
Abstract
We have previously demonstrated that in the diploid rose population 97/9 resistance to the powdery mildew race 9 is controlled by a major dominant resistance gene, Rpp1. In the study reported here, we isolated several molecular markers closely linked to Rpp1 via bulked segregant analysis, with the gene being tagged in an interval of 5 cM between the two most adjacent markers. It was possible to convert the most closely linked amplified fragment length polymorphic (AFLP) marker into a sequence-characterised amplified region (SCAR) segregating in the same manner. Indirect mapping of Rpp1 in relation to the black spot resistance gene Rdr1 revealed no linkage between the two R genes. Furthermore, the genetic model based on a single dominant resistance gene was supported by the marker data.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Podosphaera pannosa (Wallr.: Fr.), the causal agent of powdery mildew (PM) disease on roses, was first described in 1819 by Wallroth as Alphitomorpha pannosa (Horst 1983; Braun and Takamatsu 2000). The mostly obligate ectoparasitic PMs are biotrophic pathogens found on numerous economically important cultivated plants like cereals, vegetables, fruit, forest trees and ornamentals. Because cereals are the most important food source worldwide and serious yield losses up to 33% (Fried et al. 1981) are caused by infection with Blumeria graminis (syn. Erysiphe graminis) Speer each year, there have been extensive studies on the PM-cereal interaction (Wolfe and McDermott 1994; Kunoh 1995) and on the extremely high genetic diversity of mildew populations that attack cereals (Bevan et al. 1993; Braun and Turgut 1995). With respect to the resistance against B. graminis DM f. sp. tritici ten of 26 known major genes have been mapped, and these can be used for marker-assisted selection (MAS) (Huang et al. 1997, 2000; Keller et al. 2000; Rong et al. 2000). In barley no fewer than eight PM resistance genes have been linked to molecular markers (Schönfeld et al. 1996). PM resistance genes have also been mapped in vegetables like tomato (Van der Beek et al. 1994; Huang et al. 2000) and pea (Tiwari et al. 1998) or in fruit trees like apple (Urbanietz 2002). Despite the high economic value of ornamental plants, only one resistance gene against fungal pathogens has ever been mapped in them. Von Malek et al. (2000) were the first to report markers linked to a resistance gene in roses, presenting data on the molecular mapping of Rdr1, which confers resistance to black spot disease in roses. Subsequently, Kaufmann et al. (2003) assembled a contig of six large insert clones from a Rosa rugosa bacterial artificial chromosome (BAC) library around this locus that spanned a region of about 400 kb. Recently, we presented data for the isolation and identification of eight PM races using a differential set of ten rose genotypes (Linde and Debener 2003). Due to a 1:1 segregation of resistance against race 9 of P. pannosa in a backcross population of 114 individuals, we postulated that this resistance was controlled by a single dominant gene, called Rpp1. This was the first description of a resistance gene against rose PM. In the investigation reported here, we developed molecular markers linked to Rpp1 with the following goals:
-
1.
To find markers closely linked to Rpp1 which could be used for MAS in the future.
-
2.
To determine the most appropriate cut-off point between resistant and susceptible plants for the groups with low disease indices (5% and 10%).
-
3.
To locate the resistance gene on the rose molecular marker map.
Materials and methods
Plant material
The rose genotypes used in this study originated from a cross of the diploid line 88/124-46 (resistant against PM isolate 9) and the susceptible diploid male parent 82/78-1. Both genotypes are open-pollinated seedlings from a breeding programme aimed at the introgression of genes from tetraploid garden roses into Rosa multiflora (Reimann-Philipp 1981). The resulting F1 hybrid 95/13-90 was backcrossed to 82/78-1, resulting in a population of 117 plants segregating for resistance to physiological race 9. In addition to the 114 plants previously described in Linde and Debener (2003), we also present here resistance data for the remaining three individuals.
Fungal isolates, inoculation assay and disease assessment
The monoconidial PM isolate 9 was established by transferring single conidia, originating from the host genotype 97/7-13 grown in Ahrensburg (Germany) to in vitro shoots of the susceptible genotype 94/103-02 as described previously by Linde and Debener (2003). For the inoculation experiments, the isolate was multiplied by infecting in vitro shoots growing in glass vessels.
Leaflets from three rose genotypes were placed in glass petri dishes on water-agar (0.5% agar) containing 0.03% benzimidazole to prevent fungal contamination of the agar surface. From each genotype six to nine leaflets (third to fifth unfolded leaves from the shoot tip) were used. The leaflets were infected with approximately two conidia per square millimeter of leaf surface as described by Linde and Debener (2003). At 10-days post-inoculation, the percentages of leaf area covered with conidiophores were estimated in 10% steps (from 0% to 100%) using a stereomicroscope (8- to 50-fold magnification). The inoculations were repeated five to eight times for the 117 individuals. The minimum and maximum values were excluded, and the mean was calculated and taken as a disease index (DI).
Genotypes with only very few conidiophores on the whole leaf material were scored as having a DI of 5%. Although the leaf area covered with conidiophores was significantly lower than 5% in this class, a DI value of 5% was given in order to have a numerical value that could be used for further statistical analysis of the data [e.g. quantitative trait loci (QTL) analysis] and because attempts to make a more precise area determination were very unreliable. Rose genotypes showing a DI of 10% or more were considered to be susceptible. Mean DI values of less than 5% and with no single estimation over 5% were treated as resistant.
DNA extraction
Genomic DNA was extracted from 100 mg of frozen leaf tissue according to the method of Kobayashi et al. (1998) with the following modifications. After resuspending the DNA pellet in 500 μl TE, 10 μg RNAse A (Roche, Ingelheim, Germany) were added, followed by an extraction with 500 μl phenol/chloroform. The DNA was precipitated with 1/10 volume of 3 M NaAc and 0.75 volume of isopropanol, washed two times with 70% ethanol, dried briefly and resuspended in H2O.
Amplified fragment length analysis
AFLP analyses were performed as described by Von Malek et al. (2000) using the restriction enzymes MseI and HindIII and specific primers with three selective bases each. The HindIII primers were fluorescently labelled with IRD700 and IRD800 dyes at their 5′ end (MWG Biotech, Ebersberg, Germany). DNA fragments were separated and visualised with LICOR Gene ReadIR 4200 automated sequencers (MWG biotech, Ebersberg, Germany) on 25-cm-long 6% denaturing polyacrylamide gels.
Conversion of an AFLP fragment into a sequence-characterised amplified region marker
For the generation of a SCAR marker (Paran and Michelmore 1993) ten selective AFLP reactions with the primer combination producing the desired fragment were conducted in a 10 μl reaction volume. The area around the ten marker fragments (H-CAg/M-ATT-103) were cut out in five fractions from the polyacrylamide gel and the DNA eluted overnight in H2O at room temperature. After centrifugation at 10,000 rpm the solution was diluted 1:1,000 and 1:10,000 with H2O. Two microliters of each dilution were then reamplified with the selective AFLP primer combination and run on the sequencer a second time. The fraction with the lowest number of extra bands in addition to the target fragment was used to prepare ten selective AFLP reactions of 20 μl each with the H-CAg/M-ATT primer combination. The reactions were pooled, and the amplified fragments were precipitated with ethanol and resuspended in H2O. Approximately 25 ng of the eluted fragments was cloned into the pGEM-T Easy vector (Promega, Madison, Wis.) according to the manufacturer’s instructions. Using an Escherichia coli pulser transformation apparatus (Bio-Rad, Hercules, Calif.), we used 1 μl of the ligation mixture to transform 40 μl of E. coli DH10B cells (Life Technologies, Rockville, Md.) by electroporation following standard procedures (Sambrook and Russel 2001). A total of 96 white and lightly blue colonies were picked out, and selective AFLP reactions were directly made with the bacterial colonies. Five clones displaying fragments of the correct size were selected for plasmid mini-preparation and sequenced twice from both sides using the Thermo Sequenase Cycle Sequencing kit (Amersham Biosciences, Freiburg, Germany) and fluorescently labelled M13 primers. From three of the five clones we obtained sequence data and aligned these using dnasis software (Hitachi Software Engineering, San Bruno, Calif.). Based on the consensus sequences four primer pairs were designed for the specific amplification of the AFLP fragment using oligo primer analysis software (NBI, Plymouth, UK).
SCAR amplification and analysis
The reactions for the selective PCR were carried out in a total volume of 25 μl containing 20 ng of genomic DNA, 2.5 μl of a 10× Williams buffer (Williams et al. 1990), 10 pmol of each primer, 1 U Taq DNA polymerase (Invitrogen, Karlsruhe, Germany), 0.1 mM dNTPs. The amplifications were performed in an Eppendorf Mastercycler Gradient with an initial denaturation step at 94°C for 4 min, followed by 30 cycles of 30 s at 94°C, 50 s at 47°C and 20 s at 72°C and a final elongation of 6 min at 72°C. The PCR products were separated on 1.5% agarose gels.
Results
Segregation for powdery mildew resistance
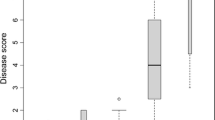
As described previously (Linde and Debener 2003) the 117 plants of population 97/9 displayed a clear 1:1 segregation (χ2=1.923, not significant) with 66 resistant versus 51 susceptible individuals (Fig. 1). This indicated that resistance against race 9 is controlled by a single dominant gene (Rpp1) in the backcross population. We considered plants to be resistant when (1) they showed no conidiophores at all (DI=0%) and (2) they had conidiophores only occasionally and only in a small number of the repeats (DI=5%).

Segregation data for rose BC1 population 97/9 inoculated with PM race 9. Disease index (DI): Percentage leaf area covered with conidiophores 10 days after infection (mean of five to eight replications)
Identification of AFLP markers linked to Rpp1
To identify markers closely linked to Rpp1 we carried out a bulked segregant analysis with bulks comprising equal amounts of DNA from ten resistant (DI=0%) and ten highly susceptible plants (DI≥50%), respectively. A total of 260 AFLP primer combinations were tested with these bulks. The 45 combinations showing a fragment present only in the resistant parent (88/124-46) and in the bulk with the resistant progeny were first tested on all individuals of the bulks. Primer combinations with a banding pattern indicating linkage to Rpp1 were then tested on the remaining 44 individuals with a DI=0 and on the 35 susceptible plants with a DI>20. Based on the first analyses individuals displaying recombination for a specific linked marker fragment were discarded from the bulks and replaced by a non-recombinant genotype. The discarded recombinants were then tested in a separate lane on the gel beside the bulks with new primer combinations. Following this approach, the putative resistance gene Rpp1 could be located at an interval of 5 cM (Fig. 2) between the AFLP markers H-CAT/M-ggC1 (3.4 cM) and H-CAg/M-ATT (1.7 cM).

Linkage maps for Rpp1 in BC1 population 97/9 segregating for PM race 9 (left side) and indirect mapping of this locus in population 94/1 using AFLP marker H-CAT/M-ggC-1 (right side)
Analysis of plants with disease scores of 5% and 10%
In a previous study, plants with a DI of 5% (plants that only occasionally developed single conidiophores on single leaves in some of the replicates) were considered to be resistant, whereas plants with a DI of 10% were considered to be susceptible (Fig. 1). After performing marker analyses using plants with a DI of either 0% or more than 20% the fraction of plants with a DI of 5% and 10% was analysed with the most closely linked marker on each side of the major locus. Individual genotypes from both classes did not show any recombination between the two markers and the target locus. All 18 plants with a DI of 5% carried the markers from the resistant parent, whereas all five plants with a DI of 10% lacked these markers, thereby supporting the classification into resistant and susceptible plants (Table 1).
Localisation of Rpp1 in comparison to Rdr1
In population 97/9, segregation for resistance to PM race 9 differs significantly from segregation of the black spot resistance gene Rdr1 in the same population, indicating that these two loci are not linked (Linde and Debener 2003). To obtain more information on the map location of Rpp1, we utilised the diploid population 94/1, which had been used previously to construct a molecular marker map. As this population does not segregate for resistance to PM race 9, an indirect approach via the mapping of markers linked to Rpp1 had to be used. Therefore, all markers closely linked to Rpp1 were tested with the parental plants (93/1-119×93/1-117) of the population 94/1 for polymorphisms. Only the AFLP marker H-CAT/M-ggC1 showed a clearly detectable polymorphism (present in 93/1-119, absent in 93/1-117) which segregated in both populations (97/9 and 94/1) with fragments of an identical size. Sixty individuals of the mapping population 94/1 were evaluated for the segregation of this AFLP marker by being run in parallel on the same polyacrylamide gel together with samples from the 97/9 population. Using joinmap 2.0 (Stam and Van Ooijen 1995) we located the fragment on linkage group A3 of the map from Debener and Mattiesch (1999), whereas the black-spot resistance gene Rdr1 is located on linkage group B1 (Fig. 2).
Conversion of H-CAg/M-ATT-103 into a SCAR marker
AFLP fragment H-CAg/M-ATT-103, which showed the closest linkage to Rpp1, was cut out from the gel, cloned and sequenced. Five clones with the correct insert length of about 103 bp were selected for further analysis. Three of these (clones 6, 8 and 18) could be properly sequenced from both sides two times each. The consensus sequences of clones 6 and 18 turned out to be identical. The sequence data from clones 6 and 8 and were then used to design an inner PCR primer pair (6in and 8in) for each clone, resulting in products of 67 bp and 65 bp, and an outer primer pair (6out and 8out), resulting in products of 99 bp and 91 bp. PCR reactions with the inner primer pairs and the parental DNAs using a temperature gradient from 44°C to 60°C gave only weak amplification products. The outer primer pairs, however, produced both clearly visible fragments of the expected size and polymorphisms between the parents. Fragments derived from primer pair 6out did not segregate in the progeny, whereas those from primer pair 8out did. The screening of all 117 individuals with the primer pair 6outforw (5′-TGAGCCCTGAGTAAATTCAGAACG-3′) and 6outrev (5′-TGCGTACCAGCTTCAGCGAG-3′) resulted in a fragment of about 99 bp with the same segregation pattern as obtained with the AFLP H-CAg/M-ATT-103 (Fig. 3). Unfortunately, this SCAR turned out to be not polymorphic within population 94/1 and therefore could not be located in the existing linkage map of this population.

Fragment patterns of the developed SCAR marker (above) and the corresponding AFLP H-CAg/M-ATT (below) from which it was converted for the resistant (P1) and susceptible (P2) parental plants and the BC1 progeny with resistant (r) and susceptible (s) genotypes. Lane 1 Sizing standards
Discussion
We describe herein the isolation of molecular markers linked to Rpp1, a gene conferring resistance to PM in rose population 97/9. To our knowledge this is the first report on molecular markers linked to a PM resistance gene in roses. The only other rose resistance gene characterised with molecular markers to date is Rdr1, a gene giving resistance to black spot (Von Malek et al. 2000).
Using a bulked segregant analysis, we located Rpp1 in a small interval of about 5 cM with AFLP markers on both sides of the putative gene and a SCAR marker at a distance of about 2 cM from the gene. This finding makes it possible to use MAS (Tanksley et al. 1989) for PM resistance in roses, which could be used also in combination with markers known to be linked to the black spot resistance gene Rdr1 (Kaufmann et al. 2003). The tight linkage of flanking markers around the locus provides a high reliability of the localisation of Rpp1 in the rose genome. However, due to the large variability that has been observed for the ratios between genetic and physical distances in different plant genomes (Schmidt et al. 1995; Paterson 1996) our markers are most probably not linked tightly enough for positional cloning of Rpp1. Screening for molecular markers linked to resistance genes rather than for disease phenotypes could significantly speed up the introgression of R genes into common cultivars. The time span required before the improved cultivar could be brought to market could be reduced by up to 50–70% using MAS (Tanksley et al. 1989; Schneider et al. 1997). This would be a great advantage for rose breeding where selection requires at least between 5 and 7 years for cut and garden roses (Chaanin 2003; Noack 2003). Unfortunately, resistance provided by single R genes is often rapidly overcome by new races of the pathogen, particularly for species grown in large monocultures like cut roses and for plants being attacked by high-risk pathogens like PMs with a high potential for gene and genotype flow (McDonald and Linde 2002). To avoid this risk, different resistance genes could be combined in one genotype (gene pyramids) to increase the likelihood of resistance durability (Schaffer and Roelfs 1985; Liu et al. 2000), or varietal mixtures with several R genes could be planted, conferring even more durable resistance (Wolfe 1985; Jones 2001). Durable resistance against PMs provided by major genes controlling all races is only known for mlo in barley (Rommens and Kishore 2000) and will probably be hard to find in roses. Based on some recently characterised PM races, additional resistance genes are currently under investigation in rose breeding investigations. These may lead to more resistance genes suitable for MAS and the pyramiding of genes in the near future. As Rpp1 also segregates in several other diploid rose populations the analysis of additional markers with a larger number of individuals could be the starting point for fine mapping and positional cloning of the putative genes. A recently established BAC library from R. rugosa containing 5.2 equivalents (Kaufmann et al. 2003) could serve as a general tool for the physical mapping and cloning of these loci which could be supported by a large number of mapped rose resistance gene analogues (Hattendorf et al. 2004).
Regarding our recent publication on the identification of eight rose PM races and the genetic analysis of the resistance gene Rpp1 (Linde and Debener 2003) the resistance classification in population 97/9 was discussed as being somehow arbitrary. Genotypes occasionally showing very few conidiophores in a fraction of the experiments, and with no single estimation of over 5%, were treated as resistant (DI=5%), whereas plants with a DI of 10% were classified as susceptible. Genotypes allowing the growth of only a small number of conidiophores are also classified as being resistant in a number of other publications on PM because they produce such a minimal number of conidia that there is no appreciable spread of the disease. This resistant classification has even been applied for genotypes showing a higher disease severity (Huang et al. 2000; Rong et al. 2000; Zeller et al. 2002). The results from our analysis with the two most closely linked AFLP markers confirmed the cut-off point between resistant and susceptible individuals. All 23 genotypes with disease indices of 5% and 10% showed the expected marker pattern of resistant or susceptible plants, respectively (Table 1). This supports our classification which was built on the results of five to eight repeated inoculations under highly reproducible conditions (Linde and Debener 2003).
The mapping of Rpp1 on the linkage map in comparison to the black spot resistance locus Rdr1 was hindered by several problems: (1) there is no segregation for resistance to PM race 9 in population 94/1, which prevents the direct mapping of Rpp1 in this population with an existing linkage map (Debener and Mattiesch 1999); (2) the SCAR marker constructed from the most closely linked AFLP H-CAg/M-ATT-103, which segregated in the same manner, turned out to be not polymorphic within population 94/1. So we had to use the AFLP markers linked to Rpp1 directly in population 94/1. (3) Screening the population 94/1 with AFLP marker H-CAT/M-ggC1 we could map this closely linked marker on linkage group A3 of the map constructed by Debener and Mattiesch in 1999, whereas the black spot resistance gene Rdr1 is located on group B1. Our results strongly support the genetic data on the independent segregation of these two resistance gene loci in diploid roses. Our current mapping of a large number of microsatellites and expressed sequence tags will hopefully circumvent these problems and enable the genomic regions around both genes in many different rose populations to be located.
References
Bevan JR, Crute IR, Clarke DD (1993) Variation for virulence in Erysiphe fischeri from Senecio vulgaris. Plant Pathol 42:622–635
Braun U, Takamatsu S (2000) Phylogeny of Erysiphe, Microsphaera, Uncinula (Erysipheae) and Cystotheca, Podosphaera, Sphaerotheca (Cystotheceae) inferred from rDNA ITS sequences—some taxonomic consequences. Schlechtendalia 4:1–33
Braun PW, Turgut I (1995) The virulence structure of mildew populations on wild barley in Turkey. J Plant Dis Prot 102:593–598
Chaanin A (2003) Breeding/selection strategies for cut roses. In: Roberts AV, Debener T, Gudin S (eds) Encyclopedia of rose science. Elsevier, Oxford, pp 33–41
Debener T, Mattiesch L (1999) Construction of a genetic linkage map for roses using RAPD and AFLP markers. Theor Appl Genet 99:891–899
Fried PM, MacKenzie DR, Nelson RR (1981) Yield loss caused by Erysiphe graminis f. sp. tritici on single culms of ‘Chancellor’ wheat and four multilines. Z Pflanzenk Pflanzen 88:256–264
Hattendorf A, Linde M, Kaufmann H, Mattiesch L, Debener T (2004) Genetic analysis of rose resistance genes and their localisation in the rose genome. Acta Hortic (in press)
Horst RK (1983) Compendium of rose diseases. American Phytopathological Society, St. Paul
Huang XQ, Hsam SLK, Zeller FJ (1997) Chromosomal location of genes for resistance to powdery mildew in common wheat (Triticum aestivum L. em. Thell.). 4. Gene Pm24 in Chinese landrace Chiyacao. Theor Appl Genet 95:950–953
Huang CC, Cui YY, Weng CR, Zabel P, Lindhout P (2000) Development of diagnostic PCR markers closely linked to the tomato powdery mildew resistance gene Ol-1 on chromosome 6 of tomato. Theor Appl Genet 101:918–924
Jones JDG (2001) Putting knowledge on disease resistance genes to work. Curr Opin Plant Biol 4:281–287
Kaufmann H, Mattiesch L, Lörz H, Debener T (2003) Construction of a bac library of Rosa rugosa Thunb. and assembly of a contig spanning Rdr1, a gene that confers resistance to black spot. Mol Genet Genomics 268:666–674
Keller B, Feuillet C, Messmer M (2000) Genetics of disease resistance: basis concepts and application in resistance breeding. In: Slusarenko AJ, Fraser RSS, Van Loon LC (eds) Mechanisms of resistance to plant diseases. Kluwer, Dordrecht, pp 101–136
Kobayashi N, Horikoshi T, Katsuyama H, Handa T, Takayanagi K (1998) A simple and efficient DNA extraction method for plants, especially woody plants. Plant Tissue Cult Biotechnol 4:77–81
Kunoh H (1995) Host-parasite specificity in powdery mildews. In: Singh US, Kohmoto K, Singh RP (eds) Pathogenesis and host specificity in plant diseases: histopathological, biochemical, genetic and molecular bases, vol. 2 Eukaryotes. Elsevier, Oxford, pp 239–250
Linde M, Debener T (2003) Isolation and identification of eight races of powdery mildew of roses [Podosphaera pannosa (Wallr.: Fr.) de Bary] and the genetic analysis of the resistance gene Rpp1. Theor Appl Genet 107:256–262
Liu J, Liu D, Tao W, Li W, Wang S, Chen P, Cheng S, Gao D (2000) Molecular marker-facilitated pyramiding of different genes for powdery mildew resistance in wheat. Plant Breed 119:21–24
McDonald BA, Linde C (2002) Pathogen population genetics, evolutionary potential and durable resistance. Annu Rev Phytopathol 40:349–379
Noack R (2003) Breeding/selection strategies for disease and pest resistance. In: Roberts AV, Debener T, Gudin S (eds) Encyclopedia of rose science. Elsevier, Oxford, pp 49–55
Paran I, Michelmore RW (1993) Development of reliable PCR-based markers linked to downy mildew resistance genes in lettuce. Theor Appl Genet 85:985–993
Paterson AH (1996) Physical mapping and map-based cloning: bridging the gap between DNA markers and genes. In: Paterson AH (ed) Genome mapping in plants. Academic, San Diego, pp 55–61
Reimann-Philipp (1981) Cytogenetics and breeding in diploid roses from the triploid hybrid R. multiflora × garden cultivars. In Eucarpia (ed): Proc Eucarpia Ornamentals Meet Rose Breed. Eucarpia, pp 27–29
Rommens CM, Kishore GM (2000) Exploiting the full potential of disease-resistance genes for agricultural use. Curr Opin Biotechnol 11:120–125
Rong JK, Millet E, Manisterski J, Feldmann M (2000) A new powdery mildew resistance gene: introgression from wild emmer into common wheat and RFLP-based mapping. Euphytica 115:121–126
Sambrook J, Russel DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Press, Cold Spring Harbor
Schaffer JF, Roelfs AP (1985) Estimated relation between numbers of urediniospores of Puccinia-graminis f. sp. tritici and rates of occurrence of virulence. Phytopathology 75:749–750
Schmidt R, West J, Love K, Lenehan Z, Lister C, Thompson H, Bouchez D, Dean C (1995) Physical map and organization of Arabidopsis thaliana chromosome 4. Science 270:480–483
Schneider KA, Brothers ME, Kelly JD (1997) Marker-assisted selection to improve drought resistance in common bean. Crop Sci 37:51–60
Schönfeld M, Ragni A, Fischbeck G, Jahoor A (1996) RFLP mapping of three new loci for resistance genes to powdery mildew (Erysiphe graminis f. sp. hordei) in barley. Theor Appl Genet 93:48–56
Stam P, Van Ooijen JW (1995) joinmap version 2.0: software for the calculation of genetic linkage maps. CPRO-DLO, Wageningen
Tanksley SD, Young ND, Paterson AH, Bonierbale MW (1989) RFLP mapping in plant breeding: new tools for an old science. Biotechnology 7:257–264
Tiwari KR, Penner GA, Warkentin TD (1998) Identification of coupling and repulsion phase RAPD markers for powdery mildew resistance gene er-1 in pea. Genome 41:440–444
Urbanietz A (2002) Genetische und molekulare Charakterisierung der Resistenz des Apfels gegen den Echten Mehltau und der Virulenz des Erregers Podosphaera leucotricha (Ell. Et Ev.) Salm. PhD thesis, University of Hannover, Germany
Van der Beek JG, Pet G, Lindhout P (1994) Resistance to powdery mildew (Oidium lycopersicon) in Lycopersicon-hirsutum is controlled by an incompletely dominant gene Ol-1 on chromosome 6. Theor Appl Genet 89:467–473
Von Malek B, Weber WE, Debener T (2000) Identification of molecular markers linked to Rdr1, a gene conferring resistance to black spot in roses. Theor Appl Genet 101:977–983
Williams JGK, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV (1990) DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res 18:6531–6535
Wolfe MS (1985) The current status and prospects of multiline cultivars and variety mixtures for disease resistance. Annu Rev Phytopathol 23:251–273
Wolfe MS, McDermott JM (1994) Population genetics of plant pathogen interactions: the example of the Erysiphe graminis–Hordeum vulgare pathosystem. Annu Rev Phytopathol 32:89–113
Zeller FJ, Kong L, Hartl L, Mohler V, Hsam SLK (2002) Chromosomal location of genes for resistance to powdery mildew in common wheat (Triticum aestivum L. em Thell.) 7. Gene Pm29 in line Pova. Euphytica 123:187–194
Acknowledgements
This project was supported by a grant from the Ministry of Economics, Technology and Transport of the Land Schleswig-Holstein and by the companies W. Kordes Söhne (Germany) and Rosen Tantau (Germany). We thank K. Sabin for valuable technical assistance and Prof. A.V. Roberts (UEL, London) for critically reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by H. Nybom
Rights and permissions
About this article
Cite this article
Linde, M., Mattiesch, L. & Debener, T. Rpp1, a dominant gene providing race-specific resistance to rose powdery mildew (Podosphaera pannosa): molecular mapping, SCAR development and confirmation of disease resistance data. Theor Appl Genet 109, 1261–1266 (2004). https://doi.org/10.1007/s00122-004-1735-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-004-1735-4