
Abstract
Sugarcane mosaic virus (SCMV) is an important virus pathogen both in European and Chinese maize production, causing serious losses in grain and forage yield in susceptible cultivars. Two major resistance loci confer resistance to SCMV, one located on chromosome 3 (Scmv2) and one on chromosome 6 (Scmv1). We developed a large isogenic mapping population segregating in the Scmv2, but not the Scmv1 region, to minimize genetic variation potentially affecting expression of SCMV resistance. We fine mapped Scmv2 to a region of 0.28 cM, covering a physical distance of 1.3426 Mb, and developed six new polymorphic SSR markers based on publicly available BAC sequences within this region. At present, we still have three recombinants left between Scmv2 and the nearest polymorphic marker on either side of the Scmv2 locus. The region showed synteny to a 1.6 Mb long sequence on chromosome 12 in rice. Analysis of the public B73 BAC library as well as the syntenic rice region did not reveal any similarity to known resistance genes. However, four new candidate genes with a possible involvement in movement of virus were detected.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sugarcane mosaic virus (SCMV) is an important virus pathogen both in European and Chinese maize production, causing serious losses in grain and forage yield in susceptible cultivars (Fuchs and Grüntzig 1995). SCMV belongs together with maize dwarf mosaic virus (MDMV), Johnsongrass mosaic virus (JGMV), sorghum mosaic virus (SrMV), and Zea mosaic virus (ZeMV) to the sugarcane mosaic subgroup of the Potyviridae, all being maize pathogens (Kuntze et al. 1995; Seifers et al. 2000). The diagnostic symptoms of SCMV include stunting, chlorosis, reduction in plant biomass, and, therefore, a reduction in grain and forage yield. Chemical control of SCMV and its vectors is not possible due to the non-persistent mode of virus transmission by aphids. Hence, the most efficient method to control SCMV infections is the cultivation of resistant maize varieties.
Kuntze et al. (1997) screened 122 early maturing European maize inbreds under both greenhouse and field conditions. Three lines (D21, D32, and FAP1360A) displayed complete resistance to SCMV and MDMV and these lines were used for further analysis. Using the cross between the resistant D32 and the susceptible line D145, three minor QTL on chromosomes 1, 5, and 10, as well as two major dominant genes on chromosomes 3 and 6 were identified (Melchinger et al. 1998; Xia et al. 1999). High-resolution mapping using progeny from the cross between FAP1360A (resistant) and F7 (susceptible) confirmed that Scmv1 on the short arm of chromosome 6 and Scmv2 near the centromere of chromosome 3 are essential for expression of complete resistance to SCMV (Xu et al. 1999). The Scmv1 region was later shown to contain most likely two closely linked resistance genes (Dussle et al. 2003; Yuan et al. 2003). Minor resistance QTL identified in the cross between D32 and D145 (Xia et al. 1999) seem to be fixed in F7 genetic background, as the major resistance genes on chromosomes 3 and 6 were sufficient to confer multiple potyvirus resistance when introgressed into the susceptible line F7 (Lübberstedt et al. 2006; Xing et al. 2006).
Resistance to SCMV, MDMV, and WSMV in US germplasm has been investigated in a number of studies (Redinbaugh et al. 2004). Inbred Pa405 has shown complete resistance to MDMV and SCMV both under field and greenhouse conditions (Louie et al. 1990) and up to five genes have been reported to be involved in resistance (Mikel et al. 1984; Rosenkranz and Scott 1984). A major gene, Mdm1, causing resistance to MDMV in Pa405, was mapped near the nor region on chromosome 6 (McMullen and Louie 1989; Simcox et al. 1995). A recent study by Jones et al. (2007) confirmed that Mdm1 is associated with resistance to MDMV in most inbreds, but that loci on chromosomes 3 and 10 also affect resistance in some lines. QTL for SCMV resistance on chromosomes 3, 6, and 10 were also found in Chinese germplasm (Zhang et al. 2003). Although clusters of QTL involved in resistance to several viruses have been found on chromosomes 3 and 6 (Wisser et al. 2006), it is still uncertain whether Scmv resistance genes are identical to other virus resistance genes such as Mdm1, mapping to the same genome regions (Quint et al. 2002). Ultimately, isolation of the respective resistance genes will solve this question.
In previous studies (Xu et al. 1999, Dussle et al. 2002, 2003, Yuan et al. 2003, 2004) the density of markers in the Scmv1 and Scmv2 genome regions was substantially increased to more than two markers per cM. This usually is a good starting point for map-based gene isolation. The major limitations so far were suboptimal populations for high-resolution mapping. In F2 and BC populations, incomplete penetrance and escapes obscure the relationship between resistance genotype and phenotype, necessary for reliable genetic fine mapping, due to simultaneous segregation of at least two resistance genes and the presence of heterozygotes with high levels of plasticity in their resistance response. Depending on environmental conditions, heterozygotes might show similar infection levels as either the homozygous resistant or susceptible inbred lines, complicating establishment of close associations between marker genotype and resistance phenotype as required for high-resolution mapping. Moreover, fine mapping and map-based gene isolation in standard mapping populations such as F2 or BC1, can be impaired by unlinked duplicated sequences (Quint et al. 2003; Frisch et al. 2004).
To minimize the impact of incomplete penetrance, phenotypic plasticity, and segregation outside the Scmv2 genome region, this region was fine mapped in isogenic genetic background fixed for the resistance allele at Scmv1. In this genetic constellation, the susceptible homozygote at the Scmv2 locus can be reliably distinguished from the genotypes carrying one or two Scmv2 resistance alleles after virus infection (Xing et al. 2006).
The objectives of this study were (a) to develop a high-resolution mapping population segregating exclusively in the Scmv2 region, (b) to assign Scmv2 to a physical contig, (c) to develop new SSR markers in the Scmv2 region to be used for genetic fine mapping of Scmv2, and (d) to evaluate synteny of this Scmv2 region to rice genomic sequence.
Materials and methods
Plant materials
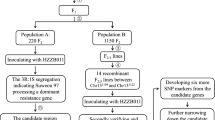
The early maturing European maize inbred lines FAP1360A (completely resistant to SCMV) and F7 (highly susceptible to SCMV) (Kuntze et al. 1997) were crossed to produce F1 offspring and were then backcrossed seven times to F7 (Dussle et al. 2003). The homozygous line F7RR/RR, was produced by three subsequent selfing steps starting from one SCMV-resistant BC7 plant. The letters left of the slash refer to the genotype at Scmv2 on chromosome 3; the letters right of the slash refer to the genotype at Scmv1 on chromosome 6. Presence of the Scmv1 and Scmv2 genome regions in F7RR/RR was confirmed by 116 SSR markers (Lübberstedt et al. 2006, unpublished data). F7RR/RR was proven to be SCMV resistant in several field and greenhouse experiments (Shi et al. 2005; Lübberstedt et al. 2006; Xing et al. 2006).
Further isogenic lines were developed from the isogenic line pair F7 and F7RR/RR. Two Scmv2-flanking SSR markers on chromosome 3 (bnlg1456 and bnlg1035) and four SSR markers in the Scmv1 genome region on chromosome 6 (bnlg161, bnlg1432, bnlg1600, and phi077) (Dussle et al. 2003), were used to screen F2 individuals derived from the cross of F7 × F7RR/RR. Selected F2 plants were selfed to produce the sublines F7RR/SS and F7SS/RR with resistance alleles (Scmv2, Scmv1) fixed in one Scmv QTL region (RR), and with F7 alleles (scmv2, scmv1) fixed in the second region (SS) (Xing et al. 2006). F2 plants from the cross F7RR/RR and F7SS/RR were used as mapping population and for screening of rare recombinants of Scmv2.
Virus inoculation and scoring
Plants were sown in the greenhouse in fully fertilised peat in 0.8 l pots. The inoculation sap was made by homogenising young leaves with typical mosaic symptoms of SCMV-infected susceptible F7 plants in five volumes of a 0.01 M phosphate buffer, pH 7.0, using a household hand blender. Carborundum was added to the sap and plants were artificially inoculated by rub inoculation at the 3-leaf stage. Plants that did not show symptoms 1 week after the first infection were inoculated a second time.
DNA extraction and PCR
Small parts of young leaves were harvested and immediately frozen in liquid nitrogen. DNA was extracted by a CTAB and phenol/chloroform extraction using an AutoGenprep 740 DNA isolation machine (AutoGen) and re-suspended in 0.2 ml R40 [40 mg/ml RNase A (Sigma-Aldrich) in TE pH 8.0]. Apart from new markers developed from public BAC sequences, all primer sequences for SSR markers were obtained from the Maize GDB (http://www.maizegdb.org). PCR amplification reactions for each of the SSR markers were performed in a 10 μl reaction volume containing 25 ng of genomic DNA, 1 μl of 10× PCR buffer, 2 mM MgCl2, 0.2 mM of each dNTP (Fermentas), 0.1 μM of each forward and reverse primer and 0.5 U Taq polymerase (Fermentas). PCR amplification was performed in a MJ Research PTC-225 Peltier Thermal Cycler (MJ Research Inc., VWR International ApS, Rødovre, Denmark) using a touchdown profile designed for the annealing temperature (T a) of the primer pair: initial denaturation, followed by 12 cycles of 30 s at 94°C, 1 min at T a + 12°C and 1 min at 72°C with a reduction of the annealing temperature of 1°C at each cycle, followed by 30 cycles of 30 s at 94°C, 1 min at T a and 1 min at 72°C, followed by a final extension. The forward primer was fluorescently labelled for detection on a Megabace™ 1000 96 capillary electrophoresis system (Amersham Biosciences, Piscataway, NJ, USA). Allele scoring was done using the software Megabace Genetic Profiler version 2.0 (Amersham Biosciences, Piscataway, NJ, USA).
Marker development
The SSR marker umc1300 was not functioning in our mapping population, as the F7 band was very weak and could not be detected in the heterozygotes. We, therefore, converted it into a cleaved amplified polymorphic sequence (CAPS) marker (umc1300caps) using the primers umc1300capsF (5′-ACCACCAGGTGTCCTTCCTT-3′) and umc1300capsR (5′-GTTGCAGCAGACGAAGAAGA-3′), with product sizes of 484 and 486 bp for F7 and FAP1360A, respectively. Primers were designed using Primer3 (http://frodo.wi.mit.edu/primer3). Restriction enzyme NsiI (New England BioLabs) cuts the FAP1360A product into two fragments of 140 and 346 bp.
SSR marker DJF001 was developed from a SSR sequence (electronic SSR no cshr00530) found in one of the BAC ends of ZMMBBb0220F13. To develop further SSR markers in the target region around Scmv2, sequences from publicly available BACs (http://www.genome.arizona.edu/fpc/maize) from the region between ZMMBBB0220F13 and bnlg1601 in contig 124 on chromosome 3 were obtained from the NCBI homepage (http://www.ncbi.nlm.nih.gov) and analysed for SSR sequences using MISA (MIcroSAtellite identification tool) developed by Thiel et al. (2003) (http://pgrc.ipk-gatersleben.de/misa). SSR search conditions were set to a minimum of six repeats of dibasic motifs, and four repeats of three to six basic motifs. The maximum distance between two SSRs was set to 50 bases. Once SSRs were found, the surrounding sequence was scanned for repetitive elements by blasting at NCBI (http://www.ncbi.nlm.nih.gov) against Zea mays nucleotide sequences. Flanking sequences without repetitive elements were used for marker development. Primers flanking the SSRs were designed using Primer3 (http://primer3.sourceforge.net/) to obtain a product size of approximately 200–300 bp. The markers were named DJF002–009.
F2 mapping population for initial mapping of Scmv2
186 F2 plants from the cross F7RR/RR and F7SS/RR were used as unselected mapping population. Plants were inoculated twice as described above in April 2005. Virus symptoms were recorded at weekly intervals at seven dates and SCMV resistance was scored as dominant marker. The following markers were mapped: CAPS marker umc1300caps as well as the SSR markers bnlg1035, bnlg1456, bnlg1601, phi053, umc1102, umc1174, umc1750. Marker orders and genetic distances were calculated with JoinMap (version 3.0.2.2; Van Ooijen and Voorrips 2001) using Kosambi’s mapping function and a LOD score of 3. However, the group was unchanged up to a LOD score of 9. The map was drawn with MapChart (Voorrips 2002).
Development of a susceptible F2 sub-population for fine mapping of Scmv2
More than 4,100 F2 plants from the cross between F7RR/RR and F7SS/RR were tested for SCMV susceptibility. Each lot of approximately 500 plants included 10 F7SS/RR and 10 F7RS/RR plants as control. The first half of the plants were tested in May–July 2005, the second half in May–June 2007. Plants were scored daily from the day of first infection until symptoms appeared in the heterozygous F7RS/RR plants. All plants showing symptoms before symptoms were visible in heterozygous F7RS/RR control plants were considered to have the genotype F7SS/RR and were chosen for further analysis. When looking for rare recombinations between marker and resistance locus, the number of plants that can be analysed is a limiting factor. By performing marker analyses exclusively on susceptible plants, we were able to analyse four times as many plants. The plants were numbered in order of symptoms appearance, making it possible to test by the markers DJF008 and bnlg1601, whether plants showing symptoms the day before symptoms were found in the F7RS/RR control plants also had the F7RS/RR genotype.
Recombinant screen and fine mapping in the susceptible sub-population
A total of 1,046 susceptible plants, including 43 plants from the mapping population, were analysed to find recombinants. The markers DJF001 and DJF008 co-segregated in the unselected mapping population (data not shown). As the physical position of DJF008 is 100–200 kbp downstream from DJF001, we used DJF008 together with bnlg1601 when screening for recombinants. To make the recombinant screen as effective as possible, a part of the susceptible plants were analysed as bulks of three plants. Bulks heterozygous at markers DJF008 or bnlg1601 were de-convoluted for further analysis.
The map distances were calculated by determining the percentage of recombinants among 1,046 susceptible plants (2,092 gametes). Values were not corrected for double crossovers. The map distance between umc1300, DJF001, and DJF008 were calculated on the non-bulked sub-fraction of the population (463 susceptible plants/926 gametes). The map was drawn with MapChart.
Minimal tiling path
Already sequenced BAC clones were used as basis to build the minimal tiling path between markers DJF003 and bnlg1601. Coordinates were taken from http://www-maizesequence.org. As the sequences of the BAC clones are not yet fully assembled, the exact position of the markers within BACs is not known, for which reason markers were placed in the centre of the BAC clones.
Maize ESTs
All sequenced public available BAC clones between the SSR markers DJF003 and bnlg1601 (c0483H04, c0087I08, c0427A03, b0239F02, c0078M13, c0023O19, b0645C1, c0281K07, c0067E08, b0175P06) were blasted against B73 ESTs in the EST database at NCBI. As the BAC clones are not fully sequenced, each partial BAC sequence was treated separately. ESTs with identities of 98–100% were used for further analyses. However, ESTs with 98% similarity were only included, if a substantial part of the sequence showed 100% identity. Based on the NCBI unigene collection, ESTs were assigned to unigenes where possible.
Synteny to rice
We used the NCBI unigene database to find homologues in rice for the unigenes mentioned above, as the database showed the highest ‘selected protein similarity’ to rice proteins. Using syntenic genes at each end of the maize BAC contig, we determined a region from rice chromosome 12 for which we extracted all predicted proteins from the NCBI Map viewer rice map (http://www.ncbi.nlm.nih.gov). The gene ontology (GO) is a controlled, species-neutral vocabulary of defined terms representing gene product properties. The ontology covers three domains; (a) cellular component that describes locations, at the levels of subcellular structures and macromolecular complexes, (b) biological process that describes series of events accomplished by one or more ordered assemblies of molecular functions, and (c) molecular function that describes activities, such as catalytic or binding activities, that occur at the molecular level (http://www.geneontology.org). Some proteins had a GO id given at the NCBI homepage. For other proteins it was possible to assign a GO id by blasting the GO page (http://amigo.geneontology.org/cgi-bin/amigo/blast.cgi). The GO id of the best plant hit was used, based on a cut off of 1.0e −10. For simplicity, only the top terms for Molecular Function and Biological Processes are given.
Results
Mapping Scmv2 in an unselected F2 population
SCMV symptoms were found from 15 to 27 days after the first inoculation in 43 out of 186 plants, giving a ratio of resistant: susceptible phenotypes of 3.3 : 1 in agreement with dominant inheritance of Scmv2. Using the markers bnlg1035, bnlg1456, bnlg1601, phi053, umc1102, umc1174, umc1750, and umc1300caps, Scmv2 was located between umc1300caps and bnlg1601 (Fig. 1a). The distance between umc1300 and Scmv2 was calculated to be 1.1 cM, whereas the distance between Scmv2 and bnlg1601 was 0.3 cM. The physical distance between both markers is 5,164,600 bp, resulting in a ratio of 368,900 bp/0.1 cM. As other SSR markers found in this area given by MaizeGDB were either monomorphic in our mapping population or not working in our lab, we developed new SSR markers between umc1300 and bnlg1601.

a Mapping of Scmv2 to bin 3.05 with eight SSR markers from MaizeGDB. Scmv2 is located in a 1.4 cM region between umc1300 and bnlg1601. Marker order and genetic distances based on 186 F2 plants were calculated with JoinMap. b Fine mapping of Scmv2 with six newly developed SSR markers. The marker DJF004 co-segregates with Scmv2. The distances to both the closest next markers were 0.14 cM. The map distance between umc1300, DJF001, and DJF008 were calculated on 463 susceptible plants (926 gametes). For all other markers, the map distances were calculated by determining the percentage of recombination among 1,046 susceptible plants (2,092 gametes). Values were not corrected for double crossovers. Both maps were drawn with MapChart
Marker development
SSR marker DJF001 was based on an electronic SSR found in one of the BAC ends of BAC clone ZMMBBb0220F13 (Table 1). Since Scmv2 was located downstream from this marker, we developed new SSR markers in the region between DJF001 and bnlg1601.
Publicly available maize BACs were found in the AGI Agarose FPC Map (http://www.genome.arizona.edu/fpc/maize) and BAC sequences were obtained from GenBank at NCBI (http://www.ncbi.nlm.nih.gov). We found from 5 to 33 SSR sequences in the analysed BAC sequences. However, many had to be discarded due to the repetitive nature of surrounding sequences. Primers for the remaining putative SSR markers were tested by PCR, and well performing markers were analysed with capillary electrophoresis using forward primers labelled with FAM, HEX, or TET. In total, we developed two monomorphic and seven polymorphic SSR markers (Table 1). The SSR marker DJF008 was found both in ZMMBBb0220F13 and ZMMBBc0417C18, supporting the expected overlap of these two BAC clones. The SSR marker DJF002 was developed based on the BAC clone ZMMBBb0531A01. This BAC is probably misplaced within contig 124, as it mapped outside the Scmv2 region introgressed from FAP1360A into F7RR/RR. Due to this, DJF002 was omitted from further analysis.
Development and analysis of a large sub-population of susceptible F2 plants
We analysed more than 4,100 plants for susceptibility to SCMV and found 1,003 plants that showed symptoms before the heterozygous F7RS/RR control plants. These, together with 43 plants from the mapping population, formed our F2 sub-population of 1,046 susceptible F2 plants.
Individual plants or bulks of three plants from the susceptible sub-population were analysed with the SSR markers DJF008 and bnlg1601 to find plants having a recombination between these two markers, flanking the Scmv2 locus. Bulks that were positive for the donor allele (from the resistant genotype FAP1360A) of either of the two markers were de-convoluted, and plants genotyped individually. Individual plants carrying a donor allele at either of the two flanking makers were analysed with DJF003, DJF004, DJF005, DJF009 in addition to DJF008 and bnlg1601. A total of 23 recombinants were found between DJF008 and bnlg1601, four of which showed a heterozygous genotype for all markers. These plants were suspected to be heterozygous for Scmv2 showing incomplete penetrance of resistance, and were excluded from further analyses. However, they might result from double-cross over events very close to the Scmv2 gene and will be re-evaluated after identification of Scmv2.
The marker DJF004 co-segregated with Scmv2, whereas the distances to the closest next markers were 0.14 cM (DJF003) and 0.14 cM (bnlg1601) (Fig. 1b). At present, three recombinants are left on either side of the Scmv2 locus, three between DJF003 and the co-segregating marker DJF004 and three between the co-segregating DJF004 and bnlg1601 (Fig. 1b). According to the physical map at http://www.maizesequence.org, our region of interest of 0.28 cM covers a physical distance of 1,342,600 bp, resulting in a ratio of 479,500 bp/0.1 cM or 0.21 cM/Mb. The order of markers was in agreement with the B73 physical map.
Minimal tiling path
The exact position of SSR markers in most BAC clones was unknown, as the sequences of the clones were given in several unordered pieces. For this reason, markers are placed in the centre of BAC clones (Fig. 2b). Thus, our region of interest (Scmv2 region) at the current stage of genetic analysis spans from the beginning of BAC clone c0483H04 (including DJF003) to the end of b0239F02 (including bnlg1601), covering a distance of 1.3426 Mb (Fig. 2a, c). A minimal tiling path over this region consists of a total of 12 BAC clones, of which 10 have already been sequenced.

Minimal tiling path between the SSR markers DJF003 and bnlg1601. a The total length in base pairs. b Above the line is shown the position of SSR markers on chromosome 3. Markers in solid boxes are polymorphic whereas markers in dashed boxes are monomorphic. Number of recombinations and distances in cM is given below the line. c Sequenced BAC clones are given in black whereas unsequenced BAC clones are grey. Four possible candidate genes found in maize are shown as stars. d The 1.58 Mb syntenic region on rice chromosome 12. The position of the 14 unigenes with equivalent 13 rice locations are shown as circles. Forty-four other rice genes are shown as triangles. Two possible candidate genes found in rice are shown as closed grey triangles, marked by arrows
Maize ESTs
To analyse the number and nature of expressed sequences present in the Scmv2 region, we blasted the individual pieces of sequence from the ten sequenced BACs against Z. mays ESTs at the NCBI database. This resulted in 161 maize sequences with a 98–100% identity to known maize ESTs. The ESTs could be assembled into 20 unigenes, whereas 6 sequences did not belong to a unigene at present (Table 2). Twenty unigenes in the 1.3426 Mb region equals an average of 67 kb per gene. However, the 20 unigenes were not equally distributed among BACs. The number of unigenes per BAC varied from zero (c0078M13) to six (c0087I08) (Table 2), giving a density per gene containing BAC clone ranging from 28 to 176 kb/gene, calculated as kb/BAC divided by number of genes/BAC. Only two well characterized genes, glutathione synthetase and auxin-binding protein1, were found among the unigenes, located at the overlap of BAC clones c0023O19/b0645C18 and b0645C18/c0281K07, respectively. The transcripts behind three unigenes were given a number by the Maize Mapping Project (Zm.81138/CL9003_1; Zm.92679/PCO079612; Zm.74126/ PCO121171; Gardiner et al. 2004) and, thus, are anchored to the BAC contig map.
Synteny to rice
For 14 out of the 20 unigenes, the NCBI unigene database showed the highest ‘selected protein similarity’ to rice proteins found on rice chromosome 12 (Table 2). From these, two different maize unigenes gave the same rice hit, indicating a duplication of this gene in maize. One unigene had no hit in rice and five unigenes had their highest hit on rice chromosomes 1 (three unigenes), 6, and 10 (Table 2). Using the 14 unigenes with hits for 13 equivalent rice locations (circles on Fig. 2d), a syntenic rice region of approx. 1.58 Mb was located at position 19.96–21.54 Mb on rice chromosome 12 (http://www.ncbi.nlm.nih.gov/mapview). According to NCBI, this rice region contains 90 protein expressing genes. Of these 90 proteins, 9 sequences were detected by RepeatMasker, 14 were listed as ‘codon recognized by tRNA scan’, and 3 were designated non-protein coding transcripts. These 26 predicted proteins were omitted from further analyses together with 7 hypothetical proteins that were predicted with EST support. This left 57 proteins (circles or triangles in Fig. 2d) for further analyses. Twenty-five proteins had a GO id given at the NCBI homepage. For ten proteins it was possible to assign a GO id by blasting at the GO page. For each of these ten proteins, the GO id of the best plant hit was used, using a cut off of 1.0e −10. Most of the proteins with a Molecular Function id were found in the categories Binding (18 proteins) and Catalytic Activity (14 proteins), whereas the rest belonged to Transporter Activity (4), Molecular Transducer Activity (2), Structural Molecule Activity (2), Antioxidant Activity (1) and Regulator Activity (1) (Fig. 3a). One of the 35 proteins with an id had no GO id for Molecular Function. Eighteen proteins belonged to the category Cellular Processes, 16 of these also belonged to Metabolic Processes (Fig. 3b). The rest fell into the categories Localization (5), Response to Stimulus (4), Biological Regulation (3), Developmental Process (2), Multicellular Organismal Process (1) and Reproduction (1) (Fig. 3b). Twelve of the 35 proteins had no GO id for biological processes.

Gene ontology identification. a Thirty-four out of 35 maize proteins had GO ids for Molecular Function. In total, 42 GO ids were identified, as 8 proteins had GO ids belonging to 2 different categories. b Twenty-three out of 35 maize proteins had GO ids for Biological Process. The 16 proteins in the Metabolic Process category also belonged to the Cellular Process category. Five other proteins belonged to 3 or more categories, giving a total of 50 GO ids
We aligned the maize and the rice sequences based on the common ESTs (Fig. 2c, d). The best alignment was obtained, when placing the rice region in opposite direction as the maize region. However, the middle part of one of the sequences has been inverted, containing a gene cluster positioned around 20.8 Mb in rice. As at least two genes, one on each side of the gene poor regions, are involved in the inversion, the breaking points seem to have occurred in the gene rich region. Two out of 14 syntenic maize genes were not found in the expected order, when compared to rice.
Candidate genes
Four maize transcripts can be considered as candidate genes for Scmv2, based on their possible role in resistance response (marked by a star in Fig. 2c). One is a heat shock protein located in b0175P06, one is a Uso1/p115 like vesicle tethering protein located in c0483H04, and the last two are a Rac GTPase activating protein, and a syntaxin/t-SNARE containing protein, located in c0281K07 and c0067E08, respectively. The syntaxin/t-SNARE protein was missing in the respective rice chromosome 12 region, whereas the other three candidate genes, the heat shock protein precursor, the Rac GTPase activating protein, and the vesicle tethering protein were present. We identified two new putative candidate genes in the rice region, encoding a NBS-LRR protein and a protein containing a protein kinase domain (closed grey triangles in Fig. 2d). Both of these genes were absent from the B73 region, but might be found in other maize lines, such as FAP1360A. As the sequenced maize BAC clones still contained gaps, we cannot completely rule out that these genes are absent from B73, although these gaps often contain repetitive regions without genes. In addition, three proteins assigned GO id Response to Stimulus might be considered as further candidate genes. One of the three proteins involved in response to stimulus is a SCF ubiquitin ligase complex protein involved in response to ethylene. The other two are coding for a protein involved in oxidative stress, and a protein involved in brassinosteroid mediated signalling, as possible response to pathogens. None of these three proteins seems to be present in the Scmv2 region of the SCMV susceptible inbred B73.
Discussion
Development of a mapping population for Scmv2 and SSR marker development
In earlier studies, F2 and BC populations were used to map the Scmv2 locus. In these populations, incomplete penetrance and escapes obscured the accuracy of genetic fine mapping due to the plasticity of heterozygotes. We divided the digenic inherited SCMV resistance found in F7RR/RR into two separate, “monogenic” populations. Fixing the Scmv1 resistance allele meant that the susceptible homozygote at the Scmv2 locus could be more reliably distinguished from the genotypes carrying one or two Scmv2 resistance alleles after virus infection (Xing et al. 2006). For co-dominant markers, recessive plants have about twice the information content of dominant plants (Hühn and Piepho 2003). As the phenotyping of SCMV-resistant plants is obtained relatively fast and cheap, we decided to focus on identification of highly susceptible recessive individuals initially, to fine map the Scmv2 locus (Huehn and Piepho 2006). In addition, we pooled plant samples to identify rare recombinants within the interval of 1.4 cM between the initially used Scmv2 flanking markers umc1300 and bnlg1601. These two strategies were efficient to shorten the region of interest from more than 5 to 1.3 Mb.
The environmental plasticity of Scmv2 heterozygotes was confirmed in this study. When infected with SCMV, F7SS/RR plants consistently showed symptoms within few weeks, whereas F7RR/RR plants were always found to be resistant. For the heterozygous F7RS/RR plants, the susceptibility was dependent on the environment and time of infection. In cooler periods or with later infection, these plants were delayed in symptom expression. This suggests that SCMV resistance mediated by Scmv2 can be considered as environment-dependent threshold character.
Consistent with other studies (e.g., Qiu et al. 2007), BAC end sequences were generally not valuable to develop new markers due to their repetitive nature. This is not surprising, as 77% of the maize genome is composed of LTR retrotransposons, DNA transposons, and tandem repeats (Meyers et al. 2001). This large amount of repetitive sequences also hampered our development of SSR markers from sequenced B73 BAC clones, as 60–70% of the putative SSR markers had to be discarded even before primer testing due to the repetitive nature of the surrounding sequences.
Genetic analysis of the Scmv2 region
In average, the maize genome has a ratio of genetic to physical distance of 2.1 cM/Mb (Yao et al. 2002). In our study, we found a ratio of 0.21–0.27 cM/Mb in the Scmv2 genome region (0.21 cM/Mb between DJF003 and bnlg1601and 0.27 cM/Mb between umc1300 and bnlg1601). Thus, this region can be considered as a recombination cold spot. In our 1.3426 Mb region, we found 20 unigenes and 6 ESTs. The number of 20 unigenes is an approximation as there might be rare transcripts in this region not found in the EST search. More proteins with predicted motifs or function were found by the maize sequence database. However, these were reduced to 20 unigenes and 6 ESTs, when all sequences of putative repetitive nature were removed. It is unclear, whether the six sequences are rare transcripts or gene fragments. An average of one gene per 67 kb found in our study is close to 65 kb/gene found in another genome region of maize (Liu et al. 2007). Thus, the lack of recombination might be due to proximity to the centromere rather than to low-gene density, as the recombination frequency in centromere areas is reported to be low (Fengler et al. 2007).
The gene density for rice is approx. 7 kb/gene, which is much higher than in maize (Feng et al. 2002; The Rice Chromosome 3 Sequencing Consortium 2005). However, Bennetzen et al. (2004) argue that there is often a mis-annotation of fragments of long-terminal repeat retrotransposons, leading to an over-estimation of gene numbers. The NCBI homepage listed 90 genes coding for rice proteins in the syntenic region, about 4× more than we found in the syntenic maize genome region. However, many of these are hypothetical proteins. We found an inversion when comparing the Scmv2 region in maize and the syntenic region in rice. Such local inversion events can hamper identification of syntenic regions (Vision 2005). However, as the inversion occurred inside the region, we were still able to detect synteny between maize and rice. In maize, repetitive sequences are located within this cluster, as the BAC clone c0078M13 with no EST hits, is inserted between two closely linked genes in rice. In rice, there is an area of low-gene density on both sides of the gene cluster at 20.8 Mb. These regions probably contain repetitive sequences. Interestingly, the region in rice is larger than the maize region (1.58 Mb in rice compared to 1.34 Mb in maize) even though the size of the rice genome is only approx. 389 Mb compared to the 2,500 Mb of maize (Arumuganathan and Earle 1991; International Rice Genome Sequencing Project 2005). This indicates that there are multiple repetitive sequences in this region of the rice genome or that there might have been deletions during the evolution of the maize region, maybe as a part of the inversion event (Fig. 2). Fourteen unigenes out of 20 were found in the syntenic region and only 2 out of these 14 genes were not found in the expected order, when the big inversion was taken into account. These two genes might have been moved around after the diversion of rice and maize. Based on this study, it seems that synteny to rice can be used to search for new candidate genes, although some genes found in rice were either lost or translocated in maize.
The publicly available sequences from B73 BACs were of enormous importance for the progress in our study. We used the available sequence to develop new markers and to search candidate genes within our region of interest. It is well known, that map-based cloning in maize might be impaired by allelic differences between maize inbred lines with respect to insertions and deletions of genes (Brunner et al. 2005). However, most of the differences might be due to partial genes or pseudogenes, carried by transposons, as shared genes are usually conserved in gene order and location relative to rice. When comparing the order of the markers between B73 and our own mapping population, all markers were located in the same order. Thus, a positional cloning strategy that relies on rice as a scaffold is not likely to be affected by inbred variation (Bortiri et al. 2006). One obstacle in our search for possible candidate genes based on the public available B73 sequence was that B73 is susceptible to SCMV. As the nature of the Scmv2 resistance gene is not known, we cannot say, whether this susceptibility is due to a mutated or missing gene. To circumvent this, we developed a BAC library based on the resistance line FAP1360A (Ingvardsen et al. 2005) and are in the process of building a FAP1360A BAC contig covering the Scmv2 region. Sequence information from this contig will verify whether or not any of the new putative candidate genes found in rice is also located within the Scmv2 region in the resistant line FAP1360A.
Virus resistance genes
Plants defend virus infection in several different ways. The best-characterized mechanism of plant antiviral defence is mediated by resistance (R) genes (Soosaar et al. 2005). These genes often result in extreme resistance (ER), where virus replication does not occur or occurs at essentially undetectable levels in infected cells, or in a hypersensitive response (HR) by which the infected cells are killed by programmed cell death (PCD). N and its homologue NH are TIR-NBS-LRR genes conferring virus resistance in tobacco (Whitham et al. 1994; Stange et al. 2004), whereas many other antiviral R genes, Sw5 from tomato and tobacco (Brommonschenkel et al. 2000; Spassova et al. 2001), HRT and RCY1 from Arabidopsis (Cooley et al. 2000; Takahashi et al. 2002), RT4-4 from common bean (Seo et al. 2006), Rx1 and Rx2 from potato (Bendahmane et al. 1999, 2000), and Tm-2 and Tm-2 2 from tomato (Lanfermeijer et al. 2003) are CC-NBS-LRR genes. NBS-LRR genes are also believed to be involved in potyvirus resistance. The CC-NBS-LRR gene 3gG2 is a good candidate for the major RSV1 gene mediating resistance to soybean mosaic virus in Soybean (Hayes et al. 2004). Further, the TIR-NBS-LRR gene Y-1, although not conferring resistance, induces a HR to PVY in potato (Vidal et al. 2002). Interestingly, so far no NBS-LRR gene providing virus resistance has been isolated from monocots. As no HR is found in SCMV resistance, and as the virus seems to be able to replicate and to some extend move inside the mesophyll of resistant plants (Lei and Agrios 1986; Pokorný and Porubová 2006), we do not believe that SCMV resistance is R gene mediated, although one NBS-LRR genes was found in the synthenic region in rice. The two NBS-LRR genes mapped by Xiao et al. (2007) to the maize bin 3.05 can be located to B73 BAC clones in contig 121 and contig 125 and can, therefore, be excluded as candidates for Scmv2, which is located also in bin 3.05 but in contig 124.
Viruses encode a very small number of genes and are, therefore, dependent on host factors to complete their life cycle. Mutations in these host factors will result in recessive inherited virus resistance. The translation initiation factor eIF-4E has repeatedly been identified as a naturally occurring recessive resistance gene. Loss-of-susceptibility mutants conferring resistance towards potyviruses due to changes in eIF-4E or its isoform have been found in lettuce, Arabidopsis, pepper, and pea (Lellis et al. 2002; Nicaise et al. 2003; Gao et al. 2004; Kang et al. 2005). Although first believed to be specific against viruses belonging to the family Potyviridae, mutated versions of eIF-4E and eIF4G have also been found to provide resistance in other families such as Carmovirus, Cucumovirus, Bymovirus, and Sobemovirus (Yoshii et al. 2004; Stein et al. 2005; Albar et al. 2006; Nieito et al. 2006). No translation initiation factor was found in the syntenic region in rice. In maize, eIFiso-4E is found on chromosome 5 and eIF-4E is located in contig 124 on chromosome 3. However, the gene is found 500 kb upstream from the Scmv2 region, and can thus be ruled out as candidate for Scmv2. We did not find any sequence in the BAC clones in the Scmv2 region that indicated the presence of any translation initiation factor. As Scmv2 is a dominant resistance gene, it is in our opinion unlikely, that eIF-4E/G or their isoforms are candidates for Scmv2.
Plant viruses need to be able to move from cell-to-cell between mesophyll cells as well as into and out of phloem tissue (long distance movement). Viruses are believed to hijack mechanisms that shuttle proteins and/or RNA to and/or through plasmodesmata (Scholthof 2005; Lucas 2006). Modifications of proteins in any of the involved mechanisms might be a way for plants to gain resistance towards plant viruses that ‘piggy-back’ the long distance transport system inside the plants. It has been found that SCMV is able to enter the plant cell and to replicate, whereas it cannot move systemically in the resistant genotypes (Lei and Agrios 1986; Pokorný and Porubová 2006). Cell-to-cell movement of the virus within the infected leaves was, although not completely blocked, slowed down in resistant lines. As it is difficult to analyse phloem loading and long distance movement independently of cell-to-cell transport between mesophyll cells, one can only guess that phloem loading might be more tightly restricted than cell-to-cell transport and thus also involve different barriers for the virus to overcome. Whether the lack of long distance movement is due to lack of delivery to the plasmodesmata pore or the phloem entry, or exit is not known. Phloem entry and exit might require different mechanisms, making plants highly capable to control macromolecular transport (Zhu et al. 2002). As cells in immature leaves seem to have a very high-size exclusion limit, the virus might have free access to infect immature leaves once it has gained access to the phloem (Oparka et al. 1999). This might explain why SCMV is only spreading upwards to young tissue whereas no mosaic symptoms are found below the infected leaves. Several different pathways for virus trafficking to and through plasmodesmata have been suggested, depending on the type of virus involved (Lucas 2006). Potyviruses are transported between cells as vRNA, probably assisted by code protein (CP) and the helper component-proteinase (HC-Pro) (Cronin et al. 1995; Dolia et al. 1995). However, the cylindrical inclusion protein (CI) and the potyvirus genome linked protein VPg have also been reported to be involved in virus movement (Schaad et al. 1997; Carrington et al. 1998). The RTM system from Arabidopsis is the only known example where resistance is due to control of virus movement in plants. The system involves two genes, RTM1, a jacalin repeat domain protein, and RTM2, a small heat shock protein, and together they control the restriction of long distance movement of the potyvirus tobacco etch virus (Whitham et al. 2000; Chrisholm et al. 2000). Both genes are expressed in sieve elements, but the molecular resistance mechanism is not understood (Chrisholm et al. 2001). Both might function as chaperones facilitating cell-to-cell protein transport. However, none of the 20 candidate genes remaining in the 1.3 Mb B73 region is an obvious candidate gene matching any of the above described mechanisms. Special attention should, however, be given to proteins that might be involved in regulation of long distance trafficking of macromolecules. Chaperones, kinases, Rho/Rac like GTPases and t-SNAREs are all involved in the movement of macromolecular complexes and can, therefore, be regarded as candidate genes (Oparka 2004; Scholthof 2005; Caviston and Holzbaur 2006). The heat shock protein, the Rac GTPase activating protein and the syntaxin/t-SNARE containing protein found in this work are all candidate genes that might be involved in movement of macromolecular complexes. Proteins that function at the plasmodesma docking sites might also be candidate genes. Uso1/p115 tethering proteins seem to be involved in ER-to-Golgi transport (Kang and Staehelin 2008), but whether or not this is important for transport of macromolecules and/or virus to and through the plasmodesmata is not clear. Two well characterized genes, glutathione synthetase and auxin-binding protein1, are located in the Scmv2 region. Nothing indicates that any of these genes are involved in resistance against virus. Finally, we cannot rule out that additional genes will be found in the allelic genome region from FAP1360A, and that susceptibility of B73 to SCMV is due to lack of the Scmv2 gene, as described for other pathosystems (Brooks et al. 2006). In conclusion map-based isolation based on FAP1360A sequence will be required to finally pinpoint Scmv2.
Pleiotropy versus clustering of resistance genes
Studies on SCMV resistance in three different genetic backgrounds all gave the same QTL for Scmv2 on chromosome 3 (D32 × D145, Xia et al. 1999; FAP1360A × F7, Dussle et al. 2000; Huangzao4 × Ye107, Zhang et al. 2003). Due to the fact that the Scmv2 QTL was detected at a later stage of plant development in the cross between FAP1360A × F7 compared to D32 × D145, Dussle et al. (2000) suggested that the Scmv2 resistance genes from D32 and FAP1360A are different. However, these two resistance genes might have a common origin (Xu et al. 2000). As we did not find any indications of a gene family being involved in SCMV resistance, likely only one gene is involved. QTL in the chromosome 3 region conferring resistance to other members of the Potyviridae family have been reported for WSMV and MDMV (McMullen et al. 1994; Jones et al. 2007). The NIL F7RR/RR used as a genetic basis for this study was also found to be resistant against MDMV and ZeMV (Xing et al. 2006). At this moment it is still unknown whether the gene involved in resistance to SCMV also gives resistance to other potyviruses. However, as some parts of the CP of potyviruses are rather conserved (Shukla and Ward 1989) and this protein seems to be involved in virus movement, it is possible that one complex of host genes can transport a range of potyviruses.
A cluster of virus resistance QTL near the centromere on chromosome 3 is also involved in resistance to the waikavirus maize chlorotic dwarf virus (MCDV), the geminivirus maize streak virus (MSV), the nucleorhabdovirus maize mosaic virus (MMV), the tenuivirus maize stripe virus (MStV), as well as the high plain virus (HPV)/WSMV disease complex (Ming et al. 1997; Welz et al. 1998; Marçon et al. 1999; Jones et al. 2004; Dintinger et al. 2005; Wisser et al. 2006). Many of these studies used very few markers, giving rather broad QTL regions, covering several million base pairs. It is, therefore, unclear whether resistance against all these different viruses is due to one pleiotropic gene or a cluster of virus resistance genes. Most likely, protein complexes are involved in systemic virus transport. The individual (and maybe interchangeable) components of such a complex might be responsible for resistance towards different virus.
Future perspectives
We are currently in the process of map-based isolation of Scmv2. This will help to resolve the question of pleiotropy versus linkage by testing appropriate genotypes with Scmv2 and performing virus tests against multiple viruses. Discovery of the Scmv2 resistance gene would make it possible to design reliable functional markers within the resistance gene itself (Andersen and Lübberstedt 2003). Using such markers, breeders could screen large populations of young plants to confirm presence of Scmv2, without need for inoculation of the plants, As resistance against potyvirus is believed to be digenic (or even polygenic in some populations), additional markers will be required for Scmv1 and any other potyvirus resistance genes to provide complete resistance by marker-assisted selection.
References
Albar L, Bangratz-Reyser M, Hébrard E, Ndjiondjop MN, Jones M, Ghesquière A (2006) Mutations in the eIF(iso)4G translation initiation factor confer high resistance of rice to rice yellow mottle virus. Plant J 47:417–426
Andersen JR, Lübberstedt T (2003) Functional markers in plants. Trends Plant Sci 8:554–560
Arumuganathan K, Earle RD (1991) Nuclear DNA content of some important plant species. Plant Mol Biol Rep 9:208–218
Bendahmane A, Kanyuka K, Baulcombe DC (1999) The Rx gene from potato controls separate virus resistance and cell death responses. Plant Cell 11:781–791
Bendahmane A, Querci M, Kanyuka K, Baulcombe DC (2000) Agrobacterium transient expression system as a tool for the isolation of disease resistance genes: application to the Rx2 locus in potato. Plant J 21:73–81
Bennetzen JL, Coleman C, Liu R, Ma J, Ramakrishna W (2004) Consistent over-estimation of gene number in complex plant genomes. Cur Opin Plant Biol 7:732–736
Bortiri E, Jackson D, Hake S (2006) Advances in maize genomics: the emergence of positional cloning. Curr Opin Plant Biol 9:164–171
Brommonschenkel SH, Frary A, Frary A, Tanksley SD (2000) The broad-spectrum Tospovirus resistance gene Sw-5 of tomato is a homolog of the root-knot nematode resistance gene Mi. Mol Plant Microbe Interact 13:1130–1138
Brooks SA, Huang L, Herbel MN, Gill BS, Brown-Guedira G, Fellers JP (2006) Structural variation and evolution of a defense-gene cluster in natural populations of Aegilops tauschii. Theor Appl Genet 112:618–626
Brunner S, Fengler K, Morgante M, Tingey S, Rafalski A (2005) Evolution of DNA sequence nonhomologies among maize inbreds. Plant Cell 17:343–360
Carrington JC, Jensen PE, Schaad MC (1998) Genetic evidence for an essential role for potyvirus Cl protein in cell-to-cell movement. Plant J 14:393–400
Caviston JP, Holzbaur ELF (2006) Microtubule motors at the intersection of trafficking and transport. Trends Cell Biol 16:530–537
Chrisholm ST, Mahajan SK, Whitham SA, Yamamoto ML, Carrington JC (2000) Cloning of the Arabidopsis RTM1 gene, which controls restriction of long-distance movement of the tobacco etch virus. Proc Natl Acad Sci USA 97:489–494
Chrisholm ST, Parra MA, Anderberg RJ, Carrington JC (2001) Arabidopsis RTM1 and RTM2 genes function in phloem to restrict long-distance movement of tobacco etch virus. Plant Physiol 127:1667–1675
Cooley MB, Pathirana S, Wu HJ, Kachroo P, Klessing DF (2000) Members of the Arabidopsis HRT/RPP8 family of resistance genes confer resistance to both viral and Oomycete pathogens. Plant Cell 12:663–676
Cronin S, Verchot J, Haldeman-Cahill R, Schaad MC, Carrington JC (1995) Long-distance movement factor: a transport function of the potyvirus helper component proteinase. Plant Cell 7:549–559
Dintinger J, Verger D, Caiveau S, Risterucci AM, Gilles J, Chiroleu F, Courtois B, Reynaud B, Hamon P (2005) Genetic mapping of maize stripe disease resistance from the Mascarene source. Theor Appl Genet 111:347–359
Dolia VV, Haldeman-Cahill R, Montgomery AE, Vandenbosch KA, Carrington JC (1995) Capsid protein determinants involved in cell-to-cell and long distance movement of tobacco etch potyvirus. Virology 206:1007–1016
Dussle CM, Melchinger AE, Kuntze L, Stork A, Lübberstedt T (2000) Molecular mapping and gene action of Scm1 and Scm2, two major QTL contributing to SCMV resistance in maize. Plant Breed 119:299–303
Dussle CM, Quint M, Xu ML, Melchinger AE, Lübberstedt T (2002) Conversion of AFLP fragments tightly linked to SCMV resistance genes Scmv1 and Scmv2 into simple PCR-based markers. Theor Appl Genet 105:1190–1195
Dussle CM, Quint M, Melchinger AE, Xu M, Lübberstedt T (2003) Saturation of two chromosome regions conferring resistance to SCMV by targeted BSA. Theor Appl Genet 106:485–493
Feng Q, Zhang Y, Hao P, Wang S, Fu G, Huang Y, Li Y, Zhu J, Liu Y, Hu X, Jia P, Zhang Y, Zhao Q, Ying K, Yu S, Tang Y, Weng Q, Zhang L, Lu Y, Mu J, Lu Y, Zhang LS, Yu Z, Fan D, Liu X, Lu T, Li C, Wu Y, Sun T, Lei H, Li T, Hu H, Guan J, Wu M, Zhang R, Zhou B, Chen Z, Chen L, Jin Z, Wang R, Yin H, Cai Z, Ren S, Lv G, Gu W, Zhu G, Tu Y, Jia J, Zhang Y, Chen J, Kang H, Chen X, Shao C, Sun Y, Hu Q, Zhang X, Zhang W, Wang L, Ding C, Sheng H, Gu J, Chen S, Ni L, Zhu F, Chen W, Lan L, Lai Y, Cheng Z, Gu M, Jiang J, Li J, Hong G, Xue Y, Han B (2002) Sequence and analysis of rice chromosome 4. Nature 420:316–320
Fengler K, Allen SM, Li B, Rafalski A (2007) Distribution of genes, recombination, and repetitive elements in the maize genome. Plant Genome 2:S83–S95
Frisch M, Quint M, Lübberstedt T, Melchinger AE (2004) Duplicate marker loci can result in incorrect locus order on linkage maps. Theor Appl Genet 109:305–316
Fuchs E, Grüntzig M (1995) Influence of sugarcane mosaic virus (SCMV) and maize dwarf mosaic virus (MDMV) on the growth and yield of two maize varieties. J Plant Dis Prot 102:44–50
Gao Z, Eyers S, Thomas C, Ellis N, Maule A (2004) Identification of markers tightly linked to sbm recessive genes for resistance to pea seed-borne mosaic virus. Theor Appl Genet 109:488–494
Gardiner J, Schroeder S, Polacco KL, Sanchez-Villeda H, Fang Z, Morgante M, Landewe T, Fengler K, Useche F, Hanafey M, Tingley S, Chou H, Wing R, Soderlund C, Coe EH Jr (2004) Anchoring 9,371 maize expressed sequence tagged unigenes to the bacterial artificial chromosome contig map by two-dimensional overgo hybridization. Plant Physiol 134:1317–1326
Hayes AJ, Jeong SC, Gore MA, Yu YG, Buss GR, Tolin SA, Saghai Maroof MA (2004) Recombination within a nucleotide-binding-site/leucine-rich-repeat gene cluster produces new variants conditioning resistance to soybean mosaic virus in soybeans. Genetics 166:493–503
Huehn M, Piepho HP (2006) A simple note on how to save money in linkage analysis. Mol Breed 18:291–300
Hühn M, Piepho HP (2003) Determining the sample size for co-dominant molecular marker-assisted linkage detection for a monogenic qualitative trait by controlling the type-I and type-II errors in a segregating F2 population. Theor Appl Genet 106:840–845
Ingvardsen C, Xing Y, Xu M, Lübberstedt T (2005) Molecular analysis of sugarcane mosaic virus resistance in maize. Parasitica 61:61–67
International Rice Genome Sequencing Project (2005) The map-based sequence of the rice genome. Nature 436:793–800
Jones MW, Redinbaugh MG, Anderson RJ, Louie R (2004) Identification of quantitative trait loci controlling resistance to maize chlorotic dwarf virus. Theor Appl Genet 110:48–57
Jones MW, Redinbaugh MG, Louie R (2007) The Mdm1 locus and maize resistance to Maize dwarf mosaic virus. Plant Dis 91:185–190
Kang B-H, Staehelin A (2008) ER-to-Golgi transport by COPII vesicles in Arabidopsis involves a ribosome-excluding scaffold that is transferred with the vesicles to the Golgi matrix. Protoplasma 234:51–64
Kang BC, Yeam I, Frantz JD, Murphy JF, Jahn MM (2005) The pvr1 locus in Capsicum encodes a translation initiation factor eIF4E that interacts with tobacco etch virus VPg. Plant J 42:392–405
Kuntze L, Fuchs E, Grüntzig M, Schulz B, Henning U, Hohmann F, Melchinger AE (1995) Evaluation of maize inbred lines for resistance to sugarcane mosaic virus SCMV and maize dwarf mosaic virus MDMV. Agronomie 15:463–467
Kuntze L, Fuchs E, Grüntzig M, Schulz B, Klein D, Melchinger AE (1997) Resistance of early-maturing European maize germplasm to sugarcane mosaic virus SCMV and maize dwarf mosaic virus MDMV. Plant Breed 116:499–501
Lanfermeijer FC, Dijkhuis J, Sturre MJG, de Haan P, Hille J (2003) Cloning and characterization of the durable tomato mosaic virus resistance gene Tm-2 2 from Lycopersicon esculentum. Plant Mol Biol 52:1037–1049
Lei JD, Agrios GN (1986) Mechanisms of resistance in corn to maize dwarf mosaic virus. Phytopathology 76:1034–1040
Lellis AD, Kasschau KD, Whitham SA, Carrington JC (2002) Loss-of-susceptibility mutants of Arabidopsis thaliana reveal an essential role for eIF(iso)4E during potyvirus infection. Curr Biol 12:1046–1051
Liu R, Vitte C, Ma J, Mahama AA, Dhliwayo T, Lee M, Bennetzen JL (2007) A GeneTrek analysis of the maize genome. Proc Natl Acad Sci USA 104:11844–11849
Louie R, Knoke JK, Findley WR (1990) Elite maize germplasm: reactions to maize dwarf mosaic and maize chlorotic dwarf viruses. Crop Sci 30:1210–1215
Lübberstedt T, Ingvardsen C, Melchinger AE, Xing Y, Salomon R, Redinbaugh MG (2006) Two chromosome segments confer multiple potyvirus resistance in maize. Plant Breed 125:352–356
Lucas WJ (2006) Plant viral movement proteins: agents for cell-to-cell trafficking of viral genomes. Virology 344:169–184
Marçon A, Kaeppler SM, Jensen SG, Senior L, Stuber C (1999) Loci controlling resistance to high plains virus and wheat streak virus in a B73 × Mo17 population of maize. Crop Sci 39:1171–1177
McMullen MD, Louie R (1989) The linkage of molecular markers to a gene controlling the symptom response in maize to maize dwarf mosaic virus. Mol Plant Microbe Interact 6:309–314
McMullen MD, Jones MW, Simcox KD, Louie R (1994) Three genetic loci control resistance to wheat streak mosaic virus in the maize inbred Pa405. Mol Plant Microbe Interact 7:708–712
Melchinger AE, Kuntze L, Gumber RK, Lübberstedt T, Fuchs E (1998) Genetic basis of resistance to sugarcane mosaic virus in European maize germplasm. Theor Appl Genet 96:1151–1161
Meyers BC, Tingey SV, Morgante M (2001) Abundance, distribution, and transcriptional activity of repetitive elements in the maize genome. Genome Res 11:1660–1676
Mikel MA, D’arcy CJ, Rhodes AM, Ford RE (1984) Genetics of resistance of two dent corn inbreds to maize dwarf mosaic virus and transfer of resistance into sweet corn. Phytopathology 74:467–473
Ming R, Brewbaker JL, Pratt RC, Musket TA, McMullen MD (1997) Molecular mapping of a major gene conferring resistance to maize mosaic virus. Theor Appl Genet 95:271–275
Nicaise V, German-Retana S, Sanjuán R, Dubrana MP, Mazier M, Maisonneuve B, Candresse T, Caranta C, LeGall O (2003) The eukaryotic translation initiation factor 4E controls lettuce susceptibility to the potyvirus lettuce mosaic virus. Plant Physiol 132:1272–1282
Nieito C, Morales M, Orjeda G, Clepet C, Monfort A, Sturbois B, Puigdomènech R, Pitrat M, Caboche M, Dogimont C, Garcia-Mas J, Aranda MA, Bendahmane A (2006) An eIF4E allele confers resistance to an uncapped and non-polyadenylated RNA virus in melon. Plant J 48:452–462
Oparka KJ (2004) Getting the message across: how do plant cells exchange macromolecular complexes? Trends Plant Sci 9:33–41
Oparka KJ, Roberts AG, Beovink P, Cruz SS, Roberts I, Pradel KS, Imlau A, Kotlizky G, Sauer N, Epel B (1999) Simple, but not branched, plasmodesmata allow the nonspecific trafficking of proteins in developing tobacco leaves. Cell 97:743–754
Pokorný R, Porubová M (2006) Movement of sugarcane mosaic virus in plants of resistant and susceptible maize lines. Cereal Res Commun 34:1109–1116
Qiu JW, Schürch AC, Yahiaoui N, Dong LL, Fan HJ, Zhang ZJ, Keller B, Ling HQ (2007) Physical mapping and the identification of a candidate for the leaf rust resistance gene Lr1 of wheat. Theor Appl Genet 115:159–168
Quint M, Mihaljevic R, Dussle CM, Xu ML, Melchinger AE, Lübberstedt T (2002) Development of RGA-CAPS markers and genetic mapping of candidate genes for sugarcane mosaic virus resistance in maize. Theor Appl Genet 105:355–363
Quint M, Dussle CM, Melchinger AE, Lübberstedt T (2003) Identification of genetically linked RGAs by BAC screening in maize and implications for gene cloning, mapping and MAS. Theor Appl Genet 106:1171–1177
Redinbaugh MG, Jones MW, Gingery RE (2004) The genetics of virus resistance in maize (Zea mays L.). Maydica 49:183–190
Rosenkranz E, Scott GE (1984) Determination of the number of genes for resistance to maize dwarf mosaic virus strain A in five corn inbred lines. Phytopathology 74:71–76
Schaad MC, Lellis AD, Carrington JC (1997) VPg of tobacco etch potyvirus is a host genotype-specific determinant for long-distance movement. J Virol 71:8624–8631
Scholthof HB (2005) Plant virus transport: motions of functional equivalence. Trends Plant Sci 10:376–382
Seifers DL, Salomon R, Marie-Jeanne V, Alliot B, Signoret P, Haber S, Loboda A, Ens W, She YM, Standing KG (2000) Characterization of a novel potyvirus isolated from maize in Israel. Phytopathology 90:505–513
Seo YS, Rojas MR, Lee JY, Lee SW, Jeon JS, Ronald P, Lucas WJ, Gilbertson RL (2006) A viral resistance gene from common bean functions across plant families and is up-regulated in a non-virus-specific manner. Proc Natl Acad Sci USA 103:11856–11861
Shi C, Ingvardsen C, Trümmler F, Melchinger AE, Wenzel G, Lübberstedt T (2005) Identification by suppression subtractive hybridisation of genes that are differentially expressed between near-isogenic maize lines in association with sugarcane mosaic virus resistance. Mol Gen Genet 273:450–461
Shukla DD, Ward CW (1989) Identification and classification of potyviruses on the basis of coat protein sequence data and serology. Arch Virol 106:171–200
Simcox KD, McMullen MD, Louie R (1995) Co-segregation of the maize dwarf mosaic virus resistance gene, Mdm1, with the nucleolus organizer region in maize. Theor Appl Genet 90:341–346
Soosaar JLM, Burch-Smith TM, Dinesh-Kumar SP (2005) Mechanisms of plant resistance to viruses. Nat Rev Microbiol 3:789–798
Spassova MI, Prins TW, Folkertsma RT, Klein-Lankhorst RM, Hile J, Goldbach RW, Prins M (2001) The tomato gene Sw5 is a member of the coiled coil, nucleotide binding, leucine-rich repeat class of plant resistance genes and confers resistance to TSWV in tobacco. Mol Breed 7:151–161
Stange C, Matus JT, Elorza A, Arce-Johnson P (2004) Identification and characterization if a novel tobacco mosaic virus resistance N gene homologue in Nicotiana tabacum plants. Funct Plant Biol 31:149–158
Stein N, Perovis D, Kumlehn J, Pellio B, Stracke S, Streng S, Ordon F, Graner A (2005) The eukaryotic translation initiation factor 4E confers multiallelic recessive Bymovirus resistance in Hordeum vulgare (L.). Plant J 42:912–922
Takahashi H, Miller J, Nozaki Y, Sukamto, Takeda M, Shah J, Hase S, Ikegami M, Ehara Y, Dinesh-Kumar SP (2002) RCY1, an Arabidopsis thaliana RPP8/HRT family resistance gene, conferring resistance to cucumber mosaic virus requires salicylic acid, ethylene and a novel signal transduction mechanism. Plant J 32:655–667
The Rice Chromosome 3 Sequencing Consortium (2005) Sequence, annotation, and analysis of synteny between chromosome 3 and diverged grass species. Genome Res 15:1284–1291
Thiel T, Michalek W, Varshney RK, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106:411–422
Van Ooijen OJW, Voorrips RE (2001) JoinMap 3.0. Software for the calculation of genetic linkage maps. Plant Research International, Wageningen
Vidal S, Cabrera H, Andersson RA, Fredriksson A, Valkonen JPT (2002) Potato gene Y-1 is an N gene homolog that confers cell death upon infection with potato virus Y. Mol Plant Microbe Interact 15:717–727
Vision TJ (2005) Gene order in plants: a slow but sure shuffle. New Phytol 168:51–60
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Welz HG, Schechert A, Pernet A, Pixley KV, Geiger HH (1998) A gene for resistance to the maize streak virus in the African CIMMYT maize inbred line CML202. Mol Breed 4:147–154
Whitham S, Dinesh-Kumar SP, Choi D, Hehl R, Corr C, Baker B (1994) The Product of the tobacco mosaic virus resistance gene N: similarity to toll and the interleukin-1 receptor. Cell 78:1101–1115
Whitham SA, Anderberg RJ, Chrisholm ST, Carrington JC (2000) Arabidopsis RTM2 gene is necessary for specific restriction of tobacco etch virus and encodes an unusual small heat shock-like protein. Plant Cell 12:569–582
Wisser RJ, Balint-Kurti PJ, Nelson RJ (2006) The genetic architecture of disease resistance in maize: a synthesis of published studies. Phytopathology 96:120–129
Xia XC, Melchinger AE, Kuntze L, Lübberstedt T (1999) Quantitative trait loci mapping of resistance to sugarcane mosaic virus in maize. Phytopathology 89:660–667
Xiao W, Zhai J, Fan S, Li L, Dai J, Xu M (2007) Mapping of genome-wide resistance gene analogs (RGAs) in maize (Zea mays L.). Theor Appl Genet 115:501–508
Xing Y, Ingvardsen C, Salomon R, Lübberstedt T (2006) Analysis of sugarcane mosaic virus resistance in maize in an isogenic dihybrid crossing scheme and implications for breeding potyvirus-resistant maize hybrids. Genome 49:1274–1282
Xu ML, Melchinger AE, Xia XC, Lübberstedt T (1999) High-resolution mapping of loci conferring resistance to sugarcane mosaic virus in maize using RFLP, SSR and AFLP markers. Mol Gen Genet 261:574–581
Xu ML, Melchinger AE, Lübberstedt T (2000) Origin of Scm1 and Scm2—two loci conferring resistance to sugarcane mosaic virus (SCMV) in maize. Theor Appl Genet 100:934–941
Yao H, Zhou Q, Li J, Smith H, Yandeau M, Nikolau BJ, Schnable PS (2002) Molecular characterization of meiotic recombination across the 140-kb multigenic a1-sh2 interval of maize. Proc Natl Acad Sci USA 99:6157–6162
Yoshii M, Nishikiori M, Tomita K, Yoshioka N, Kozuka R, Naito S, Ishikawa M (2004) The Arabidopsis Cucumovirus multiplication 1 and 2 loci encode translation initiation factors 4E and 4G. J Virol 78:6102–6111
Yuan L, Dussle CM, Melchinger AE, Utz HF, Lübberstedt T (2003) Clustering of QTL conferring SCMV resistance in maize. Maydica 48:55–62
Yuan L, Dussle CM, Muminovic J, Melchinger AE, Lübberstedt T (2004) Targeted BSA mapping of Scmv1 and Scmv2 conferring resistance to SCMV using PstI/MseI compared with EcoRI/MseI AFLP markers. Plant Breed 123:434–437
Zhang SH, Li XH, Wang ZH, George ML, Jeffers D, Wang FG, Liu XD, Li MS, Yuan LX (2003) QTL mapping for resistance to SCMV in Chinese maize germplasm. Maydica 48:307–312
Zhu Y, Qi Y, Xun Y, Owens R, Ding B (2002) Movement of potato spindle tuber viroid reveals regulatory points of phloem-mediated RNA traffic. Plant Physiol 130:138–146
Acknowledgments
This study was financially supported by the Danish Ministry of Food, Agriculture and Fisheries, Grant No. 23-03-0054.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by C. Schön.
C. R. Ingvardsen and Y. Xing contributed equally to this work.
Rights and permissions
About this article
Cite this article
Ingvardsen, C.R., Xing, Y., Frei, U.K. et al. Genetic and physical fine mapping of Scmv2, a potyvirus resistance gene in maize. Theor Appl Genet 120, 1621–1634 (2010). https://doi.org/10.1007/s00122-010-1281-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-010-1281-1