen.
Rights and permissions
About this article
Cite this article
Madrid, E., Gil, J., Rubiales, D. et al. Transcription factor profiling leading to the identification of putative transcription factors involved in the Medicago truncatula–Uromyces striatus interaction. Theor Appl Genet 121, 1311–1321 (2010). https://doi.org/10.1007/s00122-010-1390-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-010-1390-x