
Abstract
Mapping quantitative trait loci (QTL) in plants is usually conducted using a population derived from a cross between two inbred lines. The power of such QTL detection and the estimation of the effects highly depend on the choice of the two parental lines. Thus, the QTL found represent only a small part of the genetic architecture and can be of limited economical interest in marker-assisted selection. On the other hand, applied breeding programmes evaluate large numbers of progeny derived from multiple-related crosses for a wide range of agronomic traits. It is assumed that the development of statistical techniques to deal with pedigrees in existing plant populations would increase the relevance and cost effectiveness of QTL mapping in a breeding context. In this study, we applied a two-step IBD-based-variance component method to a real wheat breeding population, composed of 374 F6 lines derived from 80 different parents. Two bread wheat quality related traits were analysed by the method. Results obtained show very close agreement with major genes and QTL already known for those two traits. With this new QTL mapping strategy, inferences about QTL can be drawn across the breeding programme rather than being limited to the sample of progeny from a single cross and thus the use of the detected QTL in assisting breeding would be facilitated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Classically, quantitative trait loci (QTL) mapping in plants is conducted using a population derived from a cross between two inbred lines (see Jansen 2001 for a review). The power of such QTL detection and the accuracy of parameter estimates highly depend on the choice of the two parental lines. Thus, the QTL detected in such populations only represent a part of the genetic architecture of the trait. Besides, the effects of only two alleles are characterized, which is of limited interest to the breeder. As mentioned by Beavis (1998), ‘the integration of QTL mapping into existing breeding strategies is urgently needed’. Indeed, the breeder’s material is far from the studied bi-parental populations as breeders generally handle many small families from crosses between (often) highly related elite lines. Thus the ‘bridge’ between QTL found in bi-parental populations and their end-use in breeding is often difficult to cross, as many drawbacks exist (see also Jannink et al. 2001). First, when only two parents are considered, some markers and potential QTL are more likely to be monomorphic, even if parental lines are carefully selected for trait divergence. As QTL can only be found at polymorphic sites in the genome, the expected number of QTL detected with a bi-parental cross will be lower than that expected when analysing several crosses at a time (assuming the total number of genotypes is not the limiting factor). The second drawback is that the QTL effect is estimated as a contrast between two alleles and in one genetic background only. Therefore, in that context, the improvement of a line by the introgression of a QTL allele in a completely new genetic background is rather unpredictable, because of possible epistatic interaction between QTL and genetic background. Finally, from an economic standpoint, the cost of creation of large single cross progenies and specific trials for trait evaluation to perform QTL detection is quite high and often at the expense of other selection programmes.
All these drawbacks reduce the breeders’ interest for implementing bi-parental experimental designs when funding and work are constrained. The breeders’ focus is to characterize the effect of a wide range of alleles in his germplasm. Methods for simultaneous detection and manipulation of QTL in breeding programmes would thus enhance the applicability of marker assisted selection (MAS).
In this paper, we present results from a QTL mapping study carried on a real wheat breeding population, coming from French Nickerson (Limagrain group) programme. This population, initially composed of 391 F6 lines coming from 80 parents (each F6 lines is the result of a bi-parental cross) has all the drawbacks of a breeding population for QTL mapping: the uncertainty on pedigrees, the difficulties to check the marker data, the missing DNA for some of the parents, the unbalanced trials for phenotype evaluation and the influence of selection and genetic drift during line development.
Crepieux et al. (2004a, b) explored, by simulation, the ability to perform QTL detection on plant breeding populations. They showed that QTL detection was possible in such fragmented populations (i.e. coming from multiple bi-parental crosses, with different relationships between lines) even for small QTL accounting for only 5% of the genetic variation. They used a random effects method (Xu and Atchley 1995) called a two-step IBD-based variance component (VC) method (George et al. 2000). This kind of method has been developed and widely used for complex pedigreed populations in human genetics (see e.g. Almasy and Blangero 1998) and animal genetics (e.g. George et al. 2000), and was proposed for plants by Xie et al. (1998). The method is based upon the simple premise that individuals of similar phenotypes are more likely to share genes identical by descent (IBD) (Haseman and Elston 1972). Through the computation of IBD matrices and the fitting of mixed linear model, these methods allow to use all pedigree information together, instead of restricting the size of the studied population only to the biggest half-sibs families, as for least-squares (LS) regressions (Knott et al. 1996, see Slate et al. 2002 for a comparison of LS regression and IBD-based VC method on a pedigree composed by small families).
Crepieux et al. (2004a) showed that adding to the direct known pedigrees (half-sib or full-sib relationship) an estimate of the unknown ancestor relationships between parents estimated by pairwise marker distances to compute the IBD probabilities could increase the chance to detect QTL. The difference in the QTL detection power between the ‘classical’ IBD formula and the one adding ancestor relationships to the IBD computation was enhanced when selection occurred on the trait under study, as demonstrated by simulation (see Table 4 in Crepieux et al. 2004a). Intuitively, Malecot’s coefficients of kinship used in the ‘classical’ IBD formula are an expectation of the IBD values for random sampling, but give biased estimates when sampling occurs after directional selection.
In order to check whether the IBD-based VC method can yield accurate results on the studied breeding population and to allow sounder comparisons with published results, two bread quality related traits, for which major genes or QTL have been detected in many bi-parental populations, were studied: kernel hardness and dough strength as estimated by the W parameter of alveograph. A major locus for kernel hardness exists (Ha locus, on chromosome 5D) which is supposed to be responsible for the hard versus soft wheat classification (Symes 1965). For dough strength, the loci encoding the high-molecular-weight glutenin subunits (HMW-GS) on group-1 chromosomes have been reported to be responsible for a part of the variability of the W score (Branlard et al. 2001), even if other QTL have been detected (see Charmet and Groos 2002 for a review). Thus, because of limited resources for genotyping, the method was only checked on two groups of homeology, groups 1 and 5, which were supposed to carry the most interesting QTL.
In this paper we want to see whether relatively small breeding populations as those currently used for cereal improvement, are suitable for QTL mapping and if results are in agreement with the known literature on the underlying genes of the two studied traits. Then, in the discussion, we will try to state on the interest of such QTL mapping for breeding.
Materials and methods
Mapping population
The population chosen for QTL mapping is a part of the Nickerson wheat-breeding programmes, corresponding to the year 2002 F6 generation. In this population, 391 F6 lines were chosen. The first choice was based on the availability of the putative parents for genotyping. The second choice tried to reduce the population fragmentation (number of F6/number of parents, that is, average full-sib and half-sib family size) by choosing only F6 ear lines whose parents were declared to be genitors of at least two other F6 lines. Although some very interesting parents of large half-sib families (i.e. one parent in common at the origin of the cross) were missing for genotyping, their resulting F6 lines were kept, because little plant material would have otherwise been available at this late breeding stage. Missing marker genotypes on parents will be imputed by using marker genotypes of their offsprings and mate.
Finally, the 391 F6 lines came from crosses between 80 different parents (for which most of the pedigrees were either not available or not reliable), and they originated from different ‘end-use’ Nickerson programmes and from differently located breeders. Out of these 80 parents, 70 were available for genotyping.
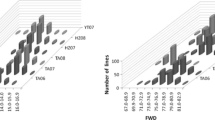
The average size of half-sib families for the ten missing parents is 10.45. The average number of full-sibs per cross is 2.35 (median 2) and the mean size of half-sib families is 9.8 (median 6). The resulting distribution of half-sib family sizes is presented in Fig. 1.

Structure of the F6 mapping population. Each bar represents the number of derived F6 progenies from a specific parent. Each of the bars could be seen as a half-sib family, even if the F6 are five generations of selfing away from the initial cross. Note that the sum of the represented family (bars) equals twice the real number of F6 as each F6 has two parents that can form separately a half-sib family. The largest F6 half-sib family has 56 lines while the smallest ones have only two lines
DNA sampling and genotyping
The 391 leaf samples for DNA extraction were taken from the F6 fixation trials and are not totally homozygous. The 70 available parents were found in breeder’s collections and genetic resources. They were planted in a greenhouse and DNA extracted from leaves. The parents were almost totally fixed, being, for most of them, already registered or coming from advanced breeding generations.
We genotyped only two homeologous groups out of seven (groups 1 and 5), which are known to contain some important QTL or known genes for quality related traits. On these two groups, 65 microsatellites markers chosen according to their map position were separated on a capillary electrophoresis sequencer (ABI prism 3100). Besides, information of the three-biochemical markers corresponding to three HMW-GS Glu-A1, Glu-B1 and Glu-D1, located on the group 1-chromosomes long arm, was available.
In order to obtain a marker-based estimate of the genetic similarities across the whole genome, we also genotyped the other 15 chromosomes but at a lower density (one marker per chromosome arm) to enable the computation of additive-relationship matrix on the whole genome, used in the estimation of the ‘polygenic’ component. These markers were chosen according to Roussel et al. (2004) for their use in diversity studies, that is, for their quality and polymorphism information content (PIC value, a synthetic parameter which summarizes both allele number and their distribution evenness).
Map construction
A genetic map, based on observed recombination rates among markers on this material was impossible to obtain. Indeed, there is a low confidence in marker recombination estimates due to the small half-sib and full-sib family sizes and the influence of selection and genetic drift between the initial cross and the F6 mapping generation. Markers were thus assigned to chromosome locations using different published maps (ITMI, Röder et al. 1998; Courtot × Chinese Spring, Sourdille et al. 2003) by comparing observed and expected amplification sizes. After this first assignment, the amount of recombination between groups of markers were checked, which allowed to confirm the order of markers. The map for these two groups covered nearly 925 cM (for comparison, the Courtot × Chinese spring map for groups 1 and 5 covers 1130 cM).
The average number of markers per chromosome for the two groups was 11.3, with a range from 10 to 13. The level of marker information across the parents, different along the chromosome, has a strong impact to discriminate for each progeny the parental origin of the alleles. We computed the number of informative markers, for each individual, as the number of polymorphic markers between its two parents. This averaged number, on the full set of progenies, is 6.10 per chromosome, with a range from 5.21 for chromosome 1A to 7.22 for chromosome 1D. This number of informative markers means that, on average, one marker every 30 cM is polymorphic between the two parents of any cross.
Table 1 contains for each chromosome the genetic length obtained from composite wheat map, the number of markers used, and the average number of informative markers.
Marker data correction and pedigree validation
Molecular data on this kind of plant material produced ‘rough’ data and presumably, biased. One concern, when dealing with such data coming from real breeding schemes, was first to remove all sources of bias that might have occurred during the line development (the initial mating was operated 6 or 7 years before obtaining the F6 and a breeder deals, each year, with thousand of lines at every developmental stage) and the marker production. These biases include (i) the uncertainty for individuals to really descend from the specified pedigree (e.g. due to undesired cross-pollination, wrong trial harvest or breeders’ notations errors), (ii) the impossibility to trace progenies alleles’ due to missing parents’ data (lost seeds...) and (iii) the marker production and analyses, including microsatellite band stuttering yielding different amplification notations between putative parents and progenies and the mistakes on computer analyses. Due to the very fragmented population structure, handmade molecular data correction, missing parents’ data reconstruction and pedigree validation on this kind of complex pedigrees is unrealistic. A software, PurPL (Joffre and Crepieux 2004), was developed to correct different possible sources of bias on this kind of very fragmented plant populations. Based on a five-step algorithm and on the computation of intuitive probability scores, PurPL corrects many different possible sources of bias by extracting the more likely information in the parent and progeny files (initially linked by putative relationships), and re-builds possible missing information. PurPL successive runs allowed us to re-perform marker analyses when the percent of errors in amplification notations was too high, and to remove individuals for which the probability that their putative parents were the right ones was too low. Finally, out of 391 lines, 374 were kept for the QTL analysis. On these 374 F6 lines, about 9% of the allele inheritances were corrected. Moreover, this correction allowed the percentage of missing data to be improved from 16 to 4% for parents. At the end, 6% of data were missing in the progenies.
Phenotypic data
A total of 362 F6 were grown in a single trial without replication near Clermont-Ferrand (France), at Nickerson-Limagrain breeding station. The 29 other F6 were grown at Chartainvilliers (close to Paris). Thirty lines in common between Clermont-Ferrand and Chartainvilliers were also analysed to remove the location main effect by including it as fixed effect in the model (the design was not optimized to evaluate the genotype × environment interaction, however, the correlation between sites for these 30 lines was 0.86 for dough strength and 0.77 for kernel hardness, suggesting a moderate level of G × E interactions, although we have no means to test their significance). Phenotypic data were obtained on F5 seed bulks (once plants are chosen on the F5 trial to go to F6, then the F5 families are harvested in bulks to establish trials the following year), which was not exactly the same generation as that used for DNA extraction (which were real F6). We have to accommodate this imprecision (the power to detect association between genotype and phenotype may be slightly reduced by this difference in fixation, which remains low in any case) as these F5 bulks are the trials currently available for F6 evaluation.
On these bulks, kernel hardness (Hard, from 1=very soft to 100=very hard) was evaluated by near-infrared reflectance spectroscopy (NIR Percon Inframatic 8620) according to AACC method 39–70A (American Association of Cereals Chemists 1995). Dough strength (W, in J 10−4) was obtained by alveograph test, performed according to the AACC method 54–30 (American Association of Cereals Chemists 1995). Kernel hardness used in this study was produced for the breeding purpose by NIR and was then used for this study. Aleograph measures, however, are generally performed at the F7 stage, for reason of cost and time. This character was thus produced at the F6 stage for the purpose of publication only. Other data produced for breeding were also analysed but results cannot be published for confidentiality reasons. Figure 2a, b shows the distribution of the two characters for the F6 mapping population.

Distribution of kernel hardness (a) and gluten strength (b) among the F6 breeding population
QTL analysis
The statistical method employed for QTL mapping is a two-step IBD-based VC analysis adapted to plant breeding material, as described in Crepieux et al. (2004a).
Fitting the linear mixed models
First, a mixed linear model was fitted under the assumption that the studied quantitative trait was controlled by a number of additive and small-effect unknown loci (also called polygenes). This model with no segregating QTL is written as
where y is the vector of phenotypes, X is the design matrix for fixed effects, β the vector of fixed effects containing the locations, Z is the incidence matrix relating records to individuals, v is the vector of additive polygenic effects, and e is the vector of residuals.
A second mixed linear model was fitted, which included the above-polygenic term plus a putative QTL effect at the location of interest. This model is written as
where u is the vector of additive QTL effects.
The random effects u, v, and e are assumed uncorrelated and distributed as multivariate normal densities: \( {\textbf{u}} \sim {\left( {0,{\textbf{G}}\sigma ^{2}_{{\textbf{u}}} } \right)};\;{\textbf{v}} \sim {\left( {0,{\textbf{A}}\sigma ^{2}_{{\textbf{v}}} } \right)};\;{\textbf{e}} \sim {\left( {0,{\textbf{I}}\sigma ^{2}_{{\textbf{e}}} } \right)}, \) with σ 2u , σ 2v , and σ 2e being, respectively the additive variance of the QTL, the polygenic variance, and the residual variance. A is the additive genetic relationship matrix (for the polygenic effects), that is, the genetic background effect; G is the IBD matrix for the QTL additive effects conditional on marker information; and I is the identity matrix. The phenotypic variance is given by
Model (1) provides an estimate of the trait’s heritability (effect of all the polygenes), in addition to a likelihood value (L1) for the REML solution while model (2) provides estimates of the polygenic heritability (h 2p ) and the putative QTL heritability (h 2QTL ), in addition to a likelihood value (L2) for the REML solution.
To test the presence of a QTL versus no QTL at a particular chromosomal position, we used the likelihood-ratio test statistic: LR=−2 ln(L1(H0, no QTL present)/L2(H1, QTL present)), where L1 and L2 represent the likelihood values of (1) and (2) evaluated at the REML solutions, respectively. ASREML (Gilmour et al. 1998) was used to solve the linear mixed models.
Computing G and A matrices
For the chromosomes of groups 1 and 5, at every marker location and at every 3 cM along the chromosome, the probabilities for each F6 individual to descend from its first or second parent (in its declared pedigree) were computed using the MDM algorithm (Servin et al. 2002). Then IBD probabilities, determined between all individuals in the pedigree were computed using these probabilities of descent, thus yielding the G matrix. The method to compute IBD probabilities between full-sibs, half-sibs and ‘unrelated’ individuals of the mapping population can be found in Crepieux et al. (2004a). We used, to build the G matrix, the IBD formula taking into account ancestor relationships estimated by markers between the parents (instead of considering the parents unrelated, as in Xie et al. 1998), as Crepieux et al. (2004a, b) showed that there could be a strong influence of the IBD formula on the power to detect QTL in breeding populations. Besides, they showed that this difference was larger if selection had occurred for the trait of interest as selection increases the chance between individuals of a same population to fix the same IBD blocks, and thus to be related. In this case the usual coefficient of kinship, which gives the expectation value, is clearly inappropriate even when available.
The A matrix to account for the polygenic component was simply computed by Nei and Li (1979) formula of genetic similarity, using one marker/chromosome arm. Thus, the same ‘weight’ in the relationship matrix was given to each chromosome, including the scanned ones. The relationship matrix is computed only once for the whole set of analysis.
Steps of the analysis
We analysed one chromosome at a time, introducing the appropriate IBD matrices into the linear mixed model, and solving it with the ASREML programme (Gilmour et al. 1998).
Once all the QTL for one character were detected on the six chromosomes, we carried on the analysis introducing the most significant QTL as a random covariate (we added a term Z w in the model, w being the BLUP values for the 374 lines at the most significant QTL). If significant QTL still remained or appeared, then the most significant one was added to the analysis and the analysis carried on until no more significant QTL appeared. This procedure is described in Almasy and Blangero (1998) and is somewhat analogous to one option of the Composite Interval Mapping proposed by Zeng (1994) for bi-parental populations.
Results are reported for a ‘comprehensive’ model that represents the situation where no more segregating QTL exists on the six scanned chromosomes.
Significance thresholds
Performing empirical threshold by data permutation was too much CPU-time-consuming. Moreover, permutation testing is problematic for such IBD-based VC analysis as it is unclear how to permute the data while retaining the association between polygenic variation and marker information (George et al. 2000). In this paper, we used Lander and Kruglyak (1995) formula, and their QTL significance definitions. They describe QTL as ‘suggestive’ if they exceed a threshold expected to be observed once on average by chance in a genome scan, and ‘significant’ if exceeding a threshold expected to be observed by chance in only 5% of genome scans. Solving the equation given in Lander and Kruglyak (1995), and assuming a map length of 925 cM covering six chromosomes, the suggestive and significance thresholds are equivalent to likelihood-ratio test statistics of 16.5 and 22.5, respectively. However, these values assume an infinitely dense map of informative markers, which is not the case in this study. Lander and Kruglyak (1995) suggest that significance thresholds be dropped by 20% for a map with 10-cM intervals to compensate for the consequent loss of QTL detection power. In this study, for the six chromosomes, the average marker interval was about 16 cM. To be conservative, we dropped the thresholds by only 20%, giving thresholds of 13.2 and 18.
Statistical significance of polygenic heritability was determined by assuming that the likelihood-ratio test statistic obtained from the polygenic model and a residuals-only model (i.e. a model without the polygenic component fitted) follows a χ 21 distribution (Lynch and Walsh 1998).
Confidence intervals
Confidence intervals (CIs) for the QTL position were calculated by the drop of one LOD unit (LOD drop-off method, Lander and Botstein 1989) converted to LR units (multiply LOD by 2×ln(10) to convert to LR units).
Results
The estimates for polygenic heritability (Eq. 1) for kernel hardness and dough strength were 0.85 (LRT=139.92, P≈0) and 0.92 (LRT=277.5, P≈0), respectively.
Figure 3 depicts the LR test profiles for the six chromosomes and the two traits for a one QTL-model (Eq. 2), for profiles exceeding at least the chromosome-wide significance level.

LR test profiles exceeding the genome-wide significance level. a Kernel hardness and b dough strength
Table 2 presents a summary of the QTL detection under a model representing the situation where no more segregating QTL were found after the QTL detected so far were introduced in the model through their appropriate IBD-matrices. For the QTL in this model, we present the position, the flanking markers, the size of the CI determined by the drop of one LOD unit, the value of the test statistic and the estimates of the QTL heritabilities (as well as for the polygene). In this ‘full’ model, 2 QTL explained most of the variation on the six chromosomes for the analysis of kernel hardness (i.e. the QTL on 1D and 5D), and 3 QTL for dough strength (i.e. the QTL detected around the three glutenin proteins on 1A, 1B, and 1D). We still notice a high heritability for the polygene (0.24 for kernel hardness, 0.26 for dough strength), meaning that other QTL can segregate on the rest of the genome.
Discussion
Many statistical methods already exist to map QTL in inbred plant material; however, most of these methods focus on a single bi-parental progeny. Other methods have been developed to address more challenging population structures (Xu 1998; Xie et al. 1998; Bink et al. 2002 for example). Nevertheless, these methods did not appear to be easily extendable to highly fragmented and unbalanced populations, at any selfed or backcrossed generation, such as in a real breeding programme. They also did not take into account the possibility for alleles to be IBD if ancestor pedigrees are not available. Other methods were proposed for QTL mapping directly in maize breeding populations (Bernardo 1998; Parisseaux and Bernardo 2004). However, the existence of heterotic group in maize and thus the existence of high-linkage disequilibrium within each heterotic group, allowed the use of fixed effects methods (with a random polygenic control) to map QTL. These methods, however, cannot lead to QTL mapping in our wheat breeding populations as such groups were difficult to define.
In this paper, we present a first attempt to map QTL in a real wheat-breeding programme. The QTL detection method used is an IBD-based VC method. This kind of methods with random QTL allelic effects are supposed to be particularly suited for complex designs (Almasy and Blangero 1998; Lynch and Walsh 1998) as they require fewer parametric assumptions than fixed effects methods. Indeed, in these methods, the number of alleles at a QTL does not need to be specified and there is no need for estimating allelic or genotypic frequencies. The resulting less parameterized statistical environment could be compulsory for very fragmented designs for mapping QTL, such as the one studied in this paper. Another advantage of VC analysis is that they provide an estimate of the additive genetic variance in the population attributable to a QTL, rather than only estimates of QTL substitution effects for specific parents (sires in half-sib designs). Finally, VC methods allow to utilize all the available relationships information, even in the case of hermaphrodite organisms (where the father of a half-sib family could be seen as a mother of another half-sib family), with a mixing of half-sib and full-sib families of different sizes. Fixed effects methods are more suited for analysis of large half-sib family sizes, and they have been successfully applied in bovine families for instance (see Zhang et al. 1998 for a comparison of the two methods on a real granddaughter design).
The VC method with REML resolution we purposed in this paper attempted to use marker information as best as possible, estimating first the relationships between the parents of the mapping population, then computing the IBD probabilities between each pair of F6 at each scanned locus, using both the direct relationships (full-sib or half-sibs) and those inferred from marker information. The assumption that parents with unknown pedigree could share IBD genes due to the structure of pedigree programmes (important use of some ‘star’ varieties for instance) was mentioned by Bernardo (1993) and confirmed by the few available pedigrees on parents.
In the following we will address some issues on the number and the existence of detected QTL, the effect of selection, the improvements that could be done and the potential to perform MAS with QTL detected on breeding populations.
Number of detected QTL
In this study, we showed that many QTL for both traits were found for the six scanned chromosomes, more than each classical bi-parental population showed on these chromosomes (see e.g. Courtot × Chinese spring population, Perretant et al. 2000; wheat × spelt population, Zanetti et al. 2001). We can suggest some explanations: First, when working on populations derived from more than two lines, there is a higher chance for a given QTL to be polymorphic. Thus, if there are many genes controlling the quantitative trait, more genes will be polymorphic (compared with a bi-parental cross), and thus the chance to detect a higher number of QTL will be enhanced. This kind of results was demonstrated by Muranty (1996) and Xu (1998), for different types of QTL effects and different number of alleles at the QTL. Nevertheless, if the majority of the parents were fixed for most of the QTL, working in a multi-parental design would still not allow QTL detection, as no variation could be explained by still segregating QTL alleles. In this multi-cross design, the F6 phenotypes showed very contrasting values for both traits (ranging from 15 to 100 for kernel hardness and from 53 to 499 J×10−4, for dough strength). These contrasting values could be explained by the fact that non-unidirectional selection on traits is generally performed during pedigree breeding, and particularly for characters such as kernel hardness and dough strength (more focus is given to yield, diseases, and direct quality tests during wheat breeding). Another explanation is that the F6 came from different end-use programmes and from different breeders, who handle different germplasm and thus altogether maintained more variability. We can finally mention that all the possible QTL for these two traits were not necessarily detected on the six chromosomes studied in this population, and that other breeding populations could allow to detect even more QTL using different germplasm (with different fixed QTL alleles for instance). Some more QTL could also be detected in larger populations by increasing the power to detect smaller QTL (for an equivalent population structure, it was demonstrated that the method should allow to detect QTL explaining 10% of the total variance in 62% of the cases, Crepieux et al. 2004b). Finally, possible interactions between QTL and environment could show different QTL in different environments even if the two traits of this study have been shown to be mostly genetically controlled (Robert and Denis 1996).
Bibliographic survey of the detected QTL
In bread wheat, kernel texture (i.e. hardness versus softness) has been extensively studied because of its influence on bread-making quality. The difference between hard and soft wheats is controlled by a major gene, Ha (Symes 1965). Monosomic analyses assigned this gene to the short arm of chromosome 5D (Mattern et al. 1973; Law et al. 1978) and using QTL analyses, Sourdille et al. (1996) mapped the Ha gene at the extremity of chromosome arm 5DS, close to loci encoding the puroindoline proteins. Recent studies have shown that wheat grain hardness is conferred either by a null allele at the puroindoline a (Pina) locus (Giroux and Morris 1998) or by specific mutations in puroindoline b (Pinb) locus (Lillemo and Morris 2000). While the genetic basis of the difference between the two major hardness classes is now well established, little is known about the residual variation within each class of hardness and its genetic components. Bettge and Morris (2000) found that among the ‘soft’ wheat samples, variation in grain texture was related to the cell-wall-associated pentosan fraction, but no similar relation has been determined in ‘hard’ wheat samples. Genetic analyses have shown the influence of many chromosomes on hardness but with minor effects between hard and soft wheat genotypes (see for instance Sourdille et al. 1996, Campbell et al. 1999).
In the present study, we found two QTL for grain hardness exceeding the significance threshold. One is likely to be the well-known QTL on chromosome 5DS associated to the Ha locus (Sourdille et al. 1996; Perretant et al. 2000). It is generally admitted that this locus explains an important part of the hardness variation in progenies from crosses between hard and soft wheat. However, these conclusions were supported by studies of bi-parental populations, which segregated for (mostly) Ha. In our broad-base population, this locus explained only 27% of the variation. This could be explained by other QTL segregating, but also by the unequal frequency of hard/soft types in our breeding material (only 13% have a hardness value lower to 50). It is known that the additive variance accounted for by a single QTL in the lines derived from a bi-parental cross is (4p(1-p)a²), where p is the frequency of one allele and a is the allele substitution effect. Thus, the value of the additive variance is maximum for p=0.5 and lower for uneven allele frequencies. To make a comparison, the QTL detected by Sourdille et al. (1996) explained 63% of the variation, with p=0.5. In our material, we could roughly estimate the frequency of Ha (soft allele) around 0.13. Thus, with the same allelic effect, this QTL will explain only 4×0.13×0.87×0.63=0.28, which is close to the value obtained under the full model.
The second QTL is located on the 1D chromosome, close to the 1D HMW-glutenin. No other study reports the presence of kernel hardness QTL on this chromosome. However, we cannot totally discard the hypothesis of an artefact caused by storage protein, either through quantitative or qualitative variation, on hardness prediction by NIR, as suggested by Groos et al. (2004), who found fewer QTL for hardness when estimated by SKCS (Single Kernel Characterization, based on mechanical properties) than by NIR spectroscopy.
For dough strength, we found three significant QTL located on chromosome 1A, 1B, and 1D, close to the HMW glutenin loci, in homeologous position. The influence of HMW glutenins on end-use quality has been widely documented (MacRitchie 1999). More specifically, HMW glutenins have been reported to be responsible for a part of the variability of the W score (Branlard and Dardevet 1985).
Effect of selection and population sampling on the QTL detection
The population used in this paper was not sampled at random. Individuals were mostly chosen according to their pedigrees (in order to reduce the number of parents), in such a way that the parents with the higher breeding values were kept through their progenies to create the mapping population. Besides, breeding occurred from the F2 to the F6, with varying selection pressure on different traits, more or less correctly estimated according to their heritability (yield, e.g. starts to be ‘correctly’ estimated at the F5–F6 generation). At the end, the resulting mapping populations were composed by lines clearly identified as coming from different end-use programmes (one was more focused on very high quality wheat, and the others were orientated toward different parts of the French market with main focus on baking quality and yield). As it is difficult to estimate the real effect of selection and of sampling, it is difficult to predict the effect it will have on parameter estimates for the QTL detection. However, it was shown that the heritability could be highly upwardly biased in such populations undergoing selection (Crepieux et al. 2004a).
The effect of historical selection on the chance to detect QTL is obvious when its consequence is the near fixation of the most favourable allele. Even when a polymorphism remains at selected loci, the IBD probabilities in their neighbourhood is far from their expectation under panmixia, as estimated, for example, by Malecot’s coefficient. This could lead to substantial bias and lack of power in QTL estimation if an inappropriate method is used. We tried to avoid this drawback with the method presented in this paper.
The main issue with this sampling effect is to know if it is likely to generate ghost QTL. Indeed, when dealing with complex populations, the question of germplasm structuration is important. In the case of linkage disequilibrium studies, we try to discern population structure in order to control for the genetic background and avoid the confounding of real QTL effect with that of the alleles of a specific population (leading to numerous spurious associations). In our mapping population, we controlled the background effect by adding a polygene term in the model, which is estimated through the additive relationship matrix. However there could be, in the material, another structuration not exactly based on the ‘allele origins’ of the lines but more on the goal of the programmes and their end use. For instance, within the present material there are two distinct hardness classes: soft and hard wheat. Soft wheat, more adapted to biscuit making, represents a small market share in France. On the contrary, ‘hard’ (medium hard and hard) wheat, which is mostly used for bread making, is the main cultivated class. Breeding soft wheat represents only a small amount of a breeding programme (explaining the low amount of soft F6 found). To be transformed, soft wheat need to have a very low W (<100–120) while for being transformed into bread, hard or medium hard wheat require higher W values (>200). At the beginning of a breeding programme, the choice of the two-pair crosses already follows an initial goal: two soft wheat crossed together, for example, are orientated for biscuit making. Some other wheat for biscuit making will be chosen during the breeding process according to their characteristics (when crossing hard × soft for instance), notably the W value. Many publications have shown the role of HMW glutenins on W and have been mentioned in the previous part. Rankings on the value of the HMW glutenins have been proposed (see Branlard et al. 2001 for instance), and these values can be used during the selection process to rank bread making and biscuit-making wheat.
We can introduce here the concept of ‘disruptive selection’, as in our wheat mapping breeding population, the lines can be divided into two almost ‘independent’ subpopulations: a part is orientated during the breeding process to soft wheat, the breeder selecting also for low W, while another part will be more suited for bread-making quality, the breeder selecting thus for higher W. At the loci of both traits, two divergent sets of alleles were thus selected for to the extent that the IBD relationships between F6 lines at some of these loci became co-linear. This co-linearity is a source of spurious QTL when not properly taken into account—some QTL of one trait having become in gametic disequilibrium with QTL of the other trait. Hence, using marker information from the whole genome did not allow us to be specific enough to be able to control for one of these QTL when searching and fitting other QTL. Yet we are left with the problem of knowing which QTL are real. This is probably where bibliographic information is useful to sort the real from the false for a given trait.
Choice of the best alleles for MAS
The IBD-based VC analysis is a random effects model. Random effect models only partition the genetic variance of quantitative traits into effects due to different chromosomal regions. They do not directly allow the estimation of effects for each of the QTL allele as fixed effect models do. However, it could be possible to estimate the effects of the alleles as defined for the markers surrounding the QTL of interest, for example, by nested ANOVA taking into account the family structures (see e.g. Lynch and Walsh 1998). Nevertheless, the fixed effects methods, including the well-known LS regression (Knott et al. 1996), and the above-mentioned ANOVA, are not well suited for such fragmented designs. They cannot easily handle the very small and uneven family sizes, the hermaphrodite status of most inbreeding species and the mixing of many different half-sibs and full-sib families. Using a fixed model approach in such designs would lead to an over-parameterization of the model leaving not enough degrees of freedom for an accurate estimation of the effects. Nevertheless, if the goal really is to estimate the effects of alleles at the QTL itself, we would adopt the haplotypic approach in a fixed effects framework, as proposed by Jansen et al. (2003) for plant breeding. In their paper, they consider that combinations of the same allele information at successive markers between two parents represent the same information (i.e. the haplotypes are IBD). We would then carry out an ANOVA, based on haplotypes information instead of marker information. A drawback of this method is that it requires a marker densification on the QTL regions in order to build haplotypes with a high degree of confidence. Another drawback is that too many haplotypes are theoretically possible with markers showing high polymorphism, finally not allowing a precise estimation of their effects. Nevertheless, the advantage of this method, if the haplotypes effects can be estimated, is to directly identify the best haplotypes for MAS in breeding schemes. More complicated haplotypes approaches combining linkage and linkage disequilibrium mapping could be envisaged for fine mapping of a QTL (see Lund et al. 2003 for instance), but to our knowledge, computer softwares have not yet been developed for the particularities of plants.
Another way to choose the best alleles in order to carry on breeding is to use the overall breeding values given by the BLUP values obtained for each of the F6 at the QTL positions. It is then quite easy to find, within a pedigree, the best alleles at the markers closest to the QTL. It could be confirmed by estimating the genetic values of the parents at the QTL according to F6’s BLUP information and their pedigrees (see Fernando and Grossman 1989). The advantage of this method is that it does not require marker densification around the QTL of interest, contrarily to the haplotypes method. The drawback of such use of BLUP values to determine the best marker alleles is that if the marker is far from the QTL of interest, the allele information would be the correct one only within a pedigree. For example, in this study, only one SSR marker was found to be in strong linkage disequilibrium with a very likely candidate for a QTL (gwm642, with Glu-1D). This shows the influence of ‘historic’ selection, structure of crosses and self-pollination on wheat.
Conclusion
Quantitative trait loci detection performed on this breeding population for the two traits of interest showed consistent results with QTL and genes already reported in the literature. An efficient use of the low linkage information spread amongst the many small breeding families was allowed by the IBD-based VC method. Such method can thus provide an alternative to the development of specifically designed recombinant populations by exploiting the genetic variation currently managed by plant breeders. However, it seems necessary, before performing such QTL detection during the selection process, to investigate the potential to implement MAS directly in breeding populations. These further studies will maybe allow defining an optimized population structure to enhance the power of QTL detection and MAS implementation (e.g. Chakraborty et al. 2002), while keeping a high potential for conventional breeding.
References
Almasy L, Blangero J (1998) Multipoint quantitative trait linkage analysis in general pedigrees. Am J Hum Genet 62:1198–1211
American Association of Cereals Chemists (1995) Approved methods of the American association of cereal chemists, 9th edn. The American Association of Cereals Chemists, Saint Paul, MN
Beavis WD (1998) QTL analysis: power, precision and accuracy. In: Paterson AH (ed) Molecular analysis of complex traits. CRC Press, Boca Raton, FL, pp 145–161
Bernardo R (1993) Estimation of coefficient of coancestry using molecular markers in maize. Theor Appl Genet 85:1055–1062
Bernardo R (1998) Predicting the performance of untested single crosses: trait and marker data. In: Lamkey KR, Staub JE (eds) Concepts and breeding of heterosis in crop plants. Publ 25, Crop Science Society of America, Madison, Wis, pp 117–127
Bettge AD, Morris CF (2000) Relationships among grain hardness, pentosan fractions, and end-use quality of wheat. Cereal Chem 77:241–247
Bink MCAM, Uimari P, Sillanpää M, Janss L, Jansen R (2002) Multiple QTL mapping in related plant populations via a pedigree-analysis approach. Theor Appl Genet 104:751–762
Branlard G, Dardevet M (1985) Diversity of grain protein and bread quality. II. Correlation between high molecular weight subunits of glutenin and flour characteristics. J Cereal Sci 3:345–354
Branlard G, Dardevet M, Saccomano R, Lagoutte F, Gourdon J (2001) Genetic diversity of wheat storage proteins and bread wheat quality. Euphytica 119:59–67
Campbell KG, Bergman CJ, Gualberto DG, Anderson JA, Giroux MJ, Hareland G, Fulcher RG, Sorrells ME, Finney PL (1999) Quantitative trait loci associated with kernel traits in a soft x hard wheat cross. Crop Sci 39:1184–1195
Chakraborty R, Moreau L, Dekkers JCM (2002) A general method to optimize selection on multiple identified trait loci. Gen Sel Evol 34:145–170
Charmet G, Groos C (2002) QTL in bread-making. In: Molecular techniques in crop improvement. Kluwer Academic Publishers, Hingham, MA, pp 601–616
Crepieux S, Lebreton C, Servin B, Charmet G (2004a) QTL detection in multi-cross inbred designs: recovering QTL-IBD status information from marker data. Genetics 168:1737–1749
Crepieux S, Lebreton C, Servin B, Charmet G (2004b) IBD-based QTL detection in inbred pedigrees: a case study of cereal breeding programs. Euphytica 137:101–109
Fernando RL, Grossman M (1989) Marker assisted selection using best linear unbiased prediction. Gen Sel Evol 21:467–477
George AW, Visscher PM, Haley CS (2000) Mapping quantitative trait in complex pedigrees: a two-step variance component approach. Genetics 156:2081–2092
Gilmour AR, Cullis BR, Welham SJ, Thompson R (1998) ASREML program user manual. Ed. Orange Agricultural Institute, New South Wales, Australia
Giroux M, Morris C (1998) Wheat grain hardness results from highly conserved mutations in the friabilin components puroindoline a and b. Proc Natl Acad Sci 95:6262–6266
Groos C, Bervas E, Charmet G (2004) Genetic analysis of grain protein content, grain hardness and dough rheology in a hard x hard bread wheat progeny. J Cereal Sci 40:93–100
Haseman JK, Elston RC (1972) The investigation of linkage between a quantitative trait and a marker locus. Behav Genet 2:3–19
Jannink J-L, Bink MCAM, Jansen RC (2001) Using complex plant pedigrees to map valuable genes. Trends Plant Sci 6:337–342
Jansen RC (2001) Quantitative trait loci in inbred lines. In: Balding DJ, et al. (ed) Handbook of statistical genetics. Wiley, New York, pp 567–597
Jansen RC, Jannink J-L, Beavis WD (2003) Mapping quantitative trait loci in plant breeding populations: use of parental haplotype sharing. Crop Sci 43:829–834
Joffre M, Crepieux S (2004) PurPL: a software for marker correction on multi-cross designs made of pure lines. Mol Ecol Notes 4:789–791
Knott SA, Elsen J-M, Haley CS (1996) Methods for multiple-marker mapping of quantitative trait loci in half-sib populations. Theor Appl Genet 93:71–80
Lander E, Botstein D (1989) Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121:185–199
Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247
Law CN, Snape JW, Worland AJ (1978) The genetical relationship between height and yield in wheat. Heredity 40:133–151
Lillemo M, Morris CF (2000) A leucine to proline mutation in puroindoline b is frequently present in hard wheats from Northern Europe. Theor Appl Genet 100:1100–1107
Lund MS, Sorensen P, Guldbransten B, Sorensen DA (2003) Multitrait fine mapping of quantitative trait loci using combined linkage disequilibria and linkage analysis. Genetics 163:405–410
Lynch M, Walsh B (1998) Genetics and analysis of quantitative traits. Sinauer Associates, Sunderland, MA
MacRitchie F (1999) Wheat proteins: characterization and role in flour functionality. Cereal Foods World 44:188–193
Mattern PJ, Morris R, Schmidt JW, Johnson VA (1973) Locations of genes for kernel properties in the wheat variety “Cheyenne” using chromosome substitution lines. In: Proceedings of 4th international wheat genetic symposium, Colombus, Missouri, pp 703–707
Muranty H (1996) Power of tests for quantitative trait loci detection using full-sib families in different schemes. Heredity 76:156–165
Nei M, Li WH (1979) Mathematical model for studying genetic variations in terms of restriction endonucleases. Proc Natl Acad Sci 76:5369–5373
Parisseaux B, Bernardo R (2004) In silico mapping of quantitative trait loci in Maize. Theor Appl Genet 109:508–514
Perretant M, Cadalen T, Charmet G, Sourdille P, Nicolas P, Boeuf C, Tixier MH, Branlard G, Bernard S, Bernard M (2000) QTL analysis of bread-making quality in wheat using a doubled haploid population. Theor Appl Genet 100:1167–1175
Robert N, Denis JB (1996) Stability of baking quality in bread wheat using several statistical parameters. Theor Appl Genet 93:172–178
Röder M, Korzun V, Wendehake K, Plaschke J, Tixier M, Leroy P, Ganal M (1998) A microsatellite map of the wheat genome. Genetics 149:2007–2023
Roussel V, Koenig J, Beckert M, Balfourier F (2004) Molecular diversity in French bread wheat accessions related to temporal trends and breeding programmes. Theor Appl Genet 108:920–930
Servin B, Dillmann C, Decoux G, Hospital F (2002) MDM: a program to compute fully informative genotype frequencies in complex breeding schemes. J Hered 93:227–228
Slate J, Visscher PM, MacGregor S, Stevens D, Tate ML, Pemberton JM (2002) A genome scan for quantitative trait loci in a wild population of red deer (Cervus elaphus). Genetics 162:1863–1873
Sourdille P, Cadalen T, Guyomarc’h H, Snape J, Perretant M, Charmet G, Boeuf C, Bernard S, Bernard M (2003) An update of the Courtot × Chinese Spring intervarietal molecular marker linkage map for the QTL detection of agronomic traits in wheat. Theor Appl Genet 106:530–538
Sourdille P, Perretant MR, Charmet G, Leroy P, Gautier MF, Joudrier P, Nelson JC, Sorrells ME, Bernard M (1996) Linkage between RFLP markers and genes affecting kernel hardness in wheat. Theor Appl Genet 93:580–586
Symes KJ (1965) The inheritance of grain hardness in wheat as measured by the particle size index. Aust J Agr Res 16:113–123
Xie C, Gessler DDG, Xu S (1998) Combining different line crosses for mapping quantitative trait loci using the identical by descent-based variance component method. Genetics 149:1139–1146
Xu S (1998) Mapping quantitative trait loci using multiple families of line crosses. Genetics 148:517–524
Xu S, Atchley WR (1995) A random model approach to interval mapping of quantitative trait loci. Genetics 141:1189–1197
Zanetti S, Winzeler M, Feuillet C, Keller B, Messmer M (2001) Genetic analysis of bread-making quality in wheat and spelt. Plant Breed 120:13–19
Zeng Z-B (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Zhang Q, Boichard D, Hoeschele I, Ernst C, Eggen A, Murkve B, Pfister-Genskow M, Witte LA, Grignola FE, Uimari P, Thaller G, Bishop MD (1998) Mapping quantitative trait loci for milk production and health of dairy cattle in a large outbred pedigree. Genetics 149:1959–1973
Acknowledgements
The authors thank the two reviewers for helpful comments on the manuscript, notably for the description of the ‘disruptive selection’. They are grateful to B. Duperrier, J. B. Beaufume, and J. Stragliatti for providing the wheat breeding material and to E. Chanliaud and F. X. Oury for providing the quantitative traits. They are also grateful to M. Joffre and B. Servin for software development. Microsatellites data were produced on the INRA Clermont-Ferrand genotyping platform. This research was supported by the Ministère de l’Economie, des Finances et de l’Industrie (ASG Program No. 01 04 90 6058) and by the “Food Quality” programme, jointly founded by the INRA, the Auvergne Region and the French ministries of Research and of Agriculture.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Charcosset
Rights and permissions
About this article
Cite this article
Crepieux, S., Lebreton, C., Flament, P. et al. Application of a new IBD-based QTL mapping method to common wheat breeding population: analysis of kernel hardness and dough strength. Theor Appl Genet 111, 1409–1419 (2005). https://doi.org/10.1007/s00122-005-0073-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-005-0073-5