
Abstract
Major gene inheritance of resistance to Potato leafroll virus (PLRV) was demonstrated in a parthenogenic population derived from the highly resistant tetraploid andigena landrace, LOP-868. This major gene or chromosome region seems to control a single mechanism for resistance to infection and virus accumulation in this source. About 149 dihaploid lines segregated in a ratio of 107 resistant to 32 susceptible, fitting the expected ratio for inheritance of a duplex gene under random chromatid segregation. A tetraploid AFLP map was constructed using as reference the ultra high density (UHD) map. All AFLP markers associated with PLRV resistance mapped to the same linkage group. Map position was confirmed by analysis of previously-mapped SSR markers. Rl adg is located on the upper arm of chromosome V, at 1 cM from its most closely linked AFLP marker, E35M48.192. This marker will be used to develop allele-specific primers or a pair of flanking PCR-based markers for their use in marker assisted selection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Potato leafroll virus (PLRV) is one of the most damaging and widespread diseases of potato (Solomon-Blackburn and Barker 2001). PLRV infection causes yield reduction of up to 95%, besides diminishing marketable yield in some cultivars due to net necrosis (Barker and Waterhouse 1999; Solomon-Blackburn and Barker 2001). However, in spite of all efforts, no sources of extreme resistance to PLRV have been confirmed (Taliansky et al. 2003). Different types of PLRV resistance have been described, including: resistance to infection by aphids, resistance to virus accumulation in infected plants, resistance to aphid vectors, resistance to movement of the virus from infected foliage to tubers and hypersensitivity or intolerance (Barker and Waterhouse 1999).
Major gene inheritance has been reported for some types of resistance to this virus. Brown and Thomas (1994) found that resistance to PLRV in Solanum chacoense Bitter followed monogenic dominant inheritance. Solomon-Blackburn et al. (2003/4) concluded that resistance to virus accumulation was controlled by one or few major genes that are not complementary. Butkiewicz (1978) reported that hypersensitivity to PLRV was controlled by one dominant gene modified by several minor genes. However, under some circumstances, the restriction of the pathogen by hypersensitivity is so ineffective that some cultivars become 100% infected within 1 year (Swiezynski 1994).
It is generally believed that resistance to PLRV infection is of a polygenic nature, with several minor genes involved (Davidson 1973). However, genes with major effects may also be involved (Brown et al. 1997).
Solanum tuberosum ssp. andigena (Juz. et Bukasov) Hawkes is a native cultivated potato species grown in the Andean region that easily crosses with Solanum tuberosum ssp. tuberosum L. A previous genetic study carried out at the International Potato Center (CIP) identified an andigena accession, LOP-868, with high levels of heritable resistance to PLRV infection by viruliferous aphids combined with low virus accumulation when its plants were graft-inoculated. This landrace, whose cultivar name is ‘Alca Tarma’, was collected under CIP-Number 702853 in the province of Huancane (Puno, Peru) at 3,850 masl.
LOP-868-derived progenies yielded a high frequency of genotypes with levels of resistance to infection as high as those observed in LOP-868. This suggested the presence of a gene with large effects controlling the resistance in this landrace. It was also demonstrated that the resistance of this accession could be readily transferred to the genetic background of commercial varieties without any negative interaction (Mihovilovich et al. 2007).
The present study characterizes and maps PLRV resistance from this new source in a dihaploid population. Resistance to infection and to virus accumulation was assessed by aphid inoculation and grafting, respectively.
Materials and methods
Plant material
A parthenogenetic population of 149 dihaploids (2n = 2x = 24) was developed from LOP-868 (2n = 4x = 48) by pollinating this landrace with the Solanum phureja Juz. et Bukasov lines IVP-101 and 596001.4, which induce haploidization and are homozygous for a dominant “embryo spot” marker that facilitates the detection of hybrids (Hermsen and Verdenius 1973; Upadhya and Cabello 1999).
Seeds without the embryo-spot were selected and grown under greenhouse conditions (mean 17°C, 90% R.H.) for 45 days. Chloroplast and chromosome counts were carried out as described in Huamán (1995) and Watanabe and Orrillo (1993) to establish the ploidy of each genotype.
The diploid genotypes, LOP-868, the haploid inducers and three infection controls, the highly resistant clone DW.84-1457 (Dziewonska and Was 1994), the moderately resistant variety “Achirana-INTA” (Huarte et al. 1990) and the susceptible variety “Perricholi” (Centro Internacional de la Papa 2004), were multiplied by stem cuttings for subsequent analyses.
PLRV infection assay
PLRV infection assays were carried out under greenhouse conditions using a RCB (randomized complete block) design of three blocks with experimental units of ten plants per genotype. The first two blocks were evaluated in spring season (August–December 2003) at La Molina, Lima (247 m above sea level, mean 17°C, 90% R.H.), while the third was assessed in rainy season (January–May 2004) at La Victoria, Huancayo (3,315 m above sea level, mean 17°C, 53% R.H.). The virus isolate used was 01 from Peru, belonging to serogroup 3 (Jayasinghe 1990). Inoculum was maintained and subsequently propagated in plants of the susceptible cv. ‘Flor Blanca’. Virus was indexed with a double antibody sandwich ELISA (DAS-ELISA) 30 days after sowing.
The dihaploid population, LOP-868 and the controls were inoculated with PLRV using the Chuquillanqui and Jones method (1980), which entails shaking PLRV-infected plants heavily-infested with Myzus persicae Sulzer aphids over the test plants. The next day, aphids were counted to record infection pressure. Seven days after the inoculation, a systemic insecticide was applied to kill the aphids. Forty days later, DAS-ELISA (as described by Salazar and Jayasinghe 1997) was carried out on individual plants to assess primary infection. Samples comprised three leaflets from fully expanded leaves of approximately the same size collected from halfway up the stem which were prepared in 2.5 ml of extraction buffer. Absorbance (A 405 nm) values were determined one and a half hours after substrate addition using a microplate reader (BIO-RAD, Model 550). Samples showing an absorbance of 0.05 units higher than that of healthy samples were considered positive (Tamada and Harrison 1980).
Plants negative to DAS-ELISA were allowed to form tubers. One daughter tuber from each plant was sown in the following spring (August–December 2004) at La Molina. Forty days later, a DAS-ELISA was performed on foliage to assess secondary infection.
Infection percentages were calculated as the number of plants that tested positive to DAS-ELISA in primary or secondary infection cycles over the total number of plants tested per genotype and replication. Average overall replicates were calculated for each progeny genotype and their frequency distribution was plotted. Pearson’s correlation coefficient was estimated among replicates for the arcsine of the percentage of infection, in order to examine the correlation between blocks.
Amplified fragment length polymorphism
DNA extraction was carried out on young leaves of healthy plants using the CTAB method (Ghislain et al. 1999). Amplified fragment length polymorphism (AFLP) analyses followed Vos et al. (1995) with minor modifications, except that the streptavidin bead selection step was omitted. Primers were labeled with 33P so that fingerprint patterns could be visualized after electrophoresis on 6% denaturing polyacrylamide gels. Twenty of the most informative AFLP primer combinations from the European Ultra High Density (UHD) map database (European Union project FAIR5-PL97-3565 2001) were used. Eight of these combinations had EcoRI and MseI as their restriction enzymes while the other 12 had PstI and MseI as theirs. The parents used to develop this UHD map, SH82-93-488 and RH89-039-16, were included in all AFLP analyses.
Their DNA was kindly provided by Herman van Eck from the Wageningen Agricultural University.
Each AFLP marker was named after its Keygene® primer combination followed by its electrophoretic mobility or gel order.
Linkage map construction
AFLP markers segregating from LOP-868 were scored for presence and absence and tested for monogenic ratios expected under autosomic tetraploid inheritance (chromosomic and chromatidic) and classified according to their inferred allelic dosage. Under random chromosome segregation (i.e., when the marker is close to the centromere), expected segregation ratios are 1:1 if the marker is in simplex dose and 5:1 if it is in duplex. Random chromatid segregation or double reduction (Bradshaw 1994) changes the expected segregation ratios to 0.86:1 for a simplex marker, 3.67:1 for a duplex and 27:1 for a triplex. When a marker fitted more than one segregation ratio, it was assigned to the class for which its test value was lower.
A genetic map was constructed using the statistical program TetraploidMap (Hackett and Luo 2003). Chromosome identification relied on having at least three markers from a linkage group that comigrated with UHD alleles from a single chromosome (Rouppe van der Voort et al. 1997). Map position of the factor that controlled PLRV resistance was confirmed with previously-mapped SSR markers (Milbourne et al. 1998). The SSR protocol used is described elsewhere (Ghislain et al. 1999).
Characterization of PLRV resistance mechanism in LOP-868
Back-test
Back-tests were carried out under greenhouse conditions at La Molina (18.8°C, 89% R.H.) during November and December 2004. Virus-free plants of cv. “Perricholi” were graft-inoculated with scions from 13 plants of LOP-868 and from 4 genotypes of its progeny whose DAS-ELISA tests for secondary infection were negative (7–18 plants were tested per genotype). Scions from five plants of infected “Perricholi” were used as controls. Thirty days after grafting, DAS-ELISA was performed on the rootstocks to detect the virus.
Resistance to virus accumulation
In order to assess the resistance of PLRV accumulation of LOP-868, 11 plants of this accession and 6 of “Perricholi” were graft-inoculated with PLRV-infected scions of the susceptible cv. “Flor Blanca” under greenhouse conditions at La Molina (October–December 2003, 18°C, 89% R.H). Three plants of “Perricholi” and three of LOP-868 were grafted with healthy “Perricholi” plants as negative controls. Fifteen days later, the scions were removed. Thirty days after grafting, quantitative DAS-ELISA (Barker and Harrison 1985) was carried out to estimate viral concentrations. Samples were collected from halfway up the stem, weighed and diluted with extraction buffer in a 1:4 ratio (w/v). Titres were extrapolated from a standard curve that correlated absorbance values and known PLRV concentrations.
Results
PLRV infection
The average number of aphids per plant was 87 for the first replicate, 95 for the second one and 40 for the third replicate of progeny individuals. In spite of these differences in infection pressure, infection percentage within genotypes showed consistency through replicates as determined by a Pearson’s correlation test (r I,II = 0.95, r I,III = 0.87, r II,III = 0.88).
The distribution of resistance to PLRV was bimodal (Fig. 1). This allowed assigning nearly all of the parthegenotes to either of the two phenotypic classes. Progeny genotypes with percentages of infected plants under 13.5% (<4/30) were considered resistant and those over 70% (>21/30) susceptible. Of the 149 genotypes evaluated, 107 were regarded as resistant, 32 as susceptible and 10 remained unclassified (having intermediate percentages of infected plants). Sixty-three of the 107 resistant genotypes did not have a single plant infected out of the 30 tested.

Distribution of the percentage of infection with PLRV of the LOP-868 derived progeny
All the genotypes classified as susceptible had high absorbance values (primary infection average = 1.711 ± 0.093). Quite the opposite, almost all of the few plants of the resistant genotypes that became infected reached only low to moderate absorbance values (primary infection average = 0.530 ± 0.465) suggesting that they were not only resistant to infection by aphid inoculation but to virus accumulation as well.
The phenotypic segregation was tested against different models for tetraploid inheritance. A χ 2 test at a significance level of P < 0.05 could not distinguish between 3:1 and 3.7:1 ratio for the inheritance of the resistance (Table 1). The former is expected for two unlinked genes in simplex dosage (χ 2 = 0.20, P < 0.65) while the latter is expected for one gene in duplex dosage assuming random chromatid segregation (α = 1/7, χ 2 = 0.13, P < 0.72). Genetic mapping was pursued to help ascertain if PLRV resistance is controlled by one or two factors.
Linkage map
A total of 610 AFLP markers segregating from LOP-868 were scored. About 272 markers (44.6%) were classified as simplex and 118 as duplex (19.3%), assuming random chromosome segregation; while 71 were classified as simplex (11.6%), 46 as duplex (7.5%) and 23 as triplex (3.8%) assuming random chromatid segregation (α = 1/7). Only 442 of these markers (72.7%) were used in the genetic map construction because markers that did not fit any of the expected monogenic segregation ratios, that only fitted random chromatid segregation, or that could not be scored with enough certainty were excluded from further analyses. No markers were identified in the progeny that were present in either of the haploid inducers (IVP-101 and 596004.1) and absent in LOP-868, suggesting that all dihaploids were formed by parthenogenesis.
The tetraploid genetic map has a total length of 1,074.8 cM comprising 13 linkage groups. Each linkage group was identified when at least three markers comigrated with UHD alleles from a single chromosome (Rouppe van der Voort et al. 1997). Chromosome assignment and polarity were established this way, except for chromosome VIII. The identity of this chromosome was inferred from the fact that this chromosome was the only one left unassigned. As expected, due to the fact that potato has 12 chromosomes, two linkage groups belonged to the same chromosome, I, as identified by their comigrating UHD alleles. A weak association was observed between the homologs of these two linkage groups but it was not possible to merge them. A linkage group might split into two if a region shows a high frequency of recombination (Rouppe van der Voort et al. 1997) or if no polymorphic markers are found.
Ninety-five AFLP markers comigrated with UHD alleles, but only 68 of them could be mapped. Nine (13.2%) of these 68 markers were homoplasic because even though they were amplified with the same primer combination and had the same electrophoretic mobility, they corresponded to UHD alleles previously mapped to different chromosomes.
Marker-trait association
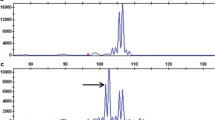
Twenty-two AFLP markers were associated with PLRV resistance as established by a test of independence (α = 0.05). Eleven were linked in coupling while the remaining 11 were linked in repulsion. All of these linked markers mapped to chromosome V. AFLP marker E35M48.192 (χ 2 = 123.68, P < 1.25E-26) is tightly linked in coupling to the resistance factor (Fig. 2). This marker showed only two recombinant plants, out of 135, i.e., it was absent in one resistant genotype whereas it was present in one susceptible genotype.

Segregation of AFLP marker E35M48.192 linked in coupling to PLRV resistance. A resistant genotype recombinant for the marker and the trait is indicated by an asterisk
Confirmation of the position of the factor that controlled PLRV resistance came from the amplification of two chromosome-V-specific microsatellites in the progeny. One of these microsatellites, STM0013, had two alleles in simplex dosage associated with PLRV resistance. One of the alleles was linked in coupling while the other was linked in repulsion.
A factor that controls PLRV infection resistance, Rl adg, maps to the upper arm of chromosome V, at 13 cM from our uppermost marker (Fig. 3). It is present in two homologs confirming its double dose or duplex stage in LOP-868 and located in coupling with AFLP marker E35M48.192 at only 1 cM.

Maternal linkage map of LOP-868 chromosome V showing markers and distances (cM) for homologues ‘a’, ‘b’, ‘c’ and ‘d’ and the combined linkage group. Rl adg is indicated in orange font. Duplex markers are marked with asterisks and previously-mapped SSR and UHD markers are in green font. Marker order was established by the simulated annealing algorithm of TetraploidMap at starting T = 10, a cooling factor of α = 0.85 and M = 100n random changes at each T°
PLRV resistance mechanism in Solanum tuberosum ssp. andigena accession LOP-868
Back-test
PLRV was not detected in any of the 55 healthy plants of “Perricholi” that were graft-inoculated with scions of LOP-868 or of the 4 resistant dihaploid genotypes. On the other hand, when infected “Perricholi” plants were used as scions, PLRV was readily detected.
Resistance to virus accumulation
Following graft-inoculation of 11 plants of LOP-868 with infected “Flor Blanca” scions, mean PLRV titer was 302.1 ± 252.5 ng of virus per gram of tissue, while the six plants of “Perricholi” treated the same way averaged 2,558.2 ± 505.6 ng. Mean virus concentration in LOP-868 was thus less than 12% of the concentration in the susceptible control. However, while ten plants of LOP-868 had PLRV titers of less than 95 ng, one plant accumulated PLRV to the same degree as was found in “Perricholi”. This LOP-868 plant had a PLRV concentration of 2,825.3 ng (Fig. 4).

Viral concentrations found in LOP-868 and in “Perricholi” after graft-inoculation with PLRV infected scions
Discussion
The PLRV resistance of LOP-868 is not a consequence of indirect resistance to aphids nor is it a form of extreme resistance. No type of resistance to aphids was detected since aphids easily colonized LOP-868 plants. Extreme resistance was ruled out because PLRV was detected when plants were graft-inoculated.
LOP-868 appears highly resistant to PLRV infection by aphids. Mihovilovich et al. (2006) were not able to infect LOP-868 with pressures of up to 100 aphids per plant. However, in the present work, virus was detected in some of LOP-868 plants by secondary infection. It has been reported that the absolute incidence of infection with PLRV rises as inoculum pressure increases (Barker and Harrison 1985; Brown et al. 1997) which might explain the results obtained. However, several characteristics support the classification of LOP-868 as highly resistant. An extremely high proportion of the dihaploid progeny of this accession (71.8%) was resistant to infection. Virus was not even detected when a back-test was performed with LOP-868 or a sample of the dihaploid genotypes negative to DAS-ELISA, ruling out the hypothesis that PLRV concentration in these plants was too low to be detected. Furthermore, 73% of the plants of the resistant control, DW.84-1457, became infected, a percentage significantly higher than that observed in LOP-868. It seems that the resistance mechanism in LOP-868 can be overcome when PLRV inoculum pressure is very high. This can be achieved either with grafting (since the scion is able to continue multiplying the virus) or with extremely high numbers of viruliferous aphid.
LOP-868 has a moderate level of resistance to PLRV accumulation. Clones that are commonly regarded as resistant to accumulation achieve PLRV titers between 1 and 10% of the concentration found in susceptible genotypes (Barker and Harrison 1985). In this study, PLRV concentration in LOP-868 was about 12% of the concentration in “Perricholi”. Barker and Harrison (1986) found that the restriction to PLRV accumulation in resistant genotypes was a result of fewer infected cells in the external phloem bundles and not of a decreased virus replication in each infected cell. Later, Derrick and Barker (1997) suggested that impairment of short distance movement of virus from sieve elements to companion cells seems to underlie this resistance. These authors proposed that a gene product in resistant genotypes may act directly against a PLRV-encoded protein—putatively a phloem-specific movement protein. The fact that one plant of LOP-868 achieved a concentration as high as that of the susceptible control after graft inoculation shows that this resistance mechanism can be overcome. It seems that if the pathogen recognition or defense response is compromised or delayed, plants of this landrace might not successfully suppress PLRV movement, allowing the virus to progress without restraint as it does in a susceptible genotype.
PLRV resistance of LOP-868 (either to infection or accumulation) seems to be controlled by a single mechanism. Genotypes that were susceptible to infection also had high mean absorbance values whereas the opposite was true for those resistant to infection. This relationship would not have been expected if there were two separate resistance mechanisms in LOP-868, unless they were controlled by two tightly linked genes on the same chromosome.
A major factor that controls the resistance to PLRV infection is present in double dose in LOP-868. The use of parthenogenetic dihaploids (2x) facilitated the elucidation of the resistance genotype of this tetraploid landrace (4x), without the influence of an additional genetic background on the resistance phenotype of the progeny (e.g., modification or even suppression), as might be present when an alternative, susceptible parent is used to make a cross (Gebhardt and Valkonen 2001). Dihaploids have been used similarly in the past to determine the breeding value or number of genes for desirable traits of tetraploid varieties (Cipar and Lawrence 1972). Parthenogenotes and anther-culture derived dihaploid potato progenies were also used, respectively, to map the H 1 gene for resistance to G. rostochiensis (Pineda et al. 1993) and to elucidate the major dominant gene inheritance of Potato virus Y (PVY) from the tetraploid cv. ‘Assia’ (Song et al. 2005).
The major factor reported here may represent either a single gene or a chromosome region with several tightly linked resistance genes. One example of two closely linked genes is the R3 locus of potato. This locus comprises two closely linked resistance genes, R3a and R3b, which confer resistance to different isolates of Phytophthora infestans (Mont.) de Bary (Huang et al. 2004).
This is the first report of the location of a factor for strong resistance to PLRV on chromosome V. Two major QTLs for resistance to PLRV accumulation have been reported on chromosome XI (PLRV.1 and PLRV.4) (Marczewski et al. 2001, 2004). There is also a QTL with small effects (PLRV.3) located in chromosome V although its position in this linkage group is unknown because the marker associated with the resistance had a skewed segregation (Marczewski et al. 2004).
The upper arm of potato chromosome V carries a cluster of resistance genes (Gebhardt and Valkonen 2001) (between RFLP markers GP21 and GP179) comprising major genes for resistance to PVX (Rx2, Ritter et al. 1991; Nb, De Jong et al. 1997) and Phytophthora infestans (R1, Leonards-Schippers et al. 1992) and major QTLs for resistance to P. infestans (Leonards-Schippers et al. 1994), Globodera pallida (Stone) Behrens and Globodera rostochiensis (Wollenweber) (Grp1, Rouppe van der Voort et al. 1998a; Gpa, Caromel et al. 2003, Kreike et al. 1994; Gpa5, Rouppe van der Voort et al. 2000; GpaV S spl , Caromel et al. 2005). The factor that controls PLRV resistance is located adjacent to these genes. It seems to be outside this disease resistance region because previous reference maps (Milbourne et al. 1998) located the GP21–GP179 interval before STM0013, while the genetic map constructed in this study places Rl adg 8 cM after STM0013.
The information from the ultra high density (UHD) map developed by the European Community helped in the identification of the linkage groups. Without this database (European Union project FAIR5-PL97-3565 2001), chromosome identification would have required the amplification of additional reference markers, e.g., microsatellites from all 12 chromosomes. Nevertheless, it should be taken into account that AFLP shows moderate homoplasy (13.2% in this study), therefore, care should be taken when assigning the same loci to comigrating AFLP markers, especially when the populations are phylogenetically distant (Rouppe van der Voort et al. 1998b).
Immediate application of this mapping information in marker-assisted selection is hampered by the comigration of markers in the controls—which are not resistant—with each of the markers associated in coupling with resistance (with exception of one of the alleles of STM0013). For instance, even though AFLP marker E35M48.192 maps at only 1 cM from the resistance factor, a band that comigrated with this marker was found in both haploid inducers, which are susceptible to PLRV infection. This highlights the importance of developing allele-specific markers for their use in future marker-assisted selection programs.
References
Barker H, Harrison BD (1985) Restricted multiplication of potato leafroll virus in resistant potato genotypes. Ann Appl Biol 107:205–212
Barker H, Harrison BD (1986) Restricted distribution of potato leafroll virus antigen in resistant potato genotypes and its effect on transmission of the virus by aphids. Ann Appl Biol 109:595–604
Barker H, Waterhouse PM (1999) The development of resistance to Luteoviruses mediated by host genes and pathogen-derived transgenes. In: Smith HG, Barker H (eds) The luteoviridae. CABI Publishing, Wallingford, pp 169–210
Bradshaw JE (1994) Quantitative genetics theory for tetrasomic inheritance. In: Bradshaw JE, Mackay GR (eds) Potato genetics. CAB International, Wallingford, pp 71–100
Brown CR, Thomas PE (1994) Resistance to potato leafroll virus derived from Solanum chacoense: characterization and inheritance. Euphytica 74:51–57
Brown CR, Corsini D, Pavek J, Thomas PE (1997) Heritability of field resistance to potato leafroll virus in cultivated potato. Plant Breed 116:585–588
Butkiewicz H (1978) (Intolerance to potato leafroll virus (PLRV) occurring in potato plants). Ziemniak 1978:5–37
Caromel B, Mugniéry D, Lefebvre V, Andrzejewski S, Ellissèche D, Kerlan MC, Rousselle P, Rousselle-Bourgeois F (2003) Mapping QTLs for resistance against Globodera pallida (Stone) Pa2/3 in a diploid potato progeny originating from Solanum spegazzinii. Theor Appl Genet 106:1517–1523
Caromel B, Mugniery D, Kerlan MC, Andrzejewski S, Palloix A, Ellisseche D, Rousselle-Bourgeois F, Lefebvre V (2005) Resistance quantitative trait loci originating from Solanum sparsipilum act independently on the sex ratio of Globodera pallida and together for developing a necrotic reaction. Mol Plant Microbe Interact 18(11):1186–1194
Centro Internacional de la Papa (CIP) (2004) CIP potato varieties. CIP, Lima, Perú. http://www.potato.riu.cip.cgiar.org/index.php
Chuquillanqui C, Jones RAC (1980) A rapid technique for assessing the resistance of families of potato seedlings to potato leaf roll virus. Potato Res 23:121–128
Cipar MS, Lawrence CH (1972) Scab resistance of haploids from two Solanum tuberosum cultivars. Am Potato J 49:117–119
Davidson TMW (1973) Assessing resistance to leaf roll in potato seedlings. Potato Res 16(2):99–108
De Jong W, Forsyth A, Leister D, Gebhardt C, Baulcombe DC (1997) A potato hypersensitive resistance gene against potato virus X maps to a resistance gene cluster on chromosome 5. Theor Appl Genet 95:246–252
Derrick PM, Barker H (1997) Short and long distance spread of potato leafroll luteovirus: effects of host genes and transgenes conferring resistance to virus accumulation in potato. J Gen Virol 78:243–251
Dziewonska M, Was M (1994) Diploid genotype DW.84-1457 highly resistant to potato leafroll virus (PLRV). Potato Res 37:217–224
European Union FAIR (Agriculture and Fisheries) program grant FAIR5-PL97–3565: Department of Plant Breeding, Wageningen Agricultural University; Department of Nematology, Wageningen Agricultural University; Institut national de la recherche agronomique, Montfavet; AZTI-CIMA; Scottish Crop Research Institute (2001) EU project Ultra High Density Map of the Potato. http://www.dpw.wau.nl/uhd/index.htm
Gebhardt C, Valkonen JPT (2001) Organization of genes controlling disease resistance in the potato genome. Annu Rev Phytopathol 39:79–102
Ghislain M, Zhang D, Herrera MR (1999) Molecular biology laboratory protocols: plant genotyping training manual, 2nd edn. International Potato Center (CIP), Lima
Hackett CA, Luo ZW (2003) TetraploidMap: construction of a linkage map in autotetraploid species. J Hered 94(4):358–359
Hermsen JGTh, Verdenius J (1973) Selection from Solanum tuberosum group Phureja of genotypes combining high-frequency haploid induction with homozygosity for embryo-spot. Euphytica 22:244–259
Huamán Z (1995) Técnicas citológicas para determinar el número cromosómico y la fertilidad de las papas. Guía de Investigación CIP 10. Centro Internacional de la Papa, Lima, pp 18
Huang S, Vleeshouwers VGAA, Werij JS, Hutten RCB, van Eck HJ, Visser RGF, Jacobsen E (2004) The R3 resistance to Phytophthora infestans in potato is conferred by two closely linked R genes with distinct specificities. Mol Plant Microbe Interact 17(4):428–435
Huarte MA, Butzonitch IP, Mendiburo AO (1990) Mejoramiento para resistencia a virus en el programa argentino de papa. In: Hidalgo OA, Rincón H (eds) Avances en el mejoramiento genético de la papa en los países del cono sur. Centro Internacional de la Papa, Lima, pp 85–93
Jayasinghe U (1990) Variability and resistance to Potato Leafroll Virus. In: Control of virus and virus-like diseases of potato and sweetpotato. Report of the 3rd Planning Conference. International Potato Center, Lima, pp 141–153
Kreike CM, de Koning JRA, Vinke JH, van Ooijen JW, Stiekema WJ (1994) Quantitatively-inherited resistance to Globodera pallida is dominated by one major locus in Solanum spegazzinii. Theor Appl Genet 88:764–769
Leonards-Schippers C, Gieffers W, Salamini F, Gebhardt C (1992) The R1 gene conferring race-specific resistance to Phytophthora infestans in potato is located on potato chromosome V. Mol Gen Genet 233:278–283
Leonards-Schippers C, Gieffers W, Schafer-Pregl R, Ritter E, Knapp SJ, Salamini F, Gebhardt C (1994) Quantitative resistance to Phytophthora infestans in potato: A case study for QTL mapping in a allogamous plant species. Genetics 137:67–77
Marczewski W, Flis B, Syller J, Schäfer-Pregl R, Gebhardt C (2001) A major quantitative trait locus for resistance to Potato leafroll virus is located in a resistance hotspot of potato chromosome XI and is tightly linked to N-gene-like markers. Mol Plant Microbe Interact 14(12):1420–1425
Marczewski W, Flis B, Syller J, Strzelczyk-Zyta D, Henning J, Gebhardt C (2004) Two allelic or tightly linked genetic factors at the PLRV.4 locus on potato chromosome XI control resistance to potato leafroll virus accumulation. Theor Appl Genet 109:1604–1609
Mihovilovich E, Alarcón L, Pérez AL, Alvarado J, Arellano C, Bonierbale M (2007) High levels of heritable resistance to Potato leafroll virus (PLRV) in Solanum tuberosum subsp. andigena. Crop Sci (in press)
Milbourne D, Meyer RC, Collins AJ, Ramsay LD, Gebhardt C, Waugh R (1998) Isolation, characterization and mapping of simple sequence repeat loci in potato. Mol Gen Genet 259:233–245
Pineda O, Bonierbale MW, Plaisted RL, Brodie BB, Tanksley SD (1993) Identification of RFLP markers linked to the H 1 gene conferring resistance to the potato cyst nematode Globodera rostochiensis. Genome 36:152–156
Ritter E, Debener T, Barone A, Salamini F, Gebhardt C (1991) RFLP mapping on potato chromosomes of two genes controlling extreme resistance to potato virus X (PVX). Mol Gen Genet 227:81–85
Rouppe van der Voort JNAM, van Zandvoort P, van Eck HJ, Folkertsma RT, Hutten RCB, Draaistra J, Gommers FJ, Jacobsen E, Helder J, Bakker J (1997) Use of allele specificity of comigrating AFLP markers to align genetic maps from different potato genotypes. Mol Gen Genet 255:438–447
Rouppe van der Voort J, Lindeman W, Folkertsma R, Hutten R, Overmars H, van der Vossen E, Jacobsen E, Bakker J (1998a) A QTL for broad-spectrum resistance to cyst nematode species (Globodera spp.) maps to a resistance gene cluster in potato. Theor Appl Genet 96:654–661
Rouppe van der Voort JNAM, van Eck HJ, Draaistra J, van Zandvoort PM, Jacobsen E, Bakker J (1998b) An online catalog of AFLP markers covering the potato genome. Mol Breed 4:73–77
Rouppe van der Voort J, van der Vossen E, Bakker E, Overmars H, van Zandvoort P, Hutten R, Klein Lankhorst R, Bakker J (2000) Two additive QTLs conferring broad-spectrum resistance in potato to Globodera pallida are localized on resistance gene clusters. Theor Appl Genet 101:1122–1130
Salazar LF, Jayasinghe U (eds) (1997) Ensayos inmunológicos con conjugados enzimáticos: doble-anticuerpo “sandwich” (DAS-ELISA) In: Técnicas en virología de plantas. Manual de capacitación. Fascículo 3.3.6. Centro Internacional de la Papa (CIP), Lima, pp 6
Solomon-Blackburn RM, Barker H (2001) Breeding virus resistant potatoes (Solanum tuberosum): a review of traditional and molecular approaches. Heredity 86:17–35
Solomon-Blackburn RM, Barker H, Bradshaw JE, de Jong W (2003/4) Evidence that resistance to potato leafroll virus accumulation in tetraploid Solanum tuberosum L is controlled by one or few major genes that are not complementary. Potato Res 46:137–145
Song YS, Hepting L, Schweizer G, Hartl L, Wenzel G, Schwarzfischer A (2005) Mapping of extreme resistance to PVY (Ry sto) on chromosome XII using anther culture-derived primary dihaploid lines. Theor Appl Genet 111(5):879–887
Swiezynski KM (1994) Inheritance of resistance to viruses. In: Bradshaw JE, Mackay GR (eds) Potato genetics. CAB International, Wallingford, pp 339–363
Taliansky M, Mayo MA, Barker H (2003) Potato leafroll virus: a classic pathogen shows some new tricks. Mol Plant Pathol 4(2):81–89
Tamada T, Harrison BD (1980) Factors affecting the detection of Potato leafroll virus in potato foliage by enzyme-linked immunosorbent assay. Ann Appl Biol 95:209–219
Upadhya MD, Cabello R (1999) Interaction of maternal clones of potato (Solanum tuberosum) with pollinator clones of S. phureja for induced parthenogenesis. In: Impact on a changing world. Program Report 1997–98. International Potato Center, Lima, pp 179–184
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23(21):4407–4414
Watanabe KN, Orrillo M (1993) An alternative pretreatment method for mitotic chromosome observation in potatoes. Am Potato J 70:543–548
Acknowledgments
Valuable assistance in development of the mapping progeny and identification of dihaploids was provided by Mahesh Upadhya, Romulo Quispe and Matilde Orrillo. We also appreciate the support from Juan Huaccachi and Julio Patilla in greenhouse management and infection assays, Genoveva Rossel in AFLP analyses and Dr. Christine Hackett in the tetraploid map construction.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. E. Bradshaw.
Rights and permissions
About this article
Cite this article
Velásquez, A.C., Mihovilovich, E. & Bonierbale, M. Genetic characterization and mapping of major gene resistance to potato leafroll virus in Solanum tuberosum ssp. andigena . Theor Appl Genet 114, 1051–1058 (2007). https://doi.org/10.1007/s00122-006-0498-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-006-0498-5