
Abstract
We have developed a transiently-expressed transposase (TET)-mediated Dissociation (Ds) insertional mutagenesis system for generating stable insertion lines in rice which will allow localized mutagenesis of a chromosomal region. In this system, a Ds containing T-DNA construct was used to produce Ds launch pad lines. Callus tissues, from single-copy Ds/T-DNA lines, were then transiently infected with Agrobacterium harbouring an immobile Ac (iAc) construct, also containing a green fluorescent protein gene (sgfpS65T) as the visual marker. We have regenerated stable Ds insertion lines at a frequency of 9–13% using selection for Ds excision and GFP counter selection against iAc and nearly half of them were unique insertion lines. Double transformants (iAc/Ds) were also obtained and their progeny yielded ~10% stable insertion lines following excision and visual marker screening with 50% redundancy. In general, more than 50% of the Ds reinsertions were within 1 cM of the launch pad. We have produced a large number of single-copy Ds/T-DNA launch pads distributed over the rice chromosomes and have further refined the Ds/T-DNA construct to enrich for “clean” single-copy T-DNA insertions. The availability of single copy “clean” Ds/T-DNA launch pads will facilitate chromosomal region-directed insertion mutagenesis. This system provides an opportunity for distribution of gene tagging tasks among collaborating laboratories on the basis of chromosomal locations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Following the completion of the genome sequences of Arabidopsis (The Arabidopsis Genome Initiative 2000) and rice (Goff et al. 2002; Yu et al. 2002; International Rice Genome Sequencing Project 2005), the international focus now is to identify the specific functions of the predicted ~30,000 plant genes. Significant advances have been made in rice, a model for cereals including the complete physical map of the japonica rice cv. Nipponbare (Chen et al. 2002), 200,000 publicly available rice expressed sequence tags (ESTs) and ~28,000 full-length cDNAs (Kikuchi et al. 2003). Various bioinformatics, gene mining and gene prediction tools are being developed to improve the sequence annotations. Genome-wide techniques such as microarrays are helping to group genes involved in specific pathways, developmental stages or tissues. Nevertheless, assigning a function to each plant gene is a slow, tedious and often difficult process.
One way of determining gene function is to isolate mutants for each gene and study the effects of those mutations on the plant. Site directed mutagenesis using homologous recombination in plants is still at its infancy (Hanin et al. 2001; Terada et al. 2002). Specific gene expression knockouts by RNA interference (RNAi) or virus-induced gene silencing (VIGS) can be used (for reviews see Wesley et al. 2001; Lu et al. 2003). Another way of producing mutants is random insertion mutagenesis. Retrotransposons (Tos17), T-DNA, iAc/Ds and En/I are being used as insertion mutagens (for a review see Hirochika et al. 2004). Nearly 50,000 Tos17 insertion lines, each containing ~10 copies of Tos17 have been generated (Hirochika 2001; Hirochika et al. 2004). However, only 3% of visible mutants could be traced back to a Tos17 insertion suggesting difficulties in using this method for forward genetics. A sizable population of T-DNA insertion lines have been generated (An et al. 2003, 2005; Chen et al. 2003; Sallaud et al. 2004). Although these resources will be useful for reverse genetics, there are limitations in obtaining T-DNA insertions into smaller genes such as single exon genes which may account for up to 40% of the genes in rice (http://www.btn.genomics.org.cn:8080/rice/annotation.php).
The two-component iAc/Ds (Chin et al. 1999) or En/I (Greco et al. 2004) system provides an advantage over T-DNA system. The transposon-based insertion mutagens can be remobilized to produce new insertion lines in order to target genes in a specific chromosomal region, e.g. corresponding to a mapped quantitative trait loci (QTL). The iAc/Ds-based gene and enhancer trap systems work in rice yielding 5–10% unique stable insertion lines (Chin et al. 1999; Nakagawa et al. 2000; Greco et al. 2001, 2004; Upadhyaya et al. 2002, 2003; Ito et al. 2004; Kim et al. 2004; Kolesnik et al. 2004). The Ds re-insertions linked to the original location of Ds within the T-DNA (the Ds launch pad) varied from 36 to 67% with the majority being within 1 cM of the Ds launch pad (Upadhyaya et al. 2002, unpublished; Kim et al. 2004).
Most of the T-DNA and transposon (Ds or I) constructs used as insertional mutagens have been modified to act as gene traps, thus, in addition to producing gene knockouts, they provide gene expression data as measured by the trap reporter activity. Gene trapping efficiencies of ~6% have been reported for these constructs (see Hirochika et al. 2004). The efficiency of T-DNA gene trapping depends on the frequency of “clean” T-DNA insertions, i.e. insertions devoid of direct or inverted T-DNA repeats or of the incorporation of vector backbone (VB) sequences derived from outside the T-DNA borders (Sallaud et al. 2004; Upadhyaya et al. unpublished). A “clean” Ds containing T-DNA is also essential for satisfactory mobilization of Ds.
Here we report a novel method of producing stable Ds insertion lines using a transiently-expressed transposase (TET) system. We have developed constructs suited for high efficiency insertional mutagenesis in general, and the TET system in particular. By super-infecting callus tissue from single-copy Ds/T-DNA lines, having both Ds excision and reinsertion markers, with Agrobacterium harbouring iAc constructs containing a visual marker sgfpS65T, we have been able to regenerate stable Ds insertion lines at a frequency of ~5%, in addition to iAc/Ds double transformants. Application of mapped single-copy Ds/T-DNA launch pads, produced using these constructs, in efficient chromosomal region-directed insertion mutagenesis is discussed.
Materials and methods
Construction of gene trap Ds and iAc vectors
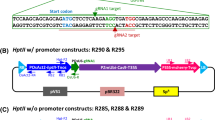
The various cloning intermediates involved in the construction of different gene trap Ds/T-DNA constructs (Fig. 1) can be obtained upon request from the authors. Sources of various components of these constructs are binary VB, T-DNA borders and selectable marker constructs—part of pCAMBIA1300 (http://www.cambia.org/main/r_et_camvec.htm) or pWBVec8 (Wang et al. 1998) or pMNDRBBin1 (Lu et al. 2001); Ds3 and GPA1 intron—pSK200 (Kumar and Narayanan 1997); bla and ColE Ori—pSP72 (Promega Corporation, Madison, USA); Ds5—a 378 bp end region of Ds5′ from pEU334BN (Eamens et al. 2004); sgfpS65—kindly supplied by J. Sheen (Chiu et al. 1996); Barnase—PCR amplified from pMT416 (Hartley 1988) as described previously (Eamens et al. 2004), CaMV35S P-hph(i)-nosT—from pWBVec8 (Wang et al. 1998).

Salient features of constructs developed during this study. a Constructs pUR224NA (GENBANK. Acc. No. DQ225746) and pUR224NB (Acc. No. DQ225747) have a unidirectional gene trap Ds (DsG) with GPA1 intron-triple splice acceptor (SA)-uidA as trap reporter, bla and pBR322 ori as the flanking sequence tag (FST) recovery cassette, and 2′ promoter-nptII-nosT as Ds tracer, inserted between the CaMV35S promoter and bar-nosT cassette. Here the bar serves as the Ds excision marker. These constructs have a CaMV35 promoter driven, intron interrupted hygromycin phosphotransferase gene [hph(i)] as the selectable marker for transformation. Orientations of different components in these constructs indicated; b constructs pNU393A1 (Acc. No. DQ225748) and pNU393B2 (Acc. No. DQ225749) have bidirectional gene trap Ds cassette (Ds3′-GPA1-SA-uidA-nosT and Ds5′-GPA1-SA-eyfp-nosT), also containing FST recovery cassette and CaMV35S P-bar-ocsT as the transformation selection marker and Ds reinsertion marker. The Ds cassette (D5′–Ds3′ orientation) is placed between the CaMV35S promoter and hph(i)-nosT cassette so that hph gene can serve as Ds excision marker. Orientations of the entire cassette within the T-DNA border sequences of these two constructs are shown; c pNU435 (Acc. No. DQ225750) has the promoter-DsG-excision marker cassette of pNU393A1. A specially designed cassette, maize ubiquitin promoter-first exon-modified intron (with LB repeat sequences incorporated)-intron interrupted barnase [bn(i)]-nosT, to serve as vector backbone counter selector as well as dormant gene activator, and (b) a promoterless intron interrupted barnase-nosT cassette placed next to RB before the promoter-DsG-excision marker cassette to serve as T-DNA direct repeat (RB–LB–RB–LB) counter selector and T-DNA gene trap counter selector. This construct also does not have any pBR322 ori in the vector backbone; d iAc constructs pKU352NA (Acc. No. DQ225751) has the Ubi1 P(I)-Ω-iAc-T and CaMV35S P-hph-CaMV35S T cassettes from the construct pSK300 (Kumar and Narayanan 1997) and a Ubi1P-sgfpS65T-nosT cassette from pSK200 (Kumar and Narayanan 1997); e iAc constructs pKU400D (Acc. No. DQ225752) has the Ubi1 P(I)-Ω-iAc-T cassette from the construct pSK300 (Kumar and Narayanan 1997) and a Ubi1P-sgfpS65T-nosT cassette from pSK200 (Kumar and Narayanan 1997). The positions of recognition sites for SalI (Sa), AflII (Af), SphI (Sp), HindIII (H), NheI (Nh), NotI (N) and XhoI (Xh) used in plasmid rescue and/or Southern blot analyses are indicated
The constructs pUR224NA (GenBank Acc. No. DQ225746) and pUR224NB (GenBank Acc. No. DQ225747) essentially have the Ds gene trap cassette from pSK200 [unidirectional gene trap Ds with GPA1 intron-triple splice acceptor-uidA as trap reporter, bla and pBR322 ori as the flanking sequence tag (FST) recovery cassette, and 2′ promoter-nptII-ocsT as Ds tracer] inserted between the CaMV35S promoter and bar-ocsT cassette (placed after T-DNA right border) so that bar gene can serve as Ds excision marker. In pUR224NA, the CaMV35S promoter is upstream of the Ds3′ end which harbours gene trap reporter and in pUR224NB it is upstream of the Ds5′ end. Both constructs have CaMV35S P-hph(i)-nosT selectable marker gene cassette placed before the T-DNA left border.
The constructs pNU393A1 (GenBank Acc. No. DQ225748) and pNU393B2 (GenBank Acc. No. DQ225749) have bidirectional gene trap Ds cassette (Ds3′-GPA1-SA-uidA-nosT and Ds5′-GPA1-SA-eyfp-nosT), also containing FST recovery cassette and CaMV35S P-bar-ocsT as the transformation selection marker and Ds reinsertion marker. The Ds cassette (D5′–Ds3′ orientation) is placed between the CaMV35S promoter and hph(i)-tmlT cassette so that hph gene can serve as Ds excision marker. In pNU393A1, the entire construct is oriented as LB-promoter-DsG-excision marker-RB where as in pNU393B2 it is oriented as RB-promoter-DsG-excision marker-LB. The second pBR322 origin of replication was removed from both binary vector constructs to increase the efficiency of FST recovery by plasmid rescue. The HindIII restriction enzyme (RE) recognition site adjacent to the LB in pNU393B2 was found to be absent due to a 4 bp deletion (possibly happened during the end-filling step of cloning).
The latest construct pNU435 (GenBank Acc. No. DQ225750) has the promoter-DsG-excision marker cassette of pNU393A1. A specially designed cassette, maize ubiquitin promoter-first exon-modified intron (with LB repeat sequences incorporated)-intron interrupted barnase-nosT was placed to serve as VB counter selector and dormant gene activator. A promoterless intron interrupted barnase-nosT cassette was placed next to RB (before the promoter-DsG-excision marker cassette) to serve as T-DNA direct repeat (RB–LB–RB–LB) counter selector and T-DNA gene trap counter selector. This construct also does not have any pBR322 ori in the VB.
The Ubi1P-sgfpS65T-nosT cassette from pSK200 was excised as SpeI/PmeI fragment, end-filled and ligated with pSK300 linearized with PmeI (just inside RB border) in direct orientation to produce pKU352NA (GenBank Acc. No. DQ225751). pCAMBIA1300 was digested with XhoI/EcoRI, end-filled and the vector part was self-ligated. A SacI/SacII fragment from the resulting plasmid (LB region) was used to replace a SacI/SacII fragment (containing iAc-CaMV35SP-hph-CamV35S T-LB) of pKU352NA. The iAc fragment (SacI fragment) was then incorporated to the resulting plasmid to produce pNU400 (GenBank Acc. No. DQ225752), which is another version of transposase construct having only egfpS65T as visual marker without any selectable marker.
Standard procedures were used for restriction endonuclease digestion, restriction fragment elution and purification, end-filling, ligation and bacterial transformation. E. coli strains DH5α (Hanahan 1983) or XL Blue MRF’ (Stratagene, La Jolla, California, USA) were used. Where required, the insert orientation following cloning was determined by sequencing or restriction endonuclease analysis.
Transformation
The steps involved in the callus induction from mature seeds of rice cv. Nipponbare, Agrobacterium-mediated transformation of embryogenic calli and subsequent regeneration of transformant lines have all been previously described (Upadhyaya et al. 2000). pUR224NA and pUR224NB transformant calli were selected with hygromycin (conferred by the CaMV35S promoter-driven hph chimeric gene), whereas pNU393A1 and pNU393B2 transformants were selected with Basta (conferred by CaMV35S promoter-driven bar gene). In both the cases T-DNA copy number was determined by Southern blot hybridization of appropriately digested DNA with radioactively labelled hph probe as described previously (Upadhyaya et al. 2002). Regenerants were grown under controlled greenhouse conditions of 25±3°C and natural light conditions.
Transiently-expressed transposase system
Calli containing single-copy Ds/T-DNA were selected based on Southern blot and FST rescued (plasmid rescue or TAIL PCR). A portion of the primary transgenic callus tissues were used to regenerate plants and produce seeds for subsequent use. Introduction of the iAc constructs was achieved by super transformation as described previously (Upadhyaya et al. 2002). A transient GFP expression gave a measure of number of host cells infected with Agrobacterium which could also express transposase transiently. Subsequent selection pressure for Ds and its excision allowed proliferation of callus cells with transposed Ds. The bar gene was used as Ds excision marker for lines derived from the use of pNR224NA or pNR224NB and pKU352NA. Hygromycin resistance gene (hph) was used as excision marker and bar as reinsertion marker for lines derived from the use of pNU393A1 or pNU393B1 and pNU400. In both the cases GFP served as a visual marker for iAc to separate putative stable insertion lines (GFP−) from possible double transformants (GFP+).
Transgene analysis
Genomic DNA was extracted from Ds/T-DNA transformants using the PureGene (Gentra Systems Inc., Minneapolis, MN, USA) total nucleic acid isolation kit according to the manufacturer’s instructions. Following the initial selection and separation based on hygromycin and/or Basta selection and GFP visualization, respective transgenes were confirmed by PCR (see supplementary Table 1 for primer list) and/or Southern blot hybridization.
Analysis of reporter gene expression
Segments of callus, leaf and root tips of regenerated plantlets and root, leaf, spikelet, mature seed and germinating T1 seed samples of primary transformants were used to screen for reporter gene expression. Screening for sGFPS65T and eYFP expressions was conducted as described previously (Upadhyaya et al. 2002) using special emission and barrier filters (Kurup et al. 2005). Leica MZ FLIII microscope with GFP2 (480/40 nm excision filter and 510 nm barrier filter) and YFP (510/20 nm excision filter and 560/40 nm barrier filter) was used to visualize GFP and YFP expressions. Screening for GUS expression using 5-bromo-4-chloro-3-indolyl-β-d-glucuronide (X-gluc) was done according to Jefferson et al. (1987).
Recovery and analysis of rice Ds/T-DNA flanking sequences
The recovery of sequences flanking Ds/T-DNA gene trap insertions was carried out using the built-in plasmid rescue system by digesting 5 μg of genomic DNA with 10 u of appropriate restriction endonucleases (supplementary Table 2) according to Upadhyaya et al. (2002). Rescued plasmids showing the correct construct-derived “footprint” following cleavage with restriction endonuclease (ApaLI) were sequenced using the appropriate primers and reagents from the ABI Prism BigDye termination cycle sequencing kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. Programs from the University of Wisconsin Genetics Computer Group (GCG) were used for initial flanking sequence analysis (Devereux et al. 1984). All sequences were BLAST searched against publicly available sequences from GenBank (http://www.ncbi.nlm.nih.gov/), China Rice GD (http://www.btn.genomics.org.cn:8080/rice/) or the Rice Genome Whole Annotated Database (http://www.tigrblast.tigr.org/euk-blast/index.cgi?project=osa1) at The Institute for Genome Research (TIGR) to map the FSTs in the rice genome.
TAIL PCR was performed to rescue the RB flanking sequences of some pUR224NA/NB transformants and all pNU393A1/B2 transformants according to previously reported protocol (Liu et al. 1995) with minor modifications. The primers used in the primary, secondary and tertiary PCR reactions were RB-TAIL1 (5′-GCTGATAGTGACCTTAGGCGAC-3′), RB-TAIL2 (5′-CGTTGCGGTTCTGTCAGTTCC-3′) and RBnest5 (5′-ATCAGATTGTCGTTTCCCGC-3′). They were used with one of the two arbitrary degenerate primers AD2 (5′-STTGNTASTNCTNTGC-3′) and AD5 (5′-RCAGNTGWTNGTNCTG-3′), respectively. The secondary and tertiary PCR reactions were loaded on 1% agarose gel side by side. Specific bands with expected size difference were excised from the gel and eluted using UltraCleanTM 15 agarose gel DNA purification kit (Cambio, Cambridge, UK) according to the manufacture’s instructions. The purified PCR products were then re-amplified using the primer combinations used in the tertiary PCR before sequencing.
Availability of the materials reported in this paper
Flanking sequence tag information on the single-copy LPs reported in this paper has been submitted to NCBI (GenBank Acc. Nos. DU711663–DU711806). A regularly updated list of mapped single-copy LPs will be posted at our project website http://www.pi.csiro.au/fgrttpub. These lines will be available for distribution under a normal biological material transfer agreement with CSIRO Plant Industry. Our wish is that people receiving one or more of these lines will keep the imported stock for at least 10 years and will provide the lines to others in their home country upon request from CSIRO Plant Industry.
Results
Constructs for production of Ds launch pads and iAc
Three Ds/T-DNA constructs and two iAc constructs developed in this study are shown in Fig. 1.
Ds/T-DNA construct with bar excision marker
A CaMV35S promoter driven bar gene cassette (Lu et al. 2001) was used in constructs pUR224NA and pUR224NB (Fig. 1a) as the Ds excision marker by placing the gene trap Ds (DsG) cassette between the promoter and the bar gene. The preferred orientation of the unidirectional DsG cassette is as in pUR224NB where the Ds 5′ end is facing the CaMV35S promoter, thus avoiding cis-activation of the gene trap reporter uidA. The gene trap Ds cassette in these constructs was from pSK200 (Kumar and Narayanan 1997; Upadhyaya et al. 2002) which has nptII as the Ds reinsertion marker. The hygromycin resistance gene cassette [CaMV35S P-hph(i)-nosT] as a transformation marker (Lu et al. 2001) was placed near the LB end. The positioning of this selectable marker at the LB end was to avoid selection of transformants with truncated T-DNAs (An et al. 2003; Eamens et al. 2004; Sallaud et al. 2004).
Bidirectional gene trap Ds/T-DNA construct with hph excision marker
The constructs pNU393A1 and pNU393B2 (Fig. 1b) have a previously proven (Upadhyaya et al. 2002; Eamens et al. 2004) bidirectional gene trap Ds cassette (Ds3′-GPA1-SA-uidA-nosT and Ds5′-GPA1-SA-eyfp-nosT). It also contains CaMV35S P-bar-ocsT cassette (Lu et al. 2001) as the transformation selection marker and Ds reinsertion marker, and the FST recovery cassette (plasmid rescue system). A previously tested CaMV35S promoter driven intron-interrupted hph-tmlT gene cassette (Wang et al. 1998) was used as the Ds excision marker by placing the DsG cassette (Ds5′–Ds3′ orientation) between the promoter and the hph(i) gene cassette. Constructs (pNU393A1 and pNU393B2) with either orientation of the entire cassette (promoter-DsG-excision marker) within the T-DNA borders were used in this study. The second pBR322 origin of replication sequence was removed from both the binary vector constructs to increase the efficiency of FST recovery by plasmid rescue (Upadhyaya et al. 2002; Eamens et al. 2004).
Further improvement of Ds/T-DNA construct design
Further construct improvements were made to increase the frequency of selecting single-copy clean T-DNA insertion lines. In the construct pNU435 (Fig. 1c), a maize ubiquitin promoter (with its own first exon and the intron and LB sequence incorporated in the intronic region) driven, intron-interrupted barnase serves as a VB counter selector as transformed cell lines with VB-containing T-DNA inserts will be eliminated by the activity of barnase gene. Moreover, with a clean T-DNA insert the ubiquitin promoter near the LB has the potential to act as a dormant gene activator.
A second copy of promoterless intron interrupted barnase-nosT [bn(i)-nosT] cassette placed within the T-DNA next to RB of the construct pNU435 (before the promoter-DsG-excision marker cassette) has the potential to serve as T-DNA direct repeat (RB–LB–RB–LB) counter selector and as a T-DNA gene trap counter selector. A T-DNA direct repeat transgene will have the strong ubiquitin promoter upstream of the RB side copy of the barnase gene and the resulting cell lines will be eliminated by the barnase gene activity. Insertion of this Ds/T-DNA within a gene with high constitutive expression has the potential to form an active gene-barnase fusion thus counter selection of such gene traps. pNU435 does not contain pBR322 ori in the VB.
iAc constructs
Two versions of iAc constructs were produced and used in this study (Fig. 1d, e). A maize ubiquitin promoter driven (also containing the first exon and intron and the Ω) iAc (Kumar and Narayanan 1997; Upadhyaya et al. 2002) was used in these constructs. In the construct pKU352NA, a Ubi1P-sgfpS65T-nosT cassette is present to serve as a visual marker for iAc as we and others have shown that this expression cassette works very well in rice (Upadhyaya et al. 2002; Kolesnik et al. 2004). Consequently, this can be used effectively as a counter selector for iAc in the TET system of generating stable insertion lines. Another version of the iAc construct (pNU400) was made by removing the selectable marker gene cassette (CaMV35S P-hph-CaMV35S T) to make it compatible with the pNU393A1, pNU393B2 and pNU435 Ds/T-DNA constructs which have hph as excision marker.
Production and mapping of Ds/T-DNA launch pads in rice
A large number of selectable marker resistant callus lines were produced with different Ds/T-DNA constructs (Table 1). We produced 106 lines using the Ds/T-DNA constructs with bar as Ds excision marker and nptII as Ds reinsertion (tracer) marker (pUR224NA/NB), and 656 lines using the Ds/T-DNA constructs with hph as the Ds excision marker and bar as Ds reinsertion marker (pNU393A1/B2).
Southern blot hybridization of Ds/T-DNA lines (see examples in Fig. 2a) indicated that 24 out of 106 lines from pUR224NA/NB and 263 out of 656 lines from pNU393A1/B2 were single-copy lines, respectively (Table 1) which accounted for 23 and 40% of the total number of lines generated, consistent with previous reports (Jeon et al. 2000; Upadhyaya et al. 2002; An et al. 2003, 2005; Kim et al. 2003; Eamens et al. 2004; Ryu et al. 2004; Sallaud et al. 2004). We have obtained T-DNA FSTs from 13 and 88 lines, respectively, from these two sets of lines. FST rescue seemed to work better with pNU393A1/B2 lines than that with pUR224NA/NB lines, presumably because of the absence of a second pBR322 ori in the VB of constructs pNU393A1/B2 as when two copies of ori one of them tends to become inactive by forced mutation (Upadhyaya et al. 2002; Eamens et al. 2004). A complete list of mapped single-copy Ds/T-DNA launch pads is presented in Table 2. The chromosomal locations of these launch pads are graphically represented in Fig. 3. We have launch pads evenly distributed in chromosome 1, the short arm of chromosome 3 and the long arms of chromosomes 4, 5 and 10. We are yet to obtain launch pads in short arms of chromosomes 2, 9 and 10. T-DNA insertion preference for chromosomes 1, 2, 3 and 6 and avoidance of chromosomes 9, 10 and 12 has been reported (An et al. 2005). We are obtaining FST information for the remainder of the single-copy lines and the updated list will be posted at http://www.pi.csiro.au/fgrttpub.

Determination of copy number of Ds/T-DNA or iAc/T-DNA in primary transgenic lines. a ApaI digested genomic DNA isolated from transgenic lines transformed with the Ds/T-DNA construct pNU393B2 and hybridized with radioactively-labelled hph gene sequences; b NotI digested genomic DNA, isolated from different transgenic lines transformed with the iAc construct pNU400, probed with radioactively-labelled iAc sequences

Mapped single-copy Ds/T-DNA launch pads—graphical representation. The BAC/PAC clones corresponding to the FST sequences of single-copy Ds/T-DNA lines were identified by BLAST searching the public rice genome sequences (NCBI). Corresponding mapping (indicated by shaded triangle) in the rice chromosomes (centromere position is indicated by shaded rectangle) was based on the whole genome annotation database of The Institute of Genomic Research (TIGR, http://www.tigr.org/tdb/e2k1/osa1/irgsp.shtml). Regularly updated information of CSIRO Ds/T-DNA launch pads will be available at http://www.pi.csiro.aau/fgrttpub. These lines will be available for distribution under a normal biological material transfer agreement with CSIRO Plant Industry
For producing mutagenic lines by crossing we have several single-copy iAc lines (Fig. 2b) with the construct pNU400 which has gfp as a visual maker but no selection marker.
Ds transposition mediated by transiently-expressed transposase
Ds transposition by TET was demonstrated by introduction of iAc into single-copy Ds/T-DNA (pUR224NA or pUR224NB) callus lines. For the first set of experiments the selection for Ds excision was delayed until 10–12 days following standard co-cultivation and washing but in later experiments selection was applied 3–5 days after co-cultivation. It was important to maintain hygromycin selection throughout. Results of the first 13 super-transformation sessions with nine different Ds/T-DNA lines are summarized in Table 3. The number of putative stable insertion (PSI) lines (GFP−, hygR and BialaphosR) and the putative double transformant (GFP+, HygR) regenerants varied from session to session. In total, out of 230 lines generated (982 plants) 64 lines yielded one or more PSI lines (89 plants), the remainder were iAc/Ds double transformants.
Subsequently, we used single-copy Ds/T-DNA T0 or T1 callus lines of the constructs pNU393A1 and pNU393B2 for the TET system of generating PSIs as they have a Ds excision marker (hph) better suited for selection at the callus stage and have a Ds reinsertion marker (bar) better suited for selection at the seedling stage. An overall strategy for generating PSI lines from these launch pads either by the TET system or by double transformant progeny screening is presented in Fig. 4. Among the 1,013 plants generated so far with the TET system 132 were PSIs and the rest were double transformants giving a PSI frequency of ~13% (data not shown). The maximum expected redundancy is 50% as no more than two PSI plants were retained per independent callus line.

Strategy for generating PSI lines using Ds/T-DNA launch pads either by the TET system (shaded) or by double transformant progeny screening (not shaded)
Identification of PSIs in the progeny of double transformants
All the iAc/Ds double transformants obtained by super transformation of pUR224NA/NB lines were grown to maturity and DtT1 seeds collected for further screening for PSI. We have optimized the screening strategy as follows: (a) GFP scoring with germinating seeds and separate mutagenic (iAc +) and non-mutagenic (iAc −) seeds, (b) hygromycin spraying of seedlings to separate LP+ (hygR) and LP− (hygs) plants, (c) Basta spray to hygR seedlings to select PSIs linked to LP (eliminating intact Ds/T-DNA which could be escapes from DtT0 selection or empty LPs) and (d) PCR analyses of hygS seedlings for transposed Ds. We have collected 6,000 g of seeds from about 1,000 mutagenic lines and identified ~3,400 PSIs (Table 1) among ~30,000 seeds screened.
We are now concentrating on the T1 progeny of double transformants (DtT1) derived from the super transformation of proven single-copy pNU393A1/B2 construct lines. A total of 868 DtT0 plants have yielded 2,639 g of seeds. We have identified ~3,500 PSI lines from among ~30,000 DtT1 seeds screened by the strategy outlined in Fig. 4. We have observed considerable variability in DtT1 populations derived from different DtT0 lines with respect to level of somatic transposition (as measured by the frequency of GUS spots and sectors in shoot and roots) and frequency of PSIs (Table 4 and Fig. 5). We did find a few lines with no GFP expression (iAc −) in the progeny (presumably chimeric primary transformants) and a few lines showing all GFP+ progeny (presumably due to multi-copy iAc). We are yet to look at the efficacy of the bidirectional gene trap feature incorporated in these constructs. Although it was technically challenging to distinguish sGFP and eYFP when both are expressing, we have managed to distinguish sGFP and eYFP in few double transformants with suitable emission and barrier filter especially when GFP expression was low to medium.

GUS expression patterns in selected mutagenic (double transformants) lines and PSI lines derived from the super transformation of pNU393B2 lines with iAc
Transposition behaviour of Ds from selected Ds/T-DNA launch pads
Transposition behaviour of Ds from three launch pads—a pSK200 construct derived launch pad in chromosome 7, a pUR224NA construct derived launch pad in chromosome 4 and a pNU393B2 derived launch pad in chromosome 5—is presented in Table 5. Ds insertions into the same chromosome as the launch pad varied from 40 to 56%. The pNU393B2 construct derived launch pad in chromosome 5 yielded the highest rate of intra-chromosomal transposition and >70% of such transpositions were within 1 cM region flanking the LP. Another set of analyses with nine different launch pads derived from the construct pNU393B2 showed that linked transpositions could be as high as 95%, as out of 179 germinated PSIs 169 were linked to the launch pad (Table 4). We followed a secondary transposition from a PSI mapping to a chromosome 5 BAC clone (GenBank Acc. No. AC108500) linked to the LP which is represented in Fig. 6. Eleven of the 17 lines had the re-transposed Ds in the same chromosome (Chr 5) with four mapping to the same BAC as the original PSI.

Mapping of Ds in new PSI lines derived from a mutagenic line having a transposed Ds (tDs) linked to the original LP (indicated by dotted line). FST sequences rescued from different PSI lines were used to identify corresponding BAC clones by BLAST searching the National Centre for Biotechnology Information (NCBI) nucleotide sequence database (http://www.ncbi.nlm.nih.gov/BLAST/) and the relative positions of the BAC clones were obtained from The Institute of Genomic Research (TIGR) BAC/PAC clone table available at http://www.tigr.org/tdb/e2k1/osa1/irgsp.shtml. GenBank accession numbers are presented along with their relative positions (physical in Mb and genetic in cM) in the TIGR chromosome 5 pseudomolecule (version 3)
Discussion
A novel TET system of producing stable Ds insertion lines was developed using constructs specially designed for efficient screening of Ds transposants linked to the initial Ds/T-DNA launch pads. We have produced large number of Ds/T-DNA launch pads and show that these launch pads can be used in efficient chromosomal region-directed insertion mutagenesis in rice.
It is well-established fact that the two-element iAc/Ds system works very well in rice (Chin et al. 1999; Nakagawa et al. 2000; Greco et al. 2001, 2004; Upadhyaya et al. 2002, 2003; Kim et al. 2004; Kolesnik et al. 2004). We and others have previously shown that the Ds transpositions linked to the original launch pad varies from 36 to 67% (Upadhyaya et al. 2002; Kim et al. 2004) which is consistent with reports both from maize—the original source of Ds and other heterologous systems. Most of the previously used Ds insertion mutagens have been the modified forms with a unidirectional gene trap facility, which provide additional gene expression data as measured by the trap reporter activity (see Hirochika et al. 2004). One way of increasing gene trapping efficiency is by having bidirectional gene traps (Eamens et al. 2004; Ryu et al. 2004). The two Ds/T-DNA constructs (pNU393A1/B2 and pNU435) described in this paper have this bidirectional gene trap facility.
Ds excision markers were incorporated in all the three Ds/T-DNA constructs to increase the screening efficiency for stable insertion lines in a TET system and in the progeny of mutagenic (iAc/Ds) lines. Excision markers in combination with Ds reinsertion markers are particularly useful for selection of Ds insertions linked to the Ds/T-DNA launch pad. Although the constructs pUR224NA/NB had 2′ promoter driven nptII as Ds reinsertion marker we later found out that this nptII expression cassette is not functional in rice (Upadhyaya et al. 2002) and we had to rely on nptII gene specific PCR while developing the TET system to detect the presence of DsG. The newer constructs pNU393A1/B2 and pNU435 have a previously tested CaMV35S promoter-driven intron-interrupted hph gene cassette as the excision marker. Again the choice was based on our experience that this cassette works well as a selectable marker in rice transformation using embryogenic calli as the target tissue (Upadhyaya et al. 2000). We found it particularly advantageous for selecting callus lines with Ds excision using the TET system.
Although the bar gene is not an ideal selectable marker at the callus stage of selection (at least in our hands) it is an excellent selectable marker at the plant stage and is the preferred positive selection marker for Ds tagging in rice (Chin et al. 1999; Kim et al. 2004; Kolesnik et al. 2004). A single spray of Basta can eliminate Ds null segregants, in this instance, lines without DsG reinsertion, thus increasing the screening efficiency. Constructs pNU393A1/B2 and pNU435 have a proven CaMV35 promoter driven bar as Ds reinsertion marker.
The efficacy of T-DNA gene trapping depends on the frequency of “clean” T-DNA insertions without any T-DNA repeats and VB sequences. T-DNA repeats and VB sequences are present in 30–60% of the existing insertion lines (Jeon et al. 2000; Upadhyaya et al. 2002; Kim et al. 2003; Eamens et al. 2004; Sallaud et al. 2004) and are prone to post-integration rearrangements and gross deletions. Furthermore, rescuing the FST from these lines is very difficult or in some cases impossible. In our bidirectional gene trap constructs (Eamens et al. 2004) we did utilize an intron-interrupted barnase gene as a counter selector for VB containing lines (Hanson et al. 1999). Although we saw a reduction in the number of VB containing Ds/T-DNA lines, ~27% lines still contained direct or inverted repeats of T-DNA. We have therefore further refined the construct design (pNU435) by placing two copies of intron-interrupted barnase—one in the VB immediately after ubiquitin promoter-LB sequence and the other immediately after the RB repeat sequence. This configuration has the capacity to counter-select transformants with either VB or direct repeat T-DNA concatermerized inserts. One additional advantage with pNU345 construct design is a potential to counter select Ds/T-DNA insertions in gene with high constitutive expression as explained before. We could not incorporate a counter selector for inverted repeat T-DNA insertions, a common feature in T-DNA integration. This could, however, be addressed by a novel binary vector design where direct and inverted RB–LB integrations will produce a hairpin RNA targeting the selectable marker gene hph (Chen et al. 2005).
During the process of Agrobacterium-mediated transformation single stranded T-DNA is chaperoned into the nucleus of the infected cell (Tzfira et al. 2003, 2004). Although the molecular mechanisms of T-DNA integration leading to stable transgenic events are yet to be fully unravelled, two models have been proposed: single-strand gap repair or double-strand break repair (Tinland 1996; Tzfira et al. 2004). From a close scrutiny of junctions of a large number of T-DNAs in our insertion lines we believe that a double-strand break repair via synthesis dependent strand annealing (SDSA) mechanism is one of the main pathways for T-DNA integration (Zhu et al. unpublished). Integration of T-DNA is most likely to be through a double stranded T-DNA intermediate (Tzfira et al. 2004).
Transient expression of introduced foreign DNA in target plant cells is a well-known phenomenon which occurs before any stable integration through illegitimate recombination or its breakdown by the plant surveillance system. A burst of transient expression of genes carried by the introduced T-DNA can be visualized by reporter gene expression within 48–72 h of co-cultivation (Yoshioka et al. 1996; Upadhyaya et al. unpublished). Using a Ds-interrupted uidA gene construct (excision reporter construct) transposase activity in stably transformed Ac transgenic lines has been measured (Takumi et al. 1999; Koprek et al. 2000). Excision of a Ds-like maize transposable element (Ac delta) by transient expression of transposase has been demonstrated in Petunia protoplasts (Houba-Herin et al. 1990). Encouraged by the above findings, we have developed a system where a TET is used to produce stable insertion lines in rice. For the TET system to work efficiently, it is essential to have single-copy Ds/T-DNA lines which are equivalent to a heterozygous line. It is also important to select Ds/T-DNA lines with no gene trap reporter gene expression. None of the single-copy Ds/T-DNA lines showed any GUS activity in this study.
The main advantage of the TET system is that PSIs can be produced as primary transformants. In contrast, with the iAc/Ds crossing system the first available screening population is F2. Selection for Ds excision (hygromycin or PPT depending on the construct used) is applied after callus co-cultivation with Agrobacterium harbouring iAc which also contains GFP as a visual marker. In theory, any resulting excision marker resistant and GFP− (subsequently confirmed by iAc specific PCR) callus lines would be PSIs (unless resulting callus lines are chimeric). We have confirmed the authenticity of these PSIs by FST rescue and later by segregation analyses. A previous work in tomato on the use of hph gene as a Ds excision marker (Rudenko et al. 1994) did show phenotypic excision despite intact structure of the T-DNA (Ds in its original LP) when the selection pressure was low (30 mg/l). We have pre-tested several single copy Ds/T-DNA lines (containing hph as excision marker) for hygromycin resistance. Although 30 mg/l hygromycin B was not enough to kill rice calli 50 mg/l was good enough to suppress the callus growth efficiently. In tomato also a higher level of hygromycin (80 mg/l) was good enough to suppress any false positive results.
One disadvantage of the TET system is the tissue culture induced somaclonal variation which has been a major drawback with T-DNA insertional mutagenesis (An et al. 2005). We are addressing this by keeping the tissue culture phase to the bare minimum.
As with the screening for PSIs in the progeny of any double transformant lines (originated from single copy Ds/T-DNA lines), the main advantage here is easy sorting of PSIs linked to the original LP (GFP− and iAc PCR−, Ds tracer/reinsertion markerR and excision markerR) and PSIs unlinked from the original LP (GFP− and iAc PCR−, Ds tracer/reinsertion markerR and excision markerS). It should however be noted that if there were to be no retranspositions in any line one would get just a single linked PSI from that line (only thing happened is iAc segregation). In our latest constructs, the excision marker is hph. Hygromycin selection works very well with calli but hygromycin application does not kill the seedlings but can easily be scored for resistance and sensitivity. Because of this, any unlinked PSIs also can be easily identified. Again, if there were to be no retransposition one would get a single PSI from a line. For all these things final confirmation would be obtaining unique FSTs.
In our hand, use of GFP as a tracer for iAc is working very well with ~90% accuracy as confirmed by iAc specific PCR. Although any GFP silenced (or undetectably expressing) plants will initially be considered as PSIs, subsequent iAc specific PCR eliminates them. This holds good in screening the progeny plants of double transformants or crosses between iAc (with GFP) and Ds/T-DNA lines. Only problem in using both yfp (one of the gene trap reporter) and gfp (iAc visual reporter) is distinguishing YFP expression from GFP expression. Although special filters can be used to differentiate these two it is very difficult to show the YFP expression superimposed in GFP background by photography (although with careful observation we can distinguish them by eye). For a large-scale use it would be better to use some other visual reporters such as DsRed.
Our strategy is to produce sizable numbers of single-copy Ds/T-DNA launch pads distributed all over the chromosomes so that subsequent localized or targeted insertional mutagenesis can be achieved by selecting Ds transposants linked to the original launch pad. Although published data indicate a great variability in the transposition behaviour of Ds in rice (Chin et al. 1999; Upadhyaya et al. 2002; Kolesnik et al. 2004), it is generally accepted that Ds tends to transpose to locations close to its original position (launch pad) in plant species tested so far (Muskett et al. 2003). Our study with PSI lines derived from three launch pads (one of each construct) also showed frequency of intra-chromosomal insertion ranging from 40 to 56%. Our pNU393B2 construct derived launch pads seemed to yield higher proportion of linked insertions (Table 4). These launch pads are in different chromosomes (Chr 3, Chr 4, Chr 5, Chr 6 and Chr 11) and our intention now is to use such mapped launch pads in trait targeted insertional mutagenesis.
In conclusion, we have produced a set of Ds and iAc constructs with features suited for targeted localized insertion mutagenesis, and using these constructs, we are producing a large number of mapped single-copy Ds/T-DNA launch pads. Using the callus tissue from some of these single-copy Ds/T-DNA launch pads we have shown that a TET can be used to produce stable insertion lines. The availability of a large number of such clean Ds/T-DNA launch pads located throughout the rice genome will facilitate chromosomal region-directed high throughput insertion mutagenesis. A similar approach has been reported for Arabidopsis (Muskett et al. 2003). Saturation insertional mutagenesis of the whole rice genome is a huge task and requires serious collaborations among rice researchers. Chromosomal region-wise distribution of gene tagging task among collaborating laboratories is feasible with such Ds launch pads.
References
An S, Park S, Jeong DH, Lee DY, Kang HG, Yu JH, Hur J, Kim SR, Kim YH, Lee M, Han S, Kim SJ, Yang J, Kim E, Wi SJ, Chung HS, Hong JP, Choe V, Lee HK, Choi JH, Nam J, Park PB, Park KY, Kim WT, Choe S, Lee CB, An G (2003) Generation and analysis of end sequence database for T-DNA tagging lines in rice. Plant Physiol 133:2040–2047
An G, Lee S, Kim SH, Kim SR (2005) Molecular genetics using T-DNA in rice. Plant Cell Physiol 46:14–22
Chen M, Presting G, Barbazuk WB, Goicoechea JL, Blackmon B, Fang G, Kim H, Frisch D, Yu Y, Sun S, Higingbottom S, Phimphilai J, Phimphilai D, Thurmond S, Gaudette B, Li P, Liu J, Hatfield J, Main D, Farrar K, Henderson C, Barnett L, Costa R, Williams B, Walser S, Atkins M, Hall C, Budiman MA, Tomkins JP, Luo M, Bancroft I, Salse J, Regad F, Mohapatra T, Singh NK, Tyagi AK, Soderlund C, Dean RA, Wing RA (2002) An integrated physical and genetic map of the rice genome. Plant Cell 14:3–545
Chen S, Jin W, Wang M, Zhang F, Zhou J, Jia Q, Wu Y, Liu F, Wu P (2003) Distribution and characterization of over 1000 T-DNA tags in rice genome. Plant J 36:105–113
Chen S, Helliwell CA, Wu L-M, Dennis ES, Upadhyaya NM, Zhang R, Waterhouse PM, Wang M-B (2005) A novel T-DNA vector design for selection of transgenic lines with simple transgene integration and stable transgene expression. Funct Plant Biol 32:671–681
Chin HG, Choe MS, Lee SH, Park SH, Koo JC, Kim NY, Lee JJ, Oh BG, Yi GH, Kim SC, Choi HC, Cho MJ, Han CD (1999) Molecular analysis of rice plants harboring an Ac/Ds transposable element-mediated gene trapping system. Plant J 19:615–623
Chiu W-l, Niwa Y, Zeng W, Hirano T, Kobayashi H, Sheen J (1996) Engineered GFP as a vital reporter for plants. Curr Biol 6:325–330
Devereux J, Haeberli P, Smithies O (1984) A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res 12:387–395
Eamens AL, Blanchard CL, Dennis ES, Upadhyaya NM (2004) A bidirectional gene trap construct for T-DNA and Ds mediated insertional mutagenesis in rice (Oryza sativa L.). Plant Biotechnol J 2:367–380
Goff SA, Ricke D, Lan TH, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, Hadley D, Hutchison D, Martin C, Katagiri F, Lange BM, Moughamer T, Xia Y, Budworth P, Zhong J, Miguel T, Paszkowski U, Zhang S, Colbert M, Sun WL, Chen L, Cooper B, Park S, Wood TC, Mao L, Quail P, Wing R, Dean R, Yu Y, Zharkikh A, Shen R, Sahasrabudhe S, Thomas A, Cannings R, Gutin A, Pruss D, Reid J, Tavtigian S, Mitchell J, Eldredge G, Scholl T, Miller RM, Bhatnagar S, Adey N, Rubano T, Tusneem N, Robinson R, Feldhaus J, Macalma T, Oliphant A, Briggs S (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296:92–100
Greco R, Ouwerkerk PB, Sallaud C, Kohli A, Colombo L, Puigdomenech P, Guiderdoni E, Christou P, Hoge JH, Pereira A (2001) Transposon insertional mutagenesis in rice. Plant Physiol 125:1175–1177
Greco R, Ouwerkerk PB, Taal AJ, Sallaud C, Guiderdoni E, Meijer AH, Hoge JH, Pereira A (2004) Transcription and somatic transposition of the maize En/Spm transposon system in rice. Mol Genet Genomics 270:514–523
Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557
Hanin M, Volrath S, Bogucki A, Briker M, Ward E, Paszkowski J (2001) Gene targeting in Arabidopsis. Plant J 28:671–677
Hanson B, Engler D, Moy Y, Newman B, Ralston E, Gutterson N (1999) A simple method to enrich an Agrobacterium-transformed population for plants containing only T-DNA sequences. Plant J 19:727–734
Hartley RW (1988) Barnase and barstar. Expression of its cloned inhibitor permits expression of a cloned ribonuclease. J Mol Biol 202:913–915
Hirochika H (2001) Contribution of the Tos17 retrotransposon to rice functional genomics. Curr Opin Plant Biol 4:118–122
Hirochika H, Guiderdoni E, An G, Hsing YI, Eun MY, Han CD, Upadhyaya N, Ramachandran S, Zhang Q, Pereira A, Sundaresan V, Leung H (2004) Rice mutant resources for gene discovery. Plant Mol Biol 54:325–334
Houba-Herin N, Becker D, Post A, Larondelle Y, Starlinger P (1990) Excision of a Ds-like maize transposable element (Ac delta) in a transient assay in Petunia is enhanced by a truncated coding region of the transposable element Ac. Mol Gen Genet 224:17–23
International Rice Genome Sequencing Project (2005) The map-based sequence of the rice genome. Nature 436:793–800
Ito Y, Eiguchi M, Kurata N (2004) Establishment of an enhancer trap system with Ds and GUS for functional genomics in rice. Mol Genet Genomics 271:639–650
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6:3901–3907
Jeon JS, Lee S, Jung KH, Jun SH, Jeong DH, Lee J, Kim C, Jang S, Yang K, Nam J, An K, Han MJ, Sung RJ, Choi HS, Yu JH, Choi JH, Cho SY, Cha SS, Kim SI, An G (2000) T-DNA insertional mutagenesis for functional genomics in rice. Plant J 22:561–570
Kikuchi S, Satoh K, Nagata T, Kawagashira N, Doi K, Kishimoto N, Yazaki J, Ishikawa M, Yamada H, Ooka H, Hotta I, Kojima K, Namiki T, Ohneda E, Yahagi W, Suzuki K, Li CJ, Ohtsuki K, Shishiki T, Otomo Y, Murakami K, Iida Y, Sugano S, Fujimura T, Suzuki Y, Tsunoda Y, Kurosaki T, Kodama T, Masuda H, Kobayashi M, Xie Q, Lu M, Narikawa R, Sugiyama A, Mizuno K, Yokomizo S, Niikura J, Ikeda R, Ishibiki J, Kawamata M, Yoshimura A, Miura J, Kusumegi T, Oka M, Ryu R, Ueda M, Matsubara K, Kawai J, Carninci P, Adachi J, Aizawa K, Arakawa T, Fukuda S, Hara A, Hashizume W, Hayatsu N, Imotani K, Ishii Y, Itoh M, Kagawa I, Kondo S, Konno H, Miyazaki A, Osato N, Ota Y, Saito R, Sasaki D, Sato K, Shibata K, Shinagawa A, Shiraki T, Yoshino M, Hayashizaki Y, Yasunishi A (2003) Collection, mapping, and annotation of over 28,000 cDNA clones from japonica rice. Science 301:376–379
Kim SR, Lee J, Jun SH, Park S, Kang HG, Kwon S, An G (2003) Transgene structures in T-DNA-inserted rice plants. Plant Mol Biol 52:761–773
Kim CM, Piao HL, Park SJ, Chon NS, Je BI, Sun B, Park SH, Park JY, Lee EJ, Kim MJ, Chung WS, Lee KH, Lee YS, Lee JJ, Won YJ, Yi G, Nam MH, Cha YS, Yun DW, Eun MY, Han CD (2004) Rapid, large-scale generation of Ds transposant lines and analysis of the Ds insertion sites in rice. Plant J 39:252–263
Kolesnik T, Szeverenyi I, Bachmann D, Kumar CS, Jiang S, Ramamoorthy R, Cai M, Ma ZG, Sundaresan V, Ramachandran S (2004) Establishing an efficient Ac/Ds tagging system in rice: large-scale analysis of Ds flanking sequences. Plant J 37:301–314
Koprek T, McElroy D, Louwerse J, Williams-Carrier R, Lemaux PG (2000) An efficient method for dispersing Ds elements in the barley genome as a tool for determining gene function. Plant J 24:253–263
Kumar SC, Narayanan KK (1997) Gene and enhancer traps for isolating genetic regions from rice. Rice Biotechnol Quart 31:17–18
Kurup S, Runions J, Kohler U, Laplaze L, Hodge S, Haseloff J (2005) Marking cell lineages in living tissues. Plant J 42:444–453
Liu YG, Mitsukawa N, Oosumi T, Whittier RF (1995) Efficient isolation and mapping of Arabidopsis thaliana T-DNA insert junctions by thermal asymmetric interlaced PCR. Plant J 8:457–463
Lu HJ, Zhou X-R, Gong Z-X, Upadhyaya NM (2001) Generation of selectable marker-free transgenic rice using double right-border (DRB) binary vectors. Aust J Plant Physiol 28:241–248
Lu R, Martin-Hernandez AM, Peart JR, Malcuit I, Baulcombe DC (2003) Virus-induced gene silencing in plants. Methods 30:296–303
Muskett PR, Clissold L, Marocco A, Springer PS, Martienssen R, Dean C (2003) A resource of mapped dissociation launch pads for targeted insertional mutagenesis in the Arabidopsis genome. Plant Physiol 132:506–516
Nakagawa Y, Machida C, Machida Y, Toriyama K (2000) Frequency and pattern of transposition of the maize transposable element Ds in transgenic rice plants. Plant Cell Physiol 41:733–742
Rudenko GN, Nijkamp HJ, Hille J (1994) Ds read-out transcription in transgenic tomato plants. Mol Gen Genet 243:426–433
Ryu CH, You JH, Kang HG, Hur J, Kim YH, Han MJ, An K, Chung BC, Lee CH, An G (2004) Generation of T-DNA tagging lines with a bidirectional gene trap vector and the establishment of an insertion-site database. Plant Mol Biol 54:489–502
Sallaud C, Gay C, Larmande P, Bes M, Piffanelli P, Piegu B, Droc G, Regad F, Bourgeois E, Meynard D, Perin C, Sabau X, Ghesquiere A, Glaszmann JC, Delseny M, Guiderdoni E (2004) High throughput T-DNA insertion mutagenesis in rice: a first step towards in silico reverse genetics. Plant J 39:450–464
Takumi S, Murai K, Mori N, Nakamura C (1999) Variations in the maize Ac transposase transcript level and the Ds excision frequency in transgenic wheat callus lines. Genome 42:1234–1241
Terada R, Urawa H, Inagaki Y, Tsugane K, Iida S (2002) Efficient gene targeting by homologous recombination in rice. Nat Biotechnol 20:1030–1034
The Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408:796–815
Tinland B (1996) The integration of T-DNA into plant genomes. Trends Plant Sci 1:178–184
Tzfira T, Frankman LR, Vaidya M, Citovsky V (2003) Site-specific integration of Agrobacterium tumefaciens T-DNA via double-stranded intermediates. Plant Physiol 133:1011–1023
Tzfira T, Li J, Lacroix B, Citovsky V (2004) Agrobacterium T-DNA integration: molecules and models. Trends Genet 20:375–383
Upadhyaya NM, Surin B, Ramm K, Gaudron J, Schünmann PHD, Taylor W, Waterhouse PM, Wang M-B (2000) Agrobacterium-mediated transformation of Australian rice cultivars Jarrah and Amaroo using modified promoters and selectable markers. Aust J Plant Physiol 27:201–210
Upadhyaya NM, Zhou X-R, Ramm K, Zhu Q-H, Wu L, Eamens AL, Sivakumar R, Kato T, Yun D-W, Kumar S, Narayanan KK, Peacock WJ, Dennis ES (2002) An iAc/Ds gene and enhancer trapping system for insertional mutagenesis in rice. Funct Plant Biol 29:547–559
Upadhyaya NM, Zhu Q-H, Eamens AL, Dennis ES (2003) Rice gene machine: a vehicle for finding functions of cereal genes. Asia Pacific Biotechnol News 6:936–942
Wang MB, Cheng Z, Keese P, Graham MW, Larkin PJ, Waterhouse PM (1998) Comparison of the coat protein, movement protein and RNA polymerase gene sequences of Australian, Chinese, and American isolates of barely yellow dwarf virus transmitted by Rhopalosiphum padi. Arch Virol 143:1005–1013
Wesley SV, Helliwell CA, Smith NA, Wang MB, Rouse DT, Liu Q, Gooding PS, Singh SP, Abbott D, Stoutjesdijk PA, Robinson SP, Gleave AP, Green AG, Waterhouse PM (2001) Construct design for efficient, effective and high-throughput gene silencing in plants. Plant J 27:581–590
Yoshioka Y, Takahashi Y, Matsuoka K, Nakamura K, Koizumi J, Kojima M, Machida Y (1996) Analysis of virulence loci in Agrobacterium tumefaciens required for T-DNA transfer by the transient gene expression system. Plant Cell Physiol 37:782–789
Yu J, Hu S, Wang J, Wong GK, Li S, Liu B, Deng Y, Dai L, Zhou Y, Zhang X, Cao M, Liu J, Sun J, Tang J, Chen Y, Huang X, Lin W, Ye C, Tong W, Cong L, Geng J, Han Y, Li L, Li W, Hu G, Li J, Liu Z, Qi Q, Li T, Wang X, Lu H, Wu T, Zhu M, Ni P, Han H, Dong W, Ren X, Feng X, Cui P, Li X, Wang H, Xu X, Zhai W, Xu Z, Zhang J, He S, Xu J, Zhang K, Zheng X, Dong J, Zeng W, Tao L, Ye J, Tan J, Chen X, He J, Liu D, Tian W, Tian C, Xia H, Bao Q, Li G, Gao H, Cao T, Zhao W, Li P, Chen W, Zhang Y, Hu J, Liu S, Yang J, Zhang G, Xiong Y, Li Z, Mao L, Zhou C, Zhu Z, Chen R, Hao B, Zheng W, Chen S, Guo W, Tao M, Zhu L, Yuan L, Yang H (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296:79–92
Acknowledgements
The authors wish to thank Celia Miller, Jane Liu for their invaluable assistance. Thanks to Drs. John Watson, Ramesh Bhat, Ming-Bo Wang, Chris Helliwell and Michael Ayliffe for their critical reading. This work was supported by Rural Industries Research and Development Corporation (RIRDC), Australia and New South Wales Agricultural Genomic Center, Australia.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by E. Guiderdoni
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Upadhyaya, N.M., Zhu, QH., Zhou, XR. et al. Dissociation (Ds) constructs, mapped Ds launch pads and a transiently-expressed transposase system suitable for localized insertional mutagenesis in rice. Theor Appl Genet 112, 1326–1341 (2006). https://doi.org/10.1007/s00122-006-0235-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-006-0235-0