
Abstract
Seed dormancy in rice interrelates to the weedy characteristics shattering, awn, black hull color, and red pericarp color. A cross between the weedy strain SS18-2 and the breeding line EM93-1 was developed to investigate the genetic basis and adaptive significance of these interrelationships. These characteristics or their components differed in average degree of dominance from −0.8 to 1.5, in heritability from 0.5 to 0.96, and in their contribution to phenotypic or genotypic variation in dormancy by up to 25%. Five dormancy, four shattering, and three awn-length quantitative trait loci (QTLs) were detected in the BC1 population replicated in 2 years. Two QTLs for hull color were identified, and the SS18-2-derived and EM93-1-derived alleles increased the intensity of black, and red or yellow pigmentations, respectively. The only QTL for pericarp color co-located with the red pericarp gene Rc, with the SS18-2-derived allele increasing the intensity of black and red pigmentations. Four of the five dormancy QTLs were flanked or bracketed by one to four QTLs for the interrelated characteristics. The QTL organization pattern indicates the central role of seed dormancy in adaptive syndromes for non-domesticated plants, implies that the elimination of dormancy from cultivars could arise from the selections against multiple interrelated characteristics, and challenges the use of dormancy genes at these loci in breeding varieties for resistance to pre-harvest sprouting (PHS). However, another QTL (qSD12) provides candidate gene(s) for PHS resistance because it has a large effect in the population and it is independent of the loci for interrelated characteristics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Weeds are plants adapted to human disturbances. A number of characteristics, also known as “adaptive syndromes”, contribute to the adaptation and persistence of weeds (Harlan and de Wet 1965). Weedy races are closely related to the evolution of many cultivated plants (Harlan 1965; Oka 1988) and are treated as part of the primary gene pool for crop breeding (Harlan et al. 1973). Many weedy characteristics, such as seed dormancy, shattering, appendages, and pigmentations, are also important domestication-related traits. Genomic tools are accelerating our understanding of the genetic bases for plant adaptation or crop domestication, such as at a quantitative trait loci (QTL) level (Doebley and Stec 1991; Paterson et al. 1995) or in molecular mechanisms by cloning the underlying genes (Frary et al. 2000). In addition, such research renders opportunities to regain genetic diversity lost from cultivars during domestication and breeding (Tanksley and McCouch 1997) and to seek new strategies for weed management.
Seed dormancy, the temporary failure of a viable seed to germinate under favorable conditions, provides resistance to pre-harvest sprouting (PHS) in cereal crops. Genetic research on crops over many years has focused mainly on cultivated varieties to evaluate dormancy gene resources for breeding varieties resistant to PHS but, unfortunately, heritability for seed dormancy is relatively low in major crops such as rice and wheat (Chang and Yen 1969; Paterson and Sorrells 1990). In contrast, weedy races provide an alternative source of dormancy genes (Gu et al. 2003). However, some characteristics, such as grain type and awns in wild oat (Avena fatua) (Johnson 1935; Simpson 1992), seed-coat color in wheat (Triticum aestivum) and proso millet (Panicum milliaceum) (Gfeller and Svejda 1960; Khan et al. 1996), and seed shattering in wild rice (Oryza rufipogan) (Oka 1988; Cai and Morishima 2000) are associated with seed dormancy. The introgression of genes from non-domesticated genomes may be hindered by these interrelated characteristics, or result in a loss of desirable characteristics in the cultivated parent (Matus et al. 2003). Thus, information on the genomic distribution of potentially important QTLs is also valuable for estimating the impact of undesirable characteristics on the use of weedy and other non-domesticated genetic resources in breeding programs.
QTL clusters for adaptive or domestication-related traits have been reported in various plant species (Doebley and Stec 1991; Paterson et al. 1995; Burke et al. 2002; Peng et al. 2003), including wild (O. rufipogan) and japonica-type weedy rice (O. sativa) (Xiong et al. 1999; Bres-Patry et al. 2001; Cai and Morishima 2002; Thomson et al. 2003). As seed dormancy was excluded as a factor in a majority of these experiments, our understanding of the adaptive significance of this characteristic in the context of plant evolution is limited. On the other hand, more than 30 putative dormancy QTLs (Lin et al. 1998; Cai and Morishima 2000; Dong et al. 2002; Gu et al. 2004; Miura et al. 2002) have been reported in rice, a model monocotyledonous species. However, comparative genetic analysis between rice and other cereal species for seed dormancy has been based on the mapping information of a few QTLs (Kato et al. 2001; Gale et al. 2002; Li et al. 2004). Thus, the confirmation of reported dormancy QTLs is crucial for precision comparisons across species for this biologically and agriculturally important trait.
In previous research we characterized the phenotypic interrelationship between seed dormancy and a set of weedy characteristics, including seed shattering, awn, black hull color, and red pericarp color (Gu et al. 2003, 2005). The research reported here allowed us to identify genetic behaviors and QTLs for the set of dormancy-interrelated characteristics. A majority of the QTLs were found in four genomic regions; this forms the basis for our discussions on the role of seed dormancy in adaptive syndromes and the usefulness of dormancy genes in breeding programs for resistance to PHS.
Materials and methods
Plant genotypes and cultivation
The rice (Oryza sativa) weedy strain SS18-2 was used as the male parent to cross with the breeding line EM93-1. The F1 as the male parent was backcrossed with EM93-1 to generate the mapping population (BC1). Both parents are indica-type, and the F1 plants displayed more than 90% seed set. For rice, a seed usually refers to a dispersal unit that consists of the hull (i.e., lemma and palea), pericarp, testa, endosperm, and embryo, and the hull tightly encloses the caryopsis (Grist 1986). The SS18-2 strain has a high degree of seed dormancy and shattering, a long awn on the lemma, a black hull and a red pericarp (and testa). These weedy characteristics are absent in EM93-1.
Two-hundred and four BC1 and five parental and F1 plants were grown in pots, with one plant per pot, in a greenhouse in June 2002. A subset of 156 plants was replicated three times using the split-tiller technique described by Gu et al. (2004). Plants were watered daily to keep about 2 cm of standing water above the soil before harvesting. After harvesting, the soil was kept wet, and the plants were cut at about 15 cm above the bottom and maintained under a 14/10-h (light/dark) photoperiod to induce the development of dormant buds. Young tillers split from 198 BC1 ratooning plants were transplanted into new pots at the same time to generate an identical population in January 2003. Ten parental and F1 plants were also grown in 2003. The plants were subjected to a 10/14-h (light/dark) photoperiod to synchronize flowering. Panicles were covered by paper bags and fixed to bamboo poles to prevent shattering. Average temperature and relative humidity during the period from the onset of flowering to the end of harvest were 26±1°C and 61±11%, respectively, in 2002, and 26±1°C and 41±8%, respectively, in 2003. Seeds were harvested at 40 days after flowering, and immature ones were removed. The seeds were air dried in the greenhouse for 3 days to about 12% moisture on a dry weight basis and then stored at −20°C to prevent loss of dormancy by dry after-ripening.
Phenotypic identification
Dormancy was measured by percentage germination after 7 days of incubation. Intact seeds were after-ripened at about 25°C for 1, 11 and 21 days prior to germination. A germination experiment consisted of three replications. About 50 seeds of each replication were placed in a 9-cm petri dish that was lined with a Whatman No.1 filter paper, wetted with 10 ml de-ionized water and incubated at 30°C and 100% relative humidity in the dark. Percentage germination (y) was transformed by sin−1 (y)−0.5 for statistical analysis.
To assess seed shattering, panicles were cut from the plant and gently shaken for about 20 s over a container to collect shattered seeds, and then hand threshed to collect non-shattered ones. Seeds were cleaned by removal of empty spikelets and dried in a greenhouse for 3 days. Shattering was evaluated by the percentage of shattered seed to the total seed weight. The awn characteristic was quantified by mean awn length using three random samples of 50 seeds from a plant. Awn length data (x) were transformed by (x+0.5)0.5 to improve normality and homogeneity.
Hull and pericarp colors were measured with a Chroma Meter (Minolta CR310), a method that has been used to quantify red grain color in wheat (Wang et al. 1999; Groos et al. 2002). The reflectance spectra were transformed into component black, red, and yellow pigmentations and then quantified with L*, a*, and b* values, respectively. The L* values range from 0 to 100, indicating completely non-reflective (black) to perfectly reflective (white). The a* values vary from −100 to 100, with negative and positive values indicating degrees of green and red pigmentations, respectively; a high, positive a* value indicates a high intensity of redness. The b* values vary from −100 to 100, with the negative and positive values indicating degrees of blue and yellow pigmentations, respectively; a high, positive b* value indicates a high intensity of yellow pigmentation. About 500 seeds or 150 caryopses harvested in 2003 were placed in a 60-mm or a 35-mm petri dish and set against a black background. Caryopses were obtained by hand removal of the hull from intact seeds. The measurement was repeated three times with seeds or caryopses being mixed after each reading. Readings were averaged for further analysis.
Data analysis
Data from the parental and F1 generations were used to estimate average degree of dominance (ADD) (Kearsey and Pooni 1996) for the characteristics seed shattering, awn and pigmentations on the hull and pericarp. Data obtained from the replicated subpopulation in 2002 were used to estimate heritability for shattering and awn length and the genetic correlations between degree of dormancy (y) and each of the two aforementioned characteristics (x). The statistics required for the estimations were obtained from analyses of variance (anova) and covariance based on the two-factor linear model (Gu et al. 2004). The heritability (h2) in the BC1 population is defined as the proportion of genotypic (i.e., between split-tiller-derived lines) to phenotypic variances. The phenotypic (rp), genotypic (rg), and environmental (re) correlation coefficients are calculated as covp/δ2pyδ2 px, covg/δ2 gyδ2 gx, and cove/δ2 eyδ2 ex, respectively, where the covg and cove are the genetic and environmental covariances, the δ2 gy and δ2 ey, or δ2 gx and δ2 ex are the component genetic and environmental variances, respectively, for the pair of characteristics x and y; and the covp, or δ2 py and δ2 px are the corresponding phenotypic covariance or variances, which equals to covg+cove or δ2 gy+ δ2 ey and δ2 gx+ δ2 ex, respectively. The significance of the correlations was tested using the formula developed by Kearsey and Pooni (1996).
Germination and shattering data from 2002 and 2003 were used to detect the effect of year (growth environment) using a two-way anova based on a two-factor (genotype and year) model. Expected mean squares from the anova were dissected into their components to estimate h2 for dormancy and shattering in the BC1 population across 2 years. Data for component pigmentations on the hull and pericarp were used to estimate their correlations with the degree of dormancy.
Linkage map and QTL analysis
An additional 25 rice microsatellite (RM) markers (Temnykh et al. 2001; McCouch et al. 2002) were added to the framework genetic map, which was constructed using 150 RM markers based on the subpopulation of 156 BC1 plants (Gu et al. 2004). The remaining 48 BC1 plants were also marker-genotyped using the same methods previously described (Gu et al. 2004). The genetic map was adjusted with the additional individuals and markers using mapmaker/exp 3.0 (Lincoln et al. 1992).
mqtl beta ver. 0.98 (http://genome.agrenv.mcgill.ca/software/MQTL) was used for the QTL analyses. The simple interval mapping (SIM) procedure was used to infer the presence of a QTL or a QTL × E (year) interaction, and simplified composite interval mapping (sCIM) procedure used to refine the QTL peak position and main effect as suggested by Tinker and Mather (1995). The procedures were run in the two-environment (year) model for germination and shattering and in the single environment model for the awn length and pigmentations. The threshold for a significant QTL or QTL × E interaction was generated by 1,000 permutations at a type-I error rate of 0.05. The minimum genetic distance in the inference file to search for linked QTLs was set at 30 cM. Likelihood intervals of QTLs for shattering, awn length, and pigmentations are given as a test statistic (TS) of 14 that was generated by the SIM single-environment model. This level is equivalent to a LOD score of three generated by mapmaker/qtl (Tinker and Mather 1995). Thus, shattering data from identical split-tiller-derived plants from the 2 years were averaged to estimate the TS support interval. The remaining parameters set to run the procedures and the methods used to estimate QTL single- and multiple-locus (full-model) effects have been described in detail in our previous research (Gu et al. 2004).
A two-way anova was used to detect epistasis between the QTLs for shattering, awn length, and pigmentation on the hull. Analysis was based on a two-factor factorial model consisting of main effects of the markers nearest to the QTLs and their interaction effect. All of the above statistical analyses were implemented using the SAS glm or corr programs (SAS Institute 1999).
Results
Seed dormancy
Two-way anova detected significant genotypic and year effects on the germination of seeds at 1, 11, and 21 days of after-ripening (DAR) based on the subpopulation of BC1 split-tiller-derived lines (data not shown). Heritabilities for degree of dormancy across 2 years were 0.49, 0.66, and 0.76 at 1, 11, and 21 DAR, respectively. Correlation coefficients between the 2 years for dormancy degree at 1, 11, and 21 DAR were 0.548, 0.694, and 0.808, respectively. The correlations suggest that about 30–65% (r2) of the phenotypic variations at 1–21 DAR could result from the same genetic and/or environmental factors across the 2 years.
Five dormancy QTLs were identified, and these were relatively constant across years (Table 1). These loci mapped to chromosomes 4, 7 (two loci), 8, and 12, and are designated as qSD4, 7-1, 7-2, 8, and 12, respectively (Fig. 1). The five QTLs together accounted for 37%, 45%, and 44% of the total phenotypic variance in germination at 1, 11, and 21 DAR, respectively, with qSD12 contributing the most to total variance at each DAR (Table 1). Alleles that repressed germination at all five loci were derived from the weedy strain SS18-2. The locus qSD6 detected in a previous experiment (Gu et al. 2004) had a relatively minor effect in this experiment, as indicated by a TS peak value of 18.8 relative to a threshold of 20.1 at 21 DAR.

Distribution of QTLs responsible for seed dormancy and its interrelated characteristics on rice chromosomes. The map was based on 204 BC1 (EM93-1//EM93-1/SS18-2) individuals with only chromosomes harboring the QTLs displayed. The RM markers are labeled to the right of chromosomes 3, 4, 7, 8, and 12. Ovals on a chromosome depict the range of the TS peaks for QTLs detected based on the germination of seeds after-ripened for 1, 11, and 21 days. The vertical boxes to the left of the chromosomes depict the 14-TS (equivalent to a LOD of 3, computed by mapmaker/qtl) support limits of QTLs for seed shattering, awn-length, and black, red, and yellow pigmentations on the hull and pericarp mapped using the mqtl SIM single-environment model (Tinker and Mather 1995). Arrows on the boxes indicate the TS peak positions for the QTLs. The horizontal bars on the empty boxes indicate the TS limits defined by the black (b), red (r), and yellow (y) pigmentations on the hull and pericarp
One dormancy QTL × E interaction was detected at 1 DAR and two interactions at 11 DAR and 21 DAR (Fig. 2a). The TS peak for the interaction on chromosome 7 at 1 DAR was close to RM234, the marker nearest to qSD7-2. The TS peak for the interactions on chromosome 12 at 11 DAR and 21 DAR was nearest to RM270, the marker closest to qSD12. Therefore, qSD7-2 and qSD12 were involved in the QTL × E interactions. These two QTLs exhibit a similar QTL × E interaction pattern in that the phenotypic values for the heterozygous genotypes decreased and the values for the EM93-1-type homozygous genotypes increased under the 2003 experimental conditions as compared with those in 2002.

Scan for QTL × E (year) interactions for seed dormancy (A) and seed shattering (B) from the BC1 (EM93-1//EM93-1/SS18-2) population using the mqtl SIM procedure. Dormancy was measured by germination of seeds at 1, 11, and 21 DAR (fine solid, thick solid, and dotted lines, respectively). Cumulative genetic distance was determined from the top of chromosome 1 to the bottom of chromosome 12. Peaks correspond to the dormancy loci qSD7-2 and qSD12 and the shattering locus qSH7
Seed shattering
The parents SS18-2 and EM93-1 differed greatly in degree of seed shattering – 91% versus 4%, respectively (Table 2). Dominance of the characteristic was incomplete (ADD=−0.05) based on the observation that the F1 was intermediate between the parents (Table 2). The BC1 population exhibited a normal distribution and was absent of SS18-2-like individuals (Fig. 3a). Both genotypic and year effect on shattering were significant in the subpopulation across 2 years (data not shown). Heritability for shattering in the BC1 population was 0.65 based on data from 2002, and 0.40 based on data across both years. The correlation coefficient for shattering between the 2 years was 0.619 (P <0.0001, r2=38.3%). Genetic correlations between seed shattering and degrees of dormancy at 1–21 DAR were significant (rg=−0.21 to −0.26) (Table 3).

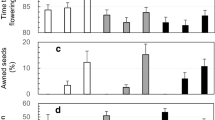
Distributions of seed shattering (A) and awn-length (B) for the 156 BC1 (EM93-1//EM93-1/SS18-2) lines. The horizontal lines on the graph indicate ranges of the parental and F1 plants
Four QTLs for seed shattering were identified based on the data collected in 2002 and 2003 (Table 4). These loci, which mapped to chromosomes 3, 4, 7, and 8, are designated as qSH3, qSH4, qSH7, and qSD8, respectively (Fig. 1). Collectively, the four QTLs accounted for 33% of the phenotypic variance, with their single-locus contributions varying from 8% to 13% (Table 4). Alleles that enhanced the degree of shattering at these loci were derived from SS18-2. A significant epistasis was not detected between the shattering loci (data not shown).
A QTL × E interaction was detected for seed shattering (Fig. 2b). The TS peak for the interaction was between markers RM118 and RM172 on chromosome 7. The TS peak for qSH7 was also located in the same interval, indicating involvement of qSH7 in the interaction. Both the heterozygotes and the EM93-1-type homozygous genotypes displayed increased shattering across years, but shattering increased more for the heterozygous genotypes resulting from the interaction.
Awn
The SS18-2 genotype has seeds with an awn length of more than 60 mm, while the EM93-1 genotype has seeds with no awn. The awn length of the seeds of the F1 generation was about 20 mm shorter than that of SS18-2 (Table 2), indicating the characteristic was partially dominant (ADD=0.35). The BC1 population segregated into awned and non-awned groups. However, the awned genotypes varied in mean awn length from less than 1 mm to about 40 mm, with the distribution dramatically skewed toward short awns (Fig. 3b). Thus, the original data were transformed using the square root. Heritability for awn length was high (0.96). A high level of genetic correlation (rg = ≈ −0.4 to −0.5) was detected between awn length and seed germination after 1–21 DAR (Table 3), indicating that genotypes with long-awned seeds tend to be more dormant.
Three QTLs were identified for awn length (Table 4). These loci, which mapped to chromosomes 4 (two loci) and 8, are designated as qAL4-1, qAL4-2, and qAL8, respectively (Fig. 1). The distance between qAL4-1 and qAL4-2 was about 32 cM according to the TS peak positions. These three loci accounted for 41% of the phenotypic variance (Table 4). The main effects of qAL4-1 and qAL4-2 were −1.4 and −1.2, respectively, based on a single-locus model, but they were −1.0 and −0.4, respectively, using the multiple-locus model. Apparently, linkage of these QTLs exaggerated single-locus effects. Alleles that increased awn length at the three loci were derived from SS18-2.
A significant digenic epistasis was detected between qAL4-1 and qAL8. The effect of qAL8 on awn-length was enhanced when the weedy form of the allele was present at qAL4-1. The component interaction effect accounted for about 5.3% (P=0.0002) of the phenotypic variance. A similar epistasis was also detected between qAL4-2 and qAL8 (data not shown), which explained 4.7% (P=0.0003) of the phenotypic variance on its own.
Hull pigmentations
The weedy strain SS18-2 contained more black but less red and yellow pigmentations on the hull than the breeding line EM93-1 as shown by the L* (a lower reading means a higher intensity of black pigmentation), a* and b* values, respectively (Table 2). Pigmentation intensities in the F1 generation were intermediate between the parents but closer to SS18-2, with ADD ranging from 0.1 to 0.5 (Table 2). The BC1 population distributed continuously and could not be unambiguously grouped based on the intensity of any of the pigmentations (Fig. 4a). Ambiguous individuals with L* values between 52 and 56 were also difficult to group using a visual scoring system. Heritability was highest for black (0.90), intermediate for yellow (0.74), and lowest for red (0.50) pigmentation in the BC1 population (Table 2). The three pigments on the hull were significantly correlated to germination of seeds after-ripened for 1–21 days (r=0.23−0.33) (Table 3). Genotypes having a high intensity of black or a low intensity of red or yellow pigments on the hull had a relatively high degree of dormancy.

Distributions of component pigmentations on the hull (A) and pericarp (B) for the BC1 (EM93-1//EM93-1/SS18-2) population. The black, red, and yellow pigmentations were quantified by the L*, a* and b* values, respectively, recorded with a Chroma Meter. The arrows indicate an increase in the intensities of the pigmentation
Two QTLs for black and red pigmentations, respectively and one for yellow pigmentation were identified (Table 4). Based on their TS peak positions and TS support limits (Fig. 1), the black and red pigmentation QTLs mapped at the same positions on chromosomes 4 and 7, and the yellow pigmentation QTL mapped at an identical position on chromosome 4 as the black or red pigmentation QTL. Thus, two QTLs for hull coloration were detected in the BC1 population and are designated as qHC4 and qHC7, respectively. These QTLs accounted for about 56% and 20% of the phenotypic variance in black and red pigmentations, respectively. The locus qHC4 explained about 50% of the total variance in yellow pigmentation (Table 4). Alleles that enhanced the intensity of black pigmentation were derived from SS18-2, and those that enhanced the red and yellow pigmentations were derived from EM93-1 (Table 4). A significant epistasis was not detected between qHC4 and qHC7.
Pericarp pigmentations
The SS18-2 genotype contained more black (i.e., lower L* values) and red and less yellow pigmentation on the pericarp than EM93-1 (Table 2). Pigmentation intensities in the F1 generation were similar to those in SS18-2, suggesting that black and red pigmentations were dominant and yellow pigmentation was recessive (Table 2). The BC1 population could be separated into relatively high and low black-pigmentation groups, as shown by the L* values (Fig. 4b). These two groups correspond to the black and white groups based on visual classification. However, BC1 genotypes could not be unambiguously grouped based on the intensity of the red or yellow pigments because they displayed continuous variations, as shown by the distribution of a* or b* values (Fig. 4b). Heritability was highest for black (0.91), intermediate for yellow (0.84), and lowest for red (0.75) pigmentation in the BC1 population (Table 2). These three pigmentations on the pericarp were significantly correlated to germination at 1, 11, and 21 DAR, with the exception of r (a*) at 1 DAR (Table 3). Genotypes having a high intensity of black and red, or a low intensity of yellow pigmentations, on the pericarp had a relatively high degree of seed dormancy.
One QTL was identified for each of black, red, and yellow pigmentations on the pericarp (Table 4). The QTLs mapped to nearly the same position on chromosome 7 and, thus, the position is designated as qPC7 (Fig. 1). This locus accounted for about 73%, 43%, and 60% of the phenotypic variance in black, red, and yellow pigmentations, respectively. The allele from SS18-2 increased the intensities of the black and red pigmentations, while the allele from EM93-1 increased the intensity of the yellow pigmentation.
Discussion
Stability of dormancy QTLs
We found the dormancy QTLs from the weedy strain to be relatively constant across the two environments (years). Five of the six dormancy QTLs previously identified by Gu et al. (2004) were confirmed in this research. We detected significant within-year and between-year environmental variations because relative humidity, temperature, and light intensity varied daily during seed development when dormancy was acquired. However, between-year variation had a larger impact on the expression of seed dormancy than within-year. For example, h2 for germination at 11 DAR was 0.66 across years and 0.81 in 2002 (Gu et al. 2004). Between-year variation changed the QTL main effects but not their relative positions on the linkage map (Fig. 1, Table 1) (see Fig. 1 and Table 5 in Gu et al. 2004). This is also true for some dormancy QTLs from barley (Oberthur et al. 1995; Han et al. 1996; Prada et al. 2004).
QTLs for seed dormancy vary in response to the environmental conditions. Dormancy QTL × E interactions have been reported for wheat, barley, and sorghum (Anderson et al. 1993; Oberthur et al. 1995; Lijavetzky et al. 2000), and such interactions may change the phenotypic effect of a QTL from one to several fold (Lijavetzky et al. 2000). The interaction effects that we detected in weedy rice were maintained only for a certain period after harvest and the period varied with the QTLs (Fig. 2a). For example, the qSD12 × E interactions were detectable at 11–21 DAR but not at 1 DAR. Further efforts are necessary to identify major environmental factors contributing to QTL × E interactions, as they are important factors when investigating individual gene expression profiles and applying weedy rice-derived dormancy alleles in breeding programs.
Genetic bases for dormancy-interrelated characteristics
Seed shattering in weedy rice is a genetically complex trait, as indicated by incomplete dominance in the F1 generation (Table 2) and a normal distribution (Fig. 3a) in the BC1 generation. Both wild and weedy races of rice are considered to have a high rate of seed shattering (Oka 1988). However, four to six QTLs for seed shattering have been identified from wild rice-derived populations (Xiong et al. 1999; Cai and Morishima 2000; Thomson et al. 2003), but only one and four shattering QTLs have been detected in populations derived from the japonica-type (Bres-Patry et al. 2001) and indica-type (Table 4) weedy strains, respectively. The QTL × E interaction (Fig. 2b) may explain some of the differences in number of QTLs between wild and weedy rice. It is also possible that genetic differentiation in this adaptive trait occurred between wild and weedy rice and among weedy races during evolution.
The high heritability estimates (h2≥0.9) for awn length and black hull color (Table 2) may explain why these morphological characteristics were treated as qualitative traits in rice genetics (Kinoshita 1984). However, the QTL analysis indicated that in addition to major gene(s), epistasis and modifiers regulate the level of expression of awn and black hull pigmentation, respectively. SS18-2 is a long-awned (more than 60 mm) genotype. Three QTLs accounted for about 41% of phenotypic variance in awn length in the BC1 population, and digenic epistasis explained only part of the remaining variation. The proportion and the lack of SS18-2-type segregates in the BC1 (Fig. 3b), which corresponded to partial dominance in the F1 generation (Table 2), suggest regulation of this trait is genetically more complex.
The loci qHC4 and qHC7 are a major gene and a modifier, respectively, as they explain a major and a minor proportion of the phenotypic variation in hull pigmentation (Table 4). The locus qHC4 is likely allelic to Bh-4 because it is the only one reported for black hull color on chromosome 4 (http://www.shigen.nig.ac.jp/rice/oryzabase/genes/). The genetic basis for pericarp pigmentation is relatively simple in the cross as only qPC7, which explained 73% of the phenotypic variance, was detected. This QTL mapped at the same position as the red pericarp color gene (Rc) (Gu et al. 2004) (Fig. 1). Groos et al. (2002) identified five QTLs for wheat grain color when it was quantified with a Chroma Meter; three QTLs co-localized with the red grain color homologous genes R1, R2, and R3. Although we did not detect additional loci for pericarp color, we identified a new locus (qHC7) where the weedy form of the allele enhanced intensities of black and red pigmentations on the hull (Table 4). Using a visual classification system (unpublished data), we were able to detect qHC4; thus, detection of qHC7 was only possible with the Chroma Meter.
Our mapping results were more comparable to those obtained from wild rice-derived populations than from a japonica-type weedy rice-derived population. For example, SS18-2 shares only two loci (qAL4-2 and qPC7) with the japonica-type weedy strain C6 (Bres-Patry et al. 2001), but most QTLs for seed shattering, awn, and pericarp color match or are similar to those from one or more of the wild rice-derived populations according to flanking markers or map positions (Xiong et al. 1999; Cai and Morishima 2002; Thomson et al. 2003). The weedy strain SS18-2 was collected from Thailand, which is part of the center of origin for Asian cultivated rice (Tang and Morishima 1997). The origin and mapping information suggest that genes for the weedy characteristics in SS18-2 were derived from its wild relative O. rufipogon.
Two groups of weed seed dormancy genes
The dormancy QTLs detected in this investigation can be classified into two groups according to their map positions relative to the QTLs responsible for seed shattering and morphological characteristics. The first group consists of four dormancy QTLs that are close to one or more of the QTLs for dormancy-interrelated characteristics. As shown by the TS peak positions, qSD4 is co-located with qHC4 and flanked by qAL4-1, qSH4, and qAL4-2; qSD7-1 is very close to qPC7; qSD8 is tightly linked to qSH8 and qAL8; qSD7-2 is proximal to qHC7 and qSH7 (Fig. 1). The non-random distribution of QTLs along a genome is similar to the QTL clusters found for domestication syndromes (Doebley and Stec 1991). The second group consists of the dormancy QTLs that are independent of the loci for shattering and seed morphological characteristics, such as qSD12 and those with a relatively minor effect on dormancy, as suggested by the secondary peaks for seed germination (Gu et al. 2004). Corresponding to these classifications there are two groups of genes regulating seed dormancy levels in weedy populations. The first group of these dormancy genes underlies the phenotypic interrelationship observed in weedy rice or weedy strain-derived populations (Oka 1988; Gu et al. 2003, 2005). These genes occur at frequencies that vary with changes in the selection (natural or artificial) pressures against the interrelated weedy characteristics or with the degrees of weediness or human disturbances (Harlan and de Wet 1965) due to clustering or linkage. Natural selection tends to increase this gene frequency as selection favors shattered seeds with awns and/or black and red pigmentations on the hull or pericarp; these characteristics allow weed seeds to escape harvest, disperse, and persist in the soil. Artificial selection through techniques such as weeding by hand in Asian countries usually selects against morphological characteristics (e.g., awns and black pigmentation) and thus reduces the frequency of the dormancy allele for linked loci. However, theoretically, only the selection(s) against multiple interrelated weedy characteristics can cause a substantial reduction in degree of dormancy.
The second group of dormancy genes is able to escape selection because they are genetically independent of dormancy-interrelated characteristics. Therefore, weedy plants that mimic the accompanying cultivars can also be carriers of these dormancy alleles. The transport of weed-infested stock seed is one of the principle means of weed dissemination. Compared with the first group of dormancy genes, the second group of genes would have a greater opportunity to transport geographically with commercial seeds when harvested together with the companion crop. Dormancy QTLs reported for cultivated varieties should belong to the second group. However, loci with a magnitude of effect on dormancy comparable to that of qSD12 have not yet been identified for cultivars (Lin et al. 1998; Cai and Morishima 2000; Dong et al. 2002; Miura et al. 2002).
Dormancy is the key adaptive syndrome in weeds as it promotes survival by distributing the time-course of seed germination in the soil seed bank over a long period (Harlan and de Wet 1965). The above-mentioned QTL organization pattern demonstrates the central role of seed dormancy in weed adaptation. Weeds may come from wild species that have long been adapted to sites of natural disturbance or from new species or varieties that have evolved since agriculture was developed (Harlan and de Wet 1965). If we consider that SS18-2 originated from a place where wild rice (O. rufipogon) was present, both groups of dormancy QTLs/genes identified in this research were most likely derived from wild rice. QTL clusters for “adaptive/domesticated syndromes” have been reported for several grass species, although seed dormancy was not one of the traits examined. Comparative genetic studies have revealed that genes for major adaptive characteristics are relatively conservative across species (Devos and Gale 2000). Therefore, it is likely that dormancy genes may also exist in QTL clusters for adaptive characteristics in other wild and weedy species.
Dormancy genes which have been eliminated from cultivated varieties by either indirect or direct selections during domestication and breeding can be regained from weedy germplasm. Selections generally have been for rapid and uniform seed germination in crops, but a moderate degree of dormancy is often desirable for cereals to resist PHS. Dormancy genes both located in and independent of QTL clusters could be useful for manipulating the onset and rate of germination in breeding programs. To utilize dormancy genes within a QTL cluster it will be necessary to determine if pleiotropy, linkage, or both are responsible for the dormancy and interrelated weedy phenotype(s). For example, red grain color has long been associated with seed dormancy in wheat, and the association could be caused by a pleiotropic effect of the R genes (Gfeller and Svejda 1960; Flintham et al. 2002; Groos at al. 2002). We are currently transferring the QTL cluster regions from SS18-2 to the EM93-1 genetic background by phenotypic selection and marker-assisted selection to fine-map genomic regions. This is the first step toward elucidating the underlying genes and the models for the clustering of seed dormancy and interrelated weedy traits or patterns for linkage disequilibrium among genes located in these regions.
References
Anderson JA, Sorrells ME, Tanksley SD (1993) RFLP analysis of genomic regions associated with resistance to pre-harvest sprouting in wheat. Crop Sci 33:453–459
Bres-Patry C, Lorieu M, Clement G, Bangratz M, Ghesquiere A (2001) Heredity and genetic mapping of domestication-related traits in a temperate japonica weedy rice. Theor Appl Genet 102:118–126
Burke JM, Tang S, Knapp SJ, Rieseberg LH (2002) Genetic analysis of sunflower domestication. Genetics 161:1257–1267
Cai HW, Morishima H (2000) Genomic regions affecting seed shattering and seed dormancy in rice. Theor Appl Genet 100:840–846
Cai HW, Morishima H (2002) QTL clusters reflect character associations in wild and cultivated rice. Theor Appl Genet 104:1217–1228
Chang TT, Yen ST (1969) Inheritance of grain dormancy in four rice crosses. Bot Bull Acad Sin 10:1–9
Devos KM, Gale MD (2000) Genome relationships: the grass model in current research. Plant Cell 12:637–646
Doebley J, Stec A (1991) Genetic analysis of the morphological differences between maize and teosinte. Genetics 129:285–295
Dong Y, Tsozuki E, Kamiunten H, Terao H, Lin D, Matsuo M, Zheng Y (2002) Identification of quantitative trait loci associated with pre-harvest sprouting resistance in rice (Oryza sativa L.). Field Crops Res 81:133–139
Flintham JE, Adlam R, Bassoi M, Holdsworth M, Gale MD (2002) Mapping genes for resistance to sprouting damage in wheat. Euphytica 126:39–45
Frary A, Nesbitt TC, Frary A, Grandillo S, Knaap E, Cong B, Liu J, Meller J, Elber R, Alpert KB, Tanksley SD (2000) fw2.2: a quantitative trait locus key to the evolution of tomato fruit size. Science 289:85–88
Gale MD, Flintham JE, Devos KM (2002) Cereal comparative genetics and preharvest sprouting. Euphytica 126:21–25
Gfeller F, Svejda F (1960) Inheritance of post-harvest seed dormancy and kernel colour in spring wheat lines. Can J Plant Sci 40:1–6
Grist DH (1986) Rice, 6th edn. Longman, London
Groos C, Gay G, Perretant MR, Gervais L, Bernard M, Dedryver F, Charmet G (2002) Study of the relationship between pre-harvest sprouting and grain color by quantitative trait loci analysis in a white × red grain bread-wheat cross. Theor Appl Genet 104:39–47
Gu X-Y, Chen Z-X, Foley ME (2003) Inheritance of seed dormancy in weedy rice. Crop Sci 43:835–843
Gu X-Y, Kianian SF, Foley ME (2004) Multiple loci and epistases control genetic variation for seed dormancy in weedy rice (Oryza sativa). Genetics 166:1503–1516
Gu X-Y, Kianian SF, Foley ME (2005) Seed dormancy imposed by covering tissues interrelates to shattering and seed morphological characteristics in weedy rice. Crop Sci 45 (in press)
Han F, Ullrich SE, Clancy JA, Jitkov V, Kilian A, Romogosa I (1996) Verification of barley seed dormancy loci via linked molecular markers. Theor Appl Genet 92:87–91
Harlan JR (1965) The possible role of weed races in the evolution of cultivated plants. Euphytica 14:173–176
Harlan JR, de Wet JMJ (1965) Some thoughts about weeds. Econ Bot 19:16–24
Harlan JR, de Wet JMJ, Price EG (1973) Comparative evolution of cereals. Evolution 27:311–325
Johnson LPV (1935) The inheritance of delayed germination in hybrids of Avena fatua and A. sativa. Can J Res 13:367–387
Kato K, Nakamura W, Tabiki T, Miura H (2001) Detection of loci controlling seed dormancy on group 4 chromosomes of wheat and comparative mapping with rice and barley genomes. Theor Appl Genet 102:980–985
Kearsey MJ, Pooni HS (1996) The genetical analysis of quantitative traits. Chapman and Hall, London
Khan M, Cavers PB, Kane M, Thompson K (1996) Role of the pigmented seed coat of proso millet (Panicum miliaceum L.) in imbibition, germination and seed persistence. Seed Sci Res 7:21–25
Kinoshita T (1984) Gene analysis and linkage map. In: Tsunoda S, Takahashi N (eds) Biology of rice. JSSP/Elsevier, Tokyo, pp 187–274
Li C, Ni P, Francki M, Hunter A, Zhang Y, Schibeci D, Li H, Tarr A, Wang J, Cakir M, Yu J, Bellgard M, Lance R, Appels R (2004) Genes controlling seed dormancy and pre-harvesting sprouting in a rice-wheat-barley comparison. Funct Integr Genomics 4:84–93
Lijavetzky D, Martinez MC, Carrari F, Hopp HE (2000) QTL analysis and mapping of pre-harvest sprouting resistance in sorghum. Euphytica 112:125–135
Lin SY, Sasaki T, Yano M (1998) Mapping quantitative trait loci controlling seed dormancy and heading date in rice. Theor Appl Genet 96:997–1003
Lincoln S, Daly M, Lander E (1992) Constructing genetic maps with mapmaker/exp 3.0, 3rd edn. Whitehead Institute, Cambridge
Matus I, Corey A, Filichkin T, Hayes PM, Vales MI, Kling J, Riera-Lizarazu O, Sato K, Powell W, Waugh R (2003) Development and characterization of recombinant chromosome substitution lines (RCSLs) using Hordeum vulgare subsp. spontaneum as a source of donor alleles in a Hordeum vulgare subsp. vulgare background. Genome 46:1010–1023
McCouch SR, Teytelman L, Xu Y, Lobos KB, Clare K, Walton M, Fu B, Maghirang R, Li Z, Xing Y, Zhang Q, Kono I, Yano M, Fjellstrom R, DeClerck G, Schneider D, Cartinhour S, Ware D, Stein L (2002) Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res 9:199–207
Miura K, Lin SY, Yano M, Nagamine T (2002) Mapping quantitative trait loci controlling seed longevity in rice (Oryza sativa L.). Theor Appl Genet 104:981–986
Oberthur L, Blake TK, Dyer WE, Ullrich SE (1995) Genetic analysis of seed dormancy in barley (Hordeum vulgare L.). J Quant Trait Loci 1:5
Oka HI (1988) Origin of cultivated rice. Japan Science Society Press, Tokyo
Paterson AH, Sorrells ME (1990) Inheritance of grain dormancy in white-kerneled wheat. Crop Sci 30:25–30
Paterson AH, Schertz KF, Lin YR, Liu SC, Chang YL (1995) The weediness of wild plants: molecular analysis of genes influencing dispersal and persistence of johnsongrass, Sorghum halepense (L) Pers. Proc Natl Acad Sci USA 92:6127–6131
Peng J, Ronin Y, Fahima T, Roder MS, Li Y, Nevo E, Korol A (2003) Domestication quantitative trait loci in Triticum dicoccoides, the progenitor of wheat. Proc Natl Acad Sci USA 100:2489–2494
Prada D, Ullrich SE, Molina-Cano JL, Cistue L, Clancy JA, Romagosa I (2004) Genetic control of dormancy in a Triumph/Morex cross in barley. Theor Appl Genet 109:62–70
SAS Institute Inc (1999) SAS/STAT user’s guide. Version 8 Cary, N.C.
Simpson GM (1992) The adaptive significance of awns and hairs in grasses. J Biol Educ 26:10–11
Tang L-H, Morishima H (1997) Genetic characterization of weedy rices and the inference on their origins. Breed Sci 47:153–160
Tanksley SD, McCouch SR (1997) Seed banks and molecular maps: unlocking genetic potential from the wild. Science 277:1063–1066
Temnykh S, DeClerck G, Lukashova A, Lipovich L, Cartinhour S, McCouch S (2001) Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res 11:441–1452
Thomson MJ, Tai TH, McClung AM, Lai XH, Hinga ME, Lobos KB, Xu Y, Martinez CP, McCouch SR (2003) Mapping quantitative trait loci for yield, yield components and morphological traits in an advanced backcross population between Oryza rufipogon and the Oryza sativa cultivar Jefferson. Theor Appl Genet 107:479–493
Tinker NA, Mather DE (1995) MQTL: software for simplified composite interval mapping of QTL in multiple environments. J Agric Genomics 1:2 http://www.ncgr.org/jag/
Wang D, Dowell FE, Lacey RE (1999) Predicting the number of dominant R alleles in single wheat kernels using visible and near-infrared reflectance spectra. Cereal Chem 76:6–8
Xiong LZ, Liu KD, Dai XK, Xu CG, Zhang Q (1999) Identification of genetic factors controlling domestication-related traits of rice using an F2 population of a cross between Oryza sativa and O. rufipogon. Theor Appl Genet 98:243–251
Acknowledgements
We thank T. Nelson, C. Kimberlin, D. Puhr, and J. Wear for their technical assistance and anonymous reviewers for their insightful comments. Funding for this work was provided by USDA-National Research Initiative (020068).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by D.J. Mackill
Rights and permissions
About this article
Cite this article
Gu, XY., Kianian, S.F., Hareland, G.A. et al. Genetic analysis of adaptive syndromes interrelated with seed dormancy in weedy rice (Oryza sativa). Theor Appl Genet 110, 1108–1118 (2005). https://doi.org/10.1007/s00122-005-1939-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-005-1939-2