
Abstract
Molecular markers for resistance of sorghum to the hemi-parasitic weed Striga hermonthica were mapped in two recombinant inbred populations (RIP-1, -2) of F3:5 lines developed from the crosses IS9830 × E36-1 (1) and N13 × E36-1 (2). The resistant parental lines were IS9830 and N13; the former is characterized by a low stimulation of striga seed germination, the latter by “mechanical” resistance. The genetic maps of RIP-1 and RIP-2 spanned 1,498 cM and 1,599 cM, respectively, with 137 and 157 markers distributed over 11 linkage groups. To evaluate striga resistance, we divided each RIP into set 1 (116 lines tested in 1997) and set 2 (110 lines evaluated in 1998). Field trials were conducted in five environments per year in Mali and Kenya. Heritability estimates for area under the striga number progress curve (ASNPC) in sets 1 and 2 were respectively 0.66 and 0.74 in RIP-1 and 0.81 and 0.82 in RIP-2. Across sites, composite interval mapping detected 11 QTL (quantitative trait loci) and nine QTL in sets 1 and 2 of RIP-1, explaining 77% and 80% of the genetic variance for ASNPC, respectively. The most significant RIP-1 QTL corresponded to the major-gene locus lgs (low stimulation of striga seed germination) in linkage group I. In RIP-2, 11 QTL and nine QTL explained 79% and 82% of the genetic variance for ASNPC in sets 1 and 2, respectively. Five QTL were common to both sets of each RIP, with the resistance alleles deriving from IS9830 or N13. Since their effects were validated across environments, years and independent RIP samples, these QTL are excellent candidates for marker-assisted selection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sorghum [Sorghum bicolor (L.) Moench] is the staple food for millions of people in the semi-arid tropics. Mean grain yield ranges from 800 kg ha−1 in Africa to 3,400 kg ha−1 in the Americas (FAO 2000). Major growing constraints to sorghum production in sub-Saharan Africa are the hemi-parasitic weeds Striga hermonthica (Del.) Benth. and S. asiatica (L.) Kuntze (Scrophulariaceae). These weeds attach to and penetrate host roots, extracting water and nutrients. Grain yield losses due to infestation of cereals in West Africa average 24% (Sauerborn 1991), but crop loss can be total when striga attack is compounded by drought. The two species also parasitize maize (Zea mays), pearl millet (Pennisetum glaucum), rice (Oryza sativa), fonio (Digitaria exilis) and teff (Eragrostis teff). A third species, S. gesnerioides (Willd.) Vatke, attacks legumes. The shifting of cultivation to continuous cropping, concomitant loss in soil fertility and the frequent cultivation of susceptible crops are responsible for increased striga infestation (Kroschel 1999).
Reducing the striga seed bank in the soil is fundamental to striga control. This may be achieved by destroying seed in the soil, reducing striga reproduction and preventing striga seed dissemination from infested to uninfested fields (Berner et al. 1994; Hess and Haussmann 1999). Striga-resistant sorghum varieties may reduce striga reproduction and the number of viable seeds in the soil, but they are often not locally adapted and agronomically inferior. Resistant, farmer-preferred varieties could be an important component of integrated striga management, but progress in breeding for striga resistance has been slow due to a limited knowledge of the genetics of striga resistance and the difficulty in evaluating resistance in the field. Field screening is hampered by heterogeneous natural infestations, high variability of soils and large environmental effects on striga emergence. However, improved field testing methodologies have recently been developed (Haussmann et al. 2000a) and validated (Omanya et al. 2004).
Several mechanisms of striga resistance exist in sorghum and its wild relatives (Ejeta et al. 2000; Gurney et al. 2002; Heller and Wegmann 2000). A current research program at Purdue University is designed to dissect the infection cycle into key events (for example, germination, haustorium formation, striga penetration and development), develop physiological assays for each event, identify specific resistance sources in breeding materials or wild relatives using these assays and, finally, to combine the resistance factors in productive genetic backgrounds (Ejeta and Butler 1993; Ejeta et al. 2000; Kapran et al. 2002; Mohamed et al. 2003). However, these are long-term research goals, and the successful development of the program requires a great deal of time and energy.
The approach of quantitative trait locus (QTL) mapping has the appealing advantage that genomic regions affecting a complex, quantitative trait like striga field resistance can be identified without prior knowledge of their function. Resistance QTL, once identified, can be transferred into adapted cultivars using marker-assisted selection. This could greatly accelerate breeding progress, also because striga seed is quarantined, confining tests to areas where striga is endemic, and because some striga resistance genes are recessive (Ramaiah et al. 1990; Vogler et al. 1996; Haussmann et al. 2001a) and difficult to select in conventional backcross schemes. A major objective of our collaborative project was therefore to identify DNA markers for striga field resistance in sorghum.
Materials and methods
Genetic materials
Two sorghum [Sorghum bicolor (L.) Moench] recombinant inbred populations (RIP-1, -2) of 226 F3:5 lines each were developed from crosses: IS9830 × E36-1 (1) and N13 × E36-1 (2). Line IS9830 is a Sudanese feterita belonging to race caudatum and striga-resistant due to its production of low amounts of striga germination stimulants, resulting in the germination of fewer striga seeds (Haussmann et al. 2000c). In contrast, line N13, an Indian durra sorghum, stimulates abundant striga seed germination but forms a mechanical barrier to parasite penetration (Maiti et al. 1984). Line E36-1, a guinea/caudatum hybrid of Ethiopian origin, possesses drought-tolerance and high yield potential but is striga-susceptible. The crosses were selfed, and 226 F2 plants per population were advanced by single-seed descent to the F4 generation. The F4 lines were multiplied by selfing 40 panicles per line, and the resulting F5 seed was bulked. These F3 -plant derived bulks in F5 are designated F3:5 lines since each F3:5 line represents the genetic makeup of a single F3 plant. One F3:5 line in RIP-1 proved to be an off-type and was removed from the final dataset.
Marker analysis
Marker analyses of the two mapping populations were performed with bulked DNA from 20 plants per F3:5 line. RIP-1 was genotyped for 235 marker loci [131 codominant and 20 dominant amplified fragment length polymorphisms (AFLPs), 67 simple sequence repeats (SSRs), 17 restriction (R)FLPs] and RIP-2 for 297 marker loci (122 codominant and 75 dominant AFLPs, 80 SSRs, 20 RFLPs). The AFLP analyses were carried out by Keygene (The Netherlands) using ten EcoRI/MseI primer combinations and the procedure described by Vos et al. (1995). RFLP data were produced by Biogenetic Services (Brookings, S.D.) according to standard procedures. The bnl (Burr and Burr 1991), csu (Gardiner et al. 1993), php (Beavis and Grant 1991), umc (Coe et al. 1990) and isu (cDNA) maize probes were selected from sorghum genetic maps published by Boivin et al. (1999), Dufour et al. (1997) and Pereira et al. (1994). The majority of SSR analyses were done by Celera AgGen (Davis, Calif.) using SSRs developed by Bhattramakki et al. (2000), Brown et al. (1996), Kong et al. (2000) and Taramino et al. (1997). PCR conditions followed recommendations from the aforementioned authors. Data were generated on an ABI PRISM 377 DNA sequencer (Perkin Elmer Biosystems, Foster City, Calif.). Additional SSR analyses were performed in the ICRISAT MS Swaminathan Applied Genomics Laboratory in India using SSR markers (Xcup) developed by Schloss et al. (2002) and SSR markers (Xisp) elaborated specifically for the project by Celera AgGen. The latter markers are not publicly available. SSR markers were amplified using PCR, and the amplified products were multiplexed post-PCR using fluorescently labeled primers. Data was generated on an ABI 3700 DNA sequencer (Perkin Elmer Biosystems). The raw data were extracted using genescan and genotyper software (Perkin Elmer Biosystems).
Phenotypic marker scores for the major low-germination stimulant (lgs) locus were obtained from the evaluation of RIP-1 for stimulation of Striga hermonthica seed germination using three geographic sources of striga (Haussmann et al. 2001a) and the agar-gel assay developed by Hess et al. (1992). In this assay about 4,500 preconditioned striga seeds were dispersed in 0.7% water-agar in a petri dish. The root of a 24-h-old sorghum seedling was inserted into the solidifying agar near one edge of the plate with the root tip pointing across the plate. After 5 days of incubation at 28°C in the dark, the maximal germination distance (distance between the host root and the most distantly germinated striga seed) was recorded under a dissecting microscope. Maximal germination distances above 10 mm refer to high-stimulant genotypes, while distances below 5 mm clearly point to low-stimulant sorghum entries. Striga sources from Samanko (Mali), Bengou (Niger) and Kibos (Kenya) were employed in different trials with six replications per trial—i.e., six F5 plants of each F3:5 line were evaluated per striga source. Bimodal frequency distributions and a chi-square test of the ratio of low- to high-stimulant plants indicated the presence of one major recessive gene for low stimulation of striga seed germination (Haussmann et al. 2001a). On the basis of this distinction between low- and high-stimulant classes across the two West African striga sources—where the difference was clearer than with Kenyan striga—we developed a dominant genotypic score (lgs_dom) with “A” lines representing the low-stimulant class and “C” lines the moderate- to high-stimulant class. In addition, a detailed analysis of the individual reactions of the six plants tested using striga seed from Samanko and Bengou resulted in two codominant scores (lgs_Bgu and lgs_Sko) where “A” denoted non-segregating, presumably homozygous low-stimulant F3:5 lines, “H” segregating F3:5 lines (i.e. the original F3 plant was heterozygous for the lgs locus) and “B” non-segregating, presumably homozygous high-stimulant F3:5 lines.
Map construction
Genetic maps were constructed using joinmap 3.0 (Van Ooijen and Voorrips 2001). The allocation of markers to linkage groups was mostly stable for a wide range of LOD grouping thresholds (from ≤4.0 to ≥7.0) in both RIPs. Markers that were attributed to a linkage group at a LOD grouping threshold of less than 4.5 were only included when it was known from other published maps that they belong to this group. In final map construction, data points with a LOD score below 0.1 or a recombination fraction (REC) above 0.49 were ignored. These non-stringent thresholds were used to retain distantly linked markers in the dataset. Some linkage groups reacted to the parameter settings (LOD and REC) with changes in marker order. More stringent parameter settings were not applied because they resulted in marker orders inconsistent with the order of “top-linked markers” determined by joinmap or with the map of Bhattramakki et al. (2000). Recombination frequencies were converted to centiMorgans (cM) with Haldane’s mapping function (Haldane 1919). Linkage groups were named according to common SSR markers selected from the map of Bhattramakki et al. (2000). Following map construction using all marker data, clustering markers (mainly AFLPs) were removed from the datasets and new maps computed for each RIP in order to obtain a more uniform marker distribution on each linkage group. These maps are presented here and were used in QTL analysis. The goodness-of-fit of the constructed maps, reflecting the discrepancy between final recombination frequencies in the map and those apparent from individual marker data pairs, was expressed as a chi-square value and computed according to Stam and van Ooijen (1995).
Evaluation of striga resistance
To evaluate striga resistance we divided both RIPs into two sets. Sets 1 and 2 of each RIP consisted of 116 F3:5 lines tested in 1997 and 110 lines evaluated in 1998, respectively. The F3:5 lines were evaluated together with checks in a 11×11 hexa-lattice design. Field trials were conducted at Samanko and Cinzana (Mali) during the rainy season. In Kenya, where rainfall is bimodal, trials were conducted at Kibos and Alupe in the Long Rains and at Alupe in the Short Rains (Table 1). Additional details on the experimental sites, sowing dates and rainfall data have been summarized by Omanya et al. (2004). The improved field testing methodology proposed by Haussmann et al. (2000a) was used in all trials. It consisted of: artificial infestation of on-station trials; six replicates; a lattice design; a two-row plot separated from the neighboring plot by one empty row; the use of novel resistance indices (Omanya et al. 2004). Plot size ranged from 2.94 m2 to 4.48 m2, depending on the location. The striga resistance trait reported here is area under the striga number progress curve (ASNPC). It was computed from four or five counts of emerged striga plants performed at 2-week intervals during the growing season through adapting the formula for calculating area under the disease progress curve (AUDPC; Shaner and Finney 1977). The ASNPC accounts for both intensity and speed of the epidemic (Haussmann et al. 2000a). It was selected for QTL mapping because of its good differentiation at all test sites and high heritability estimates in all four sets of material.
Statistical analysis
Phenotypic data of the two sets per RIP were analyzed using the computer program plabstat (Utz 1998), as described by Omanya et al. (2004). Combined within-year across-location analyses were computed with lattice-adjusted entry means from each individual environment. Test locations were regarded as fixed effects, while all other effects were considered as normally distributed random effects. Because of heterogeneity of error variances, a conservative F-test of the entry × environment interaction variance was made with t−1 and n′ degrees of freedom, where t designates the number of entries and n′ the number of error degrees of freedom in the experiment with the highest error variance (Cochran and Cox 1957). Broad-sense heritabilities were estimated on an entry mean basis (Hallauer and Miranda 1981) with 90% confidence intervals (CI) (Knapp et al. 1985).
QTL were detected by composite interval mapping (Jansen and Stam 1994; Zeng 1994) using the software plabqtl ver. 1.1 (Utz and Melchinger 2000). A purely additive model was employed. Individual cofactor sets were selected via stepwise regression for each trait and data set by plabqtl and subsequently extended or reduced by maximizing the R 2 value. Final selection was for the model that minimized Akaike’s information criterion (AIC), a measure of the goodness-of-fit of the regression model (Jansen 1993). Empirical threshold values for the LOD scores were determined by computing 2,500 permutations (Churchill and Dirge 1994), using the “permute” command of the plabqtl software. This was repeated twice, first with the cofactor set that was finally used for QTL identification in each set of material, and secondly with the automatic cofactor selection of the plabqtl software. Following a recommendation of HF Utz (personal communication), the average threshold values from the two permutation runs (fixed vs. automatic cofactor selection) are used here. For the experiment-wise error rate of α=0.25, the critical LOD scores to indicate QTL significance were therefore 2.78 and 2.90 for sets 1 and 2 of RIP-1 and 3.10 for both sets of RIP-2. QTL with a LOD score between 2.5 and these thresholds are also reported and may be regarded as suggestive. QTL positions were determined at the local maxima of the LOD-curve plot in the region under consideration. Adjacent QTL on the same chromosome were considered to be different when the curve had a minimum between peaks of at least 1 LOD unit below either peak and when the support intervals were non-overlapping. The proportion of phenotypic variance explained by a single QTL was obtained by the square of the partial correlation coefficient (R 2). Estimates of the additive effects of the QTL were computed by fitting a model including all putative QTL for a given trait. QTL × environment interaction was analyzed in each set of material as described by Utz and Melchinger (2000), and the proportion of genetic variance explained by the QTL was adjusted for QTL × environment interactions to avoid overestimation.
In the present study, QTL estimated in set 1 of each RIP (phenotypic data from 1997) were validated with set 2 of the same RIP (independent samples of F3:5 lines tested in 1998). The validation therefore refers to genotype sampling and year effects. In addition, a cross validation was performed within each dataset. In this type of validation, QTL positions and effects are estimated with 80% of the data, and a validation is performed with the remaining 20%. This can be done five times such that each 20% of the data is used in validation (Utz and Melchinger 2000). In the cross-validation summary, both the list of QTL detected in the calibration runs and the percentage ranges of phenotypic and genetic variance explained by the QTL in the calibration and validation runs reflect genotype sampling effects in the materials and provide additional information with respect to the reliability of the QTL.
Results
Genetic maps
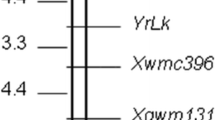
The genetic map of RIP-1 consisted of 137 markers distributed over 11 linkage groups and spanning 1,498 cM (Fig. 1a). Linkage group B was split into two. The following markers were allocated to their linkage groups at a LOD grouping threshold below 4.5: isp230 (grouped to linkage group D up to LOD=3.0) and isp258 (mapped to linkage group J up to LOD=4.0). Because isp230 equally mapped to linkage group D in RIP-2 and isp258 to linkage group J in a different population (unpublished data), they were included in the RIP-1 map. The lgs loci were mapped to linkage group I only up to a LOD grouping threshold of 4.5. In addition, linkage group I reacted to the “LOD” and “REC” parameter settings during map computation with changes in marker order. The positioning of the lgs loci should therefore be regarded as preliminary. Markers txp63, txp96 and isp200 were unlinked in RIP-1. Markers php20626 and txp273 were mapped to linkage group H at LOD 3.0 but caused a gap of more than 60 cM. Because this would have made QTL mapping problematic, these markers were excluded from the final map. Large gaps (>25 cM) occurred in linkage groups B (two gaps on B1, one gap on B2 plus missing links between B1 and B2), D (1 gap), E (2), F (1), G (2), I (2) and J (1). A high goodness-of-fit (mean χ2 <1.7, frequently <1.0, in the map construction of all linkage groups) indicated high reliability of the linkage map.

Genetic linkage maps of RIP-1 (a) and RIP-2 (b) including the positions of putative QTL for area under the striga number progress curve (ASNPC). AFLP, RFLP and SSR marker names are indicated at the right of each linkage group. Underlined markers are common to both RIPs. Linkage groups are named according to Bhattramakki et al. (2000)
The genetic map of RIP-2 consisted of 157 markers distributed over 11 linkage groups and spanning a total of 1,599 cM (Fig. 1b). Linkage group D was split in two. The upper part of linkage group B was linked only up to a LOD grouping threshold of 4.5. On the other hand, the remainder of linkage group B joined linkage group J, and the two groups could only be separated at a LOD grouping threshold of 10.5. Large gaps (>25 cM) occurred in linkage groups B (1 gap), C (1), D1 (1), E (4), G (1) and H (2). A high goodness-of-fit (mean χ2 <1.7 in the map construction of all linkage groups) indicated high reliability of the linkage map.
The two maps had 54 markers in common (Fig. 1). The conserved order of common markers also reflected the high reliability of the two maps. The only exceptions were isp344 and txp295 whose closely linked positions were interchanged on linkage group E in RIP-1 compared to RIP-2.
Phenotypic data for area under the striga number progress curve
Striga infestation was medium to high at all sites, with environmental means for the number of emerged striga plants per square meter at 90 days after sowing ranging from 16 to 91 in RIP-1 and from 15 to 56 in RIP-2 (for details see Omanya et al. 2004). Location means for ASNPC ranged from 7 to 32 in RIP-1 and from 9 to 22 in RIP-2 (Omanya et al. 2004). Repeatabilities at the individual test sites for ASNPC were moderate or high and highly significant in both RIPs, ranging from 45% to 82% in RIP-1 and from 72% to 93% in RIP-2 (Omanya et al. 2004). The combined analyses of variance across locations within years indicated highly significant genetic variances (P=0.01) among the tested F3:5 lines for ASNPC in both RIPs (Table 2). F3:5 line × location interaction variances were also highly significant (P=0.01) in both RIPs and years. Estimated heritabilities for ASNPC were moderate to high, with RIP-2 generally revealing slightly higher heritability estimates than RIP-1. Frequency distributions of the F3:5 lines for mean ASNPC were unimodal, indicating quantitative variation for striga resistance in both RIPs (Fig. 2). Transgressive segregation was apparent for susceptibility to striga, but none of the F3:5 lines was more resistant than the resistant parental lines IS9830 or N13 [the difference between the most resistant F3:5 line of set 2 in RIP-1 and parental line IS9830 (Fig. 2) was not significant at P=0.05].

Frequency distributions of the F3:5 lines of RIP-1 (IS9830 × E36-1) and RIP-2 (N13 × E36-1) for mean area under the striga number progress curve (ASNPC), averaged over five test environments per year. Filled triangle marks the classes of the respective parent lines. se standard error of F3:5-line means
QTL for area under the striga number progress curve
In the QTL analysis across locations with RIP-1 (IS9830 × E36-1), composite interval mapping detected 11 and 9 putative QTL in sets 1 and 2, explaining 77% and 86% of the genetic variance, respectively (Fig. 1a, Table 3). Five QTL on linkage groups A, B1 (two QTL), C and I were common to both sets, with the resistance alleles being contributed by the resistant parent IS9830. The shared QTL on linkage group I at 150 cM corresponded to the lgs locus and explained the largest proportion of phenotypic variance (highest R 2 values) in both sets. None of the resistance QTL alleles that were expressed in both sets was derived from the susceptible parent line E36-1. Cross-validation results reflected some genotype sampling effects in RIP-1 since several QTL appeared only in one to three of the calibration runs. The mean percentages of genetic variance explained in the calibration and validation runs of the cross-validation procedure were slightly higher in set 2 (71.6% and 58.4%, respectively) than in set 1 (56.5% and 21.0%, respectively) of RIP-1.
In RIP-2 (N13 × E36-1), 11 and 9 QTL were detected for ASNPC in sets 1 and 2 that explained 79% and 82% of the genetic variance, respectively (Fig. 1b, Table 4). Five QTL, on linkage groups A, B, I and J (two QTL) were common to both sets. For each of these stably expressed QTL, the resistance allele came from the resistant parent N13. The QTL on linkage group B fell in a large—larger than 50 cM— gap of the genetic map of RIP 2; its position could therefore not be determined precisely and should be regarded as preliminary. The majority of QTL were detected in four or even all five calibration runs of the cross-validation. In addition, the high mean percentages of genetic variance explained in the calibration (76.6% and 78.6% in sets 1 and 2, respectively) and validation runs (61.6% and 71.5% in the two sets) indicated good reliability of the detected QTL.
The QTL × location interaction within years was highly significant (P=0.01) in both sets of both RIPs and was reflected by deviating QTL effects at the different locations within each set of F3:5 lines (Table 5). In RIP-1, some QTL effects even ranged from negative to positive values—i.e. the same QTL allele contributed to resistance at one location but to susceptibility at another. Among the QTL showing strong QTL × location interaction was the QTL on linkage group I at 150 cM—i.e. the lgs locus—in set 1 of RIP-1. In RIP-2, where resistance is not due to the low-stimulant character, none of the resistance QTL alleles showed such a strong interaction with the environment.
Discussion
Genetic maps of the two populations
Powerful QTL mapping requires a genetic map with good genome coverage. Due to the low genetic polymorphism between the parental lines used in this investigation, it took several years to develop the present genetic maps of the two mapping populations. The recent addition of selected txp, cup and isp SSR markers and the phenotypic marker score for the low-stimulant character (in RIP-1 only) extended the maps of RIP-1 and RIP-2 by 207 cM and 145 cM, respectively, compared to an earlier version (Haussmann et al. 2002a, b). However, compared to the map of Bhattramakki et al. (2000), about 130 cM in RIP-1 and 95 cM in RIP-2 are still not represented. In RIP-1 the missing regions include 10 cM each at the lower ends of linkage groups A and C, a 30-cM section linking the two parts of linkage group B, 25 cM at the top of linkage group E, 15 cM at the lower end of linkage group G and an approximately 40-cM link to txp273 in linkage group H. In RIP-2 the missing regions include 25 cM at the lower end of linkage group A, a 35-cM missing link between the two groups of linkage group D, 20 cM at the upper end of linkage group H and 15 cM at the lower end of linkage group I.
Position of the low-germination stimulant locus in RIP-1
Positioning of the lgs locus in linkage group I should be regarded as preliminary since phenotypic marker scores for the lgs locus in RIP-1 were linked to the linkage group only up to a LOD grouping threshold of 4.5. In addition, this linkage group reacted with marker order changes to the LOD and REC parameter settings during the computation of the map. The positioning of the lgs locus therefore needs to be confirmed through the addition of more markers to the genetic map of RIP-1, especially to linkage group I. However, positioning the lgs locus on linkage group I was supported by (unpublished data) QTL analysis of the maximal germination distance data obtained in the agar-gel assay: when the phenotypic marker score from the RIP-1 map was excluded, there was only one QTL which consistently affected maximal germination distance in both sets of material, and this QTL equally mapped to linkage group I.
Genomic regions affecting striga resistance under field conditions
The present study clearly demonstrates that striga field resistance is a complex, quantitative trait influenced by multiple genomic regions, depending on test site and year. In fact, all of the linkage groups were involved in the expression of the trait when both RIPs, sets/years and all test environments were considered simultaneously. This is not surprising since a variety of interactions between striga and its host determine the reproductive success of the parasite. Following seed shed and a period of dormancy, striga seed will germinate only when exposed to moisture and a favorable temperature for several days (preconditioning). A germination stimulant is required, which is usually exuded by the roots of the host. Subsequent development (haustoria formation, attachment, penetration and growth) of the parasite also requires signals and resource commitment from the host plant (Ejeta et al. 2000; Haussmann et al. 2000a). These interactions provide opportunities to the host to resist the parasite. The following resistance mechanisms have been proposed in addition to low production of the germination stimulant (Ejeta and Butler 1993; Haussmann et al. 2000a; Heller and Wegmann 2000; Mohamed et al. 2003):
-
mechanical barriers (e.g. lignification of cell walls);
-
inhibition of germ-tube exoenzymes by root exudates;
-
phytoalexin synthesis;
-
post-attachment hypersensitive reactions or incompatibility;
-
antibiosis (e.g. reduced striga growth through unfavorable phytohormone supply by the host);
-
insensitivity to striga toxin (e.g., maintenance of stomatal opening and photosynthetic efficiency);
-
avoidance through root growth habit (e.g., fewer roots in the upper 15–20 cm).
In each of the two RIPs, five QTL were stably expressed across test sites, years and independent mapping population samples (sets). These QTL may affect key events in the striga infection cycle. A proven example is the QTL on linkage group I at 150 cM in RIP-1. This QTL corresponds to the lgs locus for low stimulation of striga seed germination and explained 23% and 40% of the phenotypic variance for ASNPC in sets 1 and 2 of RIP-1. The fact that four additional stable QTL were identified in both sets of RIP-1 indicates that the resistant parent of this population, line IS9830, possesses other resistance mechanisms in addition to the low stimulation of striga seed germination. Line N13, the resistant parent of RIP-2, appears also to possess several resistance mechanisms as multiple QTL were identified in both sets of this population as well. A detailed physiological study using near-isogenic lines carrying different QTL alleles in a homozygous stage would be required to identify the precise resistance mechanisms encoded by resistance QTL alleles of the two parental lines.
The main reason for our success in mapping striga resistance QTL lies in the careful conduct of field evaluations. The employment of six replications, lattice design, specific plot layout, a comprehensive resistance measure (ASNPC) and multilocational tests produced the high heritabilities essential for QTL mapping. In addition, the testing of the two independent mapping population samples (the two sets per RIP) in different years allowed for comprehensive QTL validation and identification of the most reliable QTL. Those QTL that were stable across ten test environments (five sites × 2 years) and the two independent mapping population sets should therefore be biological realities. Flanking markers to these stable QTL could be candidates for marker-assisted transfer of striga resistance alleles into adapted, farmer-preferred local cultivars.
QTL × environment interaction
The highly significant QTL × environment interaction in both RIPs was reflected by the variable effects of QTL alleles at individual test locations. While in RIP-1 (IS9830 × E36-1) two of the five QTL identified in both sets showed changes in the sign of their effects, all five QTL identified in both sets of RIP-2 (N13 × E36-1) contributed to resistance at all sites in both years. Resistance derived from N13 therefore may be more stable than resistance derived from IS9830. In field trials across diverse geographic regions, the total QTL × environment interaction variance comprises interaction effects between QTL alleles and locations (soil, climate), between QTL and putative striga biotypes as well as the three-way interaction. Striga hermonthica, due to its outcrossing behavior, possesses high genetic variability and can adapt to new hosts (Ejeta and Butler 1993; Koyama 2000). Striga populations at different locations could differ in virulence level or specificity as a result of adaptation to different host plant resistance mechanisms. Thus, the factor striga population most likely contributed to the QTL × environment interaction. In order to determine the contribution of a geographical striga population to the total QTL × environment interaction, sorghum materials would have to be evaluated at each location against striga populations of different geographic origin. For reasons of quarantine, this clearly cannot be done in Africa.
The lgs locus contributed the most to resistance at Cinzana (Mali) and had a slightly negative effect at Alupe (Kenya) in the 1997 Short Rains. These observations correspond to the coefficients of phenotypic correlation between maximal germination distance in the agar-gel assay and ASNPC at the individual test sites observed in the same genetic materials. The correlation coefficients were highest at Cinzana (0.63 and 0.51 in sets 1 and 2, respectively) and close to zero (and insignificant) for Alupe in the Short Rains (Omanya et al. 2004). Variable correlations between maximal germination distance and resistance under field conditions were also observed in diallel crosses of sorghum (Haussmann et al. 2000b). It therefore appears that the contribution of the lgs locus to striga field resistance depends on the materials under investigation as well as on the test environments, as previously suggested by Vasudeva Rao (1984). K. Wegmann (personal communication) hypothesized that abiotic stress followed by ethylene production by micro-organisms in the soil could be one possible mechanism rendering the low-stimulant character less effective in certain site/season combinations. Ethylene has been shown to efficiently stimulate striga seed germination (Bebawi and Eplee 1986; Eplee 1975).
The reported QTL × environment interactions confirm the pertinence of recommendations that the testing of breeding materials should occur at various locations with different striga populations in order to identify genotypes with stable resistance (Haussmann et al. 2000a, b, 2001b; Ramaiah 1987).
Mapping population size, power of QTL detection and importance of QTL validation
Theoretically, the simultaneous evaluation of all 226 F3:5 lines per mapping population should have resulted in a greater likelihood of QTL detection due to larger population size (Beavis 1998). However, from a practical standpoint it would have been extremely difficult, if not impossible, to simultaneously evaluate 226 genotypes for striga resistance under field conditions with the same precision as was achieved with half that number. In addition, our experimental design permitted QTL validation across genotype samples (sets of each RIP) and years. The limited number of common QTL across locations, years and independent mapping population sets for ASNPC under field conditions (five in each RIP) underlines the tremendous importance of QTL validation across genotype samples and environments. QTL studies based on a small mapping population and only one test environment should therefore be interpreted with great caution. QTL validation can consist of:
-
QTL analysis in an independent mapping population sample as done in the present study and by Bohn et al. (2001);
-
QTL analysis in a second mapping population derived from the same donor parent but a different recipient (e.g. Haussmann et al. 2002b);
-
Statistical cross validation procedures as offered by the plabqtl software, most useful in large mapping populations (n>200) ( Bohn et al. 2001; Utz and Melchinger 2000; Utz et al. 2000); or
-
Verification of QTL effects in near isogenic lines.
Utz et al. (2000) conducted extensive maize experiments investigating different agronomic traits using cross-validation and validation with independent genotypic samples. They observed highly variable numbers of QTL in calibration and validation runs as well as high bias and sampling error of the estimated proportion of genotypic variance explained by QTL. The authors concluded that it is extremely difficult to obtain reliable, unbiased QTL estimates for complex quantitative traits. Huge population sizes (n>500) and large numbers of environments would have to be employed, which would surpass the financial and logistic capacities of a breeding program (Bohn et al. 2001). Our use of QTL validation techniques addresses these concerns and should help realize gains from future marker-assisted selection.
Conclusion and outlook
The present experiments have yielded reliable QTL for striga field resistance. In a follow-up project, markers flanking the stable resistance QTL of N13 will be used in a marker-assisted foreground selection to improve local sorghum cultivars from Eritrea, Kenya, Mali and the Sudan. The present results may also facilitate the detection of striga resistance genes in syntenic genomic regions of maize, pearl millet, or rice.
References
Beavis WD (1998) QTL analyses: power, precision, and accuracy. In: Paterson AH (ed) Molecular dissection of complex traits. CRC Press, Boca Raton/New York, pp 145–162
Beavis WD, Grant D (1991) A linkage map based on information from four F2 populations of maize (Zea mays L.). Theor Appl Genet 82:636–644
Bebawi FF, Eplee RE (1986) Efficacy of ethylene as a germination stimulant of Striga hermonthica seed. Weed Sci 34:694–698
Berner DK, Cardwell KF, Faturoti BO, Ikie FO, Williams OA (1994) Relative roles of wind, crop seeds, and cattle in dispersal of Striga spp. Plant Dis 78:402–406
Bhattramakki D, Dong J, Chhabra AK, Hart GE (2000) An integrated SSR and RFLP linkage map of Sorghum bicolor (L.) Moench. Genome 43:988–1002
Bohn M, Groh S, Khairallah MM, Hoisington DA, Utz HF, Melchinger AE (2001) Re-evaluation of the prospects of marker-assisted selection for improving insect resistance against Diatraea spp. in tropical maize by cross validation and independent validation. Theor Appl Genet 103:1059–1067
Boivin K, Deu M, Rami JF, Trouche G, Hamon P (1999) Towards a saturated sorghum map using RFLP and AFLP markers. Theor Appl Genet 98:320–328
Brown SM, Hopkins MS, Mitchell SE, Senior ML, Wang TY, Duncan RR, Gonzales-Candelas F, Kresovitch S (1996) Multiple methods for the identification of polymorphic simple sequence repeats (SSRs) in sorghum [Sorghum bicolor (L.) Moench]. Theor Appl Genet 93:190–198
Burr B, Burr FA (1991) Recombinant inbreds for molecular mapping in maize: theoretical and practical considerations. Trends Genet 7:55–60
Churchill GA, Dirge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Cochran WG, Cox GM (1957) Experimental designs, 2nd edn. Wiley, London
Coe EH, Hoisington DA, Neuffer MG (1990) Linkage map of corn (maize) (Zea mays L.). In: O’Brien SJ (ed) Genetic maps. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, pp 39–67
Dufour P, Deu M, Grivet L, D’Hont A, Paulet F, Bouet A, Lanaud C, Glaszmann JC, Hamon P (1997) Construction of a composite sorghum genome map and comparison with sugarcane, a related complex polyploid. Theor Appl Genet 94:409–418
Ejeta G, Butler LG (1993) Host plant resistance to Striga. In: Buxton DR, Shibles R, Forsberg RA, Blad BL, Asay KH, Paulsen GM, Wilson RF (eds) International crop science I. Crop Science Society of America, Madison, Wis.
Ejeta G, Mohammed A, Rich P, Melake-Berhan A, Housley TL, Hess DE (2000) Selection for specific mechanisms of resistance to striga in sorghum. In: Haussmann BIG, Hess DE, Koyama ML, Grivet L, Rattunde HFW, Geiger HH (eds) Breeding for striga resistance in cereals. Margraf Verlag, Weikersheim, pp 29–37
Eplee RE (1975) Ethylene: a witchweed seed germination stimulant. Weed Sci 23:433–436
FAO (2000) Production yearbook. Food and Agriculture Organization of the United Nations, Rome
Gardiner JM, Coe EH, Melia-Hancock S, Hoisington DA, Chao S (1993) Development of a core RFLP map in maize using an immortalized F2 population. Genetics 134:917–930
Gurney AL, Press MC, Scholes JD (2002) Can wild relatives of sorghum provide new sources of resistance or tolerance against Striga species? Weed Res 42:317–324
Haldane JBS (1919) The combination of linkage values, and the calculation of distance between the loci of linked factors. J Genet 8:299–309
Hallauer AR, Miranda JB (1981) Quantitative genetics in maize breeding. Iowa State University Press, Ames
Haussmann BIG, Hess DE, Welz HG, Geiger HH (2000a) Improved methodologies for breeding striga-resistant sorghums (review article). Field Crops Res 66:195–201
Haussmann BIG, Hess DE, Reddy BVS, Mukuru SZ, Kayentao M, Welz HG, Geiger HH (2000b) Quantitative-genetic parameters of sorghum growth under striga infestation in Mali and Kenya. Plant Breed 120:49–56
Haussmann BIG, Hess DE, Reddy BVS, Welz HG, Geiger HH (2000c) Analysis of resistance to Striga hermonthica in diallel crosses of sorghum. Euphytica 116:33–40
Haussmann BIG, Hess DE, Omanya GO, Reddy BVS, Welz HG, Geiger HH (2001a) Major and minor genes for stimulation of Striga hermonthica seed germination in sorghum, and interaction with different striga populations. Crop Sci 41:1507–1512
Haussmann BIG, Hess DE, Reddy BVS, Mukuru SZ, Kayentao M, Welz HG, Geiger HH (2001b) Pattern analysis of genotype × environment interaction for striga resistance and grain yield in African sorghum trials. Euphytica 122:297–308
Haussmann BIG, Hess DE, Seetharama N, Welz HG, Geiger HH (2002a) Construction of a combined sorghum linkage map from two recombinant inbred populations using AFLP, SSR, RFLP, and RAPD markers, and comparison with other sorghum maps. Theor Appl Genet 105:629–637
Haussmann BIG, Mahalakshmi V, Reddy BVS, Seetharama N, Hash CT, Geiger HH (2002b) QTL mapping of stay-green in two sorghum recombinant inbred populations. Theor Appl Genet 106:133–142
Heller R, Wegmann K (2000) Mechanisms of resistance to Striga hermonthica (Del.) Benth. in Sorghum bicolor (L.) Moench. In: Haussmann BIG, Hess DE, Koyama ML, Grivet L, Rattunde HFW, Geiger HH (eds) Breeding for striga resistance in cereals. Margraf Verlag, Weikersheim, pp 19–28
Hess DE, Haussmann BIG (1999) Status quo of Striga control: prevention, mechanical and biological control methods and host plant resistance. In: Kroschel J, Mercer-Quarshie H, Sauerborn J (eds) Advances in parasitic weed control at on-farm level. Joint action to control Striga in Africa, vol 1. Margraf Verlag, Weikersheim, pp 75–87
Hess DE, Ejeta G, Butler LG (1992) Selecting sorghum genotypes expressing a quantitative biosynthetic trait that confers resistance to Striga. Phytochemistry 31:493–497
Jansen RC (1993) Interval mapping of multiple quantitative trait loci. Genetics 135:205–211
Jansen RC, Stam P (1994) High resolution of quantitative traits into multiple loci via interval mapping. Genetics 136:1447–1455
Kapran I, Grenier C, Elliot A, Touré A, Gutema Z, Babiker A, Sadaan H, Ejeta G (2002) Introgression of genes for Striga resistance into African landraces of sorghum. In: Biotechnol Breed Seed Systems African Crops. (Abstract)http://www.africancrops.net/Breeding%20abstracts/breeding%20kapran.htm
Knapp SJ, Stroup WW, Ross WM (1985) Exact confidence intervals for heritability on a progeny mean basis. Crop Sci 25:192–196
Kong L, Dong J, Hart GE (2000) Characteristics, linkage map positions, and allelic differentiation of Sorghum bicolor (L.) Moench DNA simple-sequence repeats (SSRs). Theor Appl Genet 101:438–448
Koyama ML (2000) Genetic variability of Striga hermonthica and effect of resistant cultivars on striga population dynamics. In: Haussmann BIG, Hess DE, Koyama ML, Grivet L, Rattunde HFW, Geiger HH (eds) Breeding for striga resistance in cereals. Margraf Verlag, Weikersheim, pp 247–260
Kroschel J (1999) Analysis of the Striga problem, the first step towards future joint action. In: Kroschel J (ed) Advances in parasitic weed control at on-farm level. Joint action to control Striga in Africa, vol 1. Margraf Verlag, Weikersheim, pp 3–25
Maiti RK, Ramaiah KV, Bisen SS, Chidley VL (1984) A comparative study of the haustorial development of Striga asiatica (L.) Kuntze on sorghum cultivars. Ann Bot 54:447–457
Mohamed A, Ellicott A, Housley TL, Ejeta G (2003) Hypersensitive response to Striga infection in sorghum. Crop Sci 43:1320–1324
Omanya GO, Haussmann BIG, Hess DE, Reddy BVS, Kayentao M, Welz HG, Geiger HH (2004) Utility of indirect and direct selection traits for improving striga resistance in two sorghum recombinant inbred populations. Field Crops Res http://dx.doi.org/10.1016.j.fcr.2004.02.003
Pereira MG, Lee M, Bramel-Cox P, Woodman W, Doebley J, Whitkus R (1994) Construction of an RFLP map in sorghum and comparative mapping in maize. Genome 37:236–243
Ramaiah KV (1987) Breeding cereal grains for resistance to witchweed. In: Musselman LJ (ed) Parasitic weeds in agriculture, vol 1. Striga. CRC Press, Boca Raton, pp 227–242
Ramaiah KV, Chidley VL, House LR (1990) Inheritance of Striga seed-germination stimulant in sorghum. Euphytica 45:33–38
Sauerborn J (1991) The economic importance of the phytoparasites Orobanche and Striga. In: Ransom JK, Musselman LJ, Worsham AD, Parker C (eds) Fifth Int Symp Parasitic Weeds. CIMMYT, Nairobi, pp 137–143
Schloss SJ, Mitchell SE, White GM, Kukatla R, Bowers JE, Paterson AH, Kresovitch S (2002) Characterization of RFLP probes sequences for gene discovery and SSR development in Sorghum bicolor (L.) Moench. Theor Appl Genet 105:912–920
Shaner G, Finney RE (1977) The effect of Nitrogen fertilization on the expression of slow-mildewing resistance in Knox wheat. Phytopathology 67:1051–1056
Stam P, Van Ooijen JW (1995) joinmap ver. 2.0: Software for the calculation of genetic linkage maps. CPRO-DLO Wageningen
Taramino G, Tarchini R, Ferrario S, Lee M, Pe ME (1997) Characterization and mapping of simple sequence repeats (SSRs) in Sorghum bicolor. Theor Appl Genet 95:66–72
Utz HF (1998) plabstat: a computer program for the statistical analysis of plant breeding experiments. Version 2 N. Institute of Plant Breeding, Seed Science, and Population Genetics, University of Hohenheim, Stuttgart http://www.uni-hohenheim.de/~ipspwww/soft.html
Utz HF, Melchinger AE (2000) plabstat: a computer program to map QTL. Version 1.1 from 29 May 2000. Institute of Plant Breeding, Seed Science, and Population Genetics, University of Hohenheim, Stuttgart http://www.uni-hohenheim.de/~ipspwww/soft.html
Utz HF, Melchinger AE, Schoen CC (2000) Bias and sampling error of the estimated proportion of genotypic variance explained by quantitative trait loci determined from experimental data in maize using cross validation and validation with independent samples. Genetics 154:1839–1849
Van Ooijen JE, Voorips RE (2001) joinmap 3.0. Software for the calculation of genetic linkage maps. Plant Research International, Wageningen
Vasudeva Rao MJ (1984) Patterns of resistance to Striga asiatica in sorghum and millets, with special reference to Asia. In: Proc Int Workshop Biol Control Striga. ICSU Press, Paris, pp 93–111
Vogler RK, Ejeta G, Butler LG (1996) Inheritance of low production of Striga germination stimulant in sorghum. Crop Sci 36:1185–1191
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414
Zeng Z-B (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Acknowledgements
We thank the Institut d’Economie Rurale (IER, Mali) and the Kenya Agricultural Research Institute (KARI) for support of the project. Special thanks are due to I. Sissoko, M. Kayentao, E. Manyasa, A. Oswald, G. Odhiambo, and G.O. Abayo for their capable assistance. We greatly appreciate the skilled technical assistance of S. Dembelé, the late I. Sangaré, B.A. Diallo, the late A. Koné, Mrs. Traoré, the late G. Mounkoro, N. Keïta, S. Bakayoko, A. Tembely, J. Kibuka, T. Mboya, J. Were, I. Absalom, and all others who carefully contributed to the field work. Prof. G. Hart deserves special thanks for providing pre-publication information on SSR primer combinations and map positions. Prof. HF Utz is thanked for the always kind and prompt statistical advice. The generous financial support of the German Federal Ministry for Economic Cooperation and Development (BMZ) is gratefully acknowledged (Grant no. 94.7860.3-01.100). Many thanks also go to the German Academic Exchange Service (DAAD) that supported G.O. Omanya during the field work, and to the German Research Foundation (DFG) that supported B.I.G. Haussmann during map extension and final data analyses.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by G. Wenzel
Rights and permissions
About this article
Cite this article
Haussmann, B.I.G., Hess, D.E., Omanya, G.O. et al. Genomic regions influencing resistance to the parasitic weed Striga hermonthica in two recombinant inbred populations of sorghum. Theor Appl Genet 109, 1005–1016 (2004). https://doi.org/10.1007/s00122-004-1706-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-004-1706-9