
Abstract
Buffalograss [Buchloe dactyloides (Nutt.) Englem.] is the only native grass that is being used extensively as a turfgrass in the Great Plains region. Its low-growth habit, drought resistance, and low-maintenance requirement make it attractive as a turfgrass species. Our objective was to obtain an overview on the genetic relatedness among and within seeded and vegetative biotype buffalograsses using inter-simple sequence repeats (ISSRs), random amplified polymorphic DNA (RAPDs), sequence-related amplified polymorphisms (SRAPs), and simple sequence repeats (SSRs) markers that were derived from related species (maize, pearl millet, sorghum, and sugarcane). Twenty individuals per cultivar were genotyped using 30 markers from each marker system. All buffalograss cultivars were uniquely fingerprinted by all four marker systems. Mean genetic similarities were estimated at 0.52, 0.51, 0.62, and 0.57 using SSRs, ISSRs, SRAPs, and RAPDs, respectively. Two main clusters separating the seeded-biotype from the vegetative-biotype cultivars were produced using UPGMA analysis. Further subgroupings were unequivocal. The Mantel test resulted in a very good fit (SRAP=0.92, ISSR=0.90) to good fit (RAPD=0.86, SSR=0.88) of cophenetic values. Comparing the four marker systems to each other, RAPD and SRAP similarity indices were highly correlated (r=0.73), while Spearman’s rank correlation coefficient between RAPDs and SSRs was r=0.24 and between ISSRs and SSRs was r=0.66. A genotype-assignment analytical approach might be useful for cultivar identification and property rights protection. Polymorphic SRAPs were abundant and demonstrated genetic diversity among closely related cultivars.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Buffalograss [Buchloe dactyloides (Nutt.) Englem.] is a native North American C4 turfgrass species with drought resistance, heat tolerance, low-growth habit, and low-management requirements. It is a member of the Chlorideae tribe and has potential to increase the genetic variability for turfgrass improvement.
Buffalograss is an open-pollinated, dioecious turfgrass species and is highly heterogeneous. It can be propagated vegetatively by stolons or sexually by seed (Riordan et al. 1993). Seeded type bufalograsses can be developed by creating synthetics. There are diploid, tetraploid, pentaploid, and hexaploid types of buffalograss in the Great Plains of North America (Huff et al. 1993; Johnson et al. 2001; Budak et al. 2004), but they are often morphologically indistinguishable, especially at higher ploidy levels. This has led to the mislabeling of some cultivars (Budak et al. 2004). For instance, the high correlation between cold hardness and higher ploidy levels for genotypes collected in the northern Great Plains might not be found in the southern regions (Johnson et al. 2001).
Due to its dioecious nature, each cultivar is a heterogeneous population of genotypes. This makes cultivar identification and property rights protection difficult. Improved methods to differentiate cultivars for genetic identification and property rights protection are needed. Historically, cultivar identification and property rights protection has been based on morphological characteristics. Morpho-agronomic characterization of buffalograss is time consuming and labor intensive. In lieu of that, the aid of the molecular markers may be an appropriate tool for cultivar identification. However, informative molecular markers that are applicable to genetic improvements of buffalograss are limited. In addition, researchers are faced with the problem of which technique offers the best sensitivity for identification of vegetative and seeded cultivars and for marker-assisted selection. Hence, it is very important for researchers to compare these marker techniques to identify an appropriate one that best address these concerns.
Simple sequence repeats (SSRs), or microsatellites and inter-simple sequence repeats (ISSRs) have been recognized as useful molecular markers in marker-assisted selection, the analysis of genetic diversity, population genetic analysis, and other purposes in various species (Gupta and Varshney 2000; Budak et al. 2003). However, these marker systems have never been used in a buffalograss breeding program. Since comparative mapping among grass genomes supports the synteny of gene content and gene order (Van Deynze et al. 1998), integrating hypervariable markers from closely related species to a buffalograss breeding program could serve as a conduit to enhance turfgrass improvements. In most cases, microsatellite markers were shown to be transferable across taxa and cross transferability within the same genus reaches 76.4% (Rossetto 2001). There have been no SSRs markers developed in buffalograss genome to date. Randomly amplified polymorphic DNA (RAPDs) have been used to characterize genetic diversity in buffalograss (Huff 1993; Wu and Lin 1994; Peakall et al. 1995). Sequence-related amplified polymorphism (SRAP) (Li and Quiros 2001) has been recognized as a new and useful molecular-marker system in fingerprinting of Buchloe (Budak et al. 2004).
The objectives of this study were to: (1) uniquely fingerprint and better characterize the distribution of genetic diversity within and among buffalograss cultivars using all marker systems; (2) compare different marker techniques; and (3) investigate the effects of probability of population identity and the sampling.
Materials and methods
Plant materials and DNA extraction
Twenty seedlings from each of eight seeded and 20 clones from each of seven vegetative biotypes of buffalograsses maintained in the Department of Agronomy and Horticulture research greenhouses at the University of Nebraska-Lincoln were used in DNA fingerprinting research (Table 1). All these genotypes were fingerprinted with 30 markers from each marker system used. Genomic DNA of these genotypes from each cultivar was extracted using the CTAB method outlined by Wagner et al. (1987).
Primers, PCR conditions, and gel electrophoresis
ISSRs (Blair et al. 1999); SSRs from maize (Senior and Heun 1993), sorghum (Bhattramakki et al. 2000), pearl millet (Allouis et al. 2001; Qi et al. 2001; Budak et al. 2003 ) and sugar cane (Cordeiro et al. 2000); RAPDs (Operon Technology, Alameda, Calif.); and SRAPs markers (Li and Quiros 2001; Budak et al. 2004) were employed. In all methods, the PCR-reaction mixtures and electrophoresis conditions were carried out as described by Budak et al. (2004). Amplifications were carried out using an MJ Research PTC-100 thermocycler programmed for 32 cycles of 1 min at 94°C; 1 min at 47°C for SRAP, 50°C for SSRs and ISSRs, and 37°C for RAPD analyses; 1 min at 72°C; followed by a final extension at 72°C for 5 min before cooling to 24°C. The PCR products (25 μl) were fractionated on 12% polyacrylamide using a Hoefer vertical-gel apparatus (SE600).
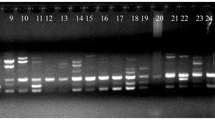
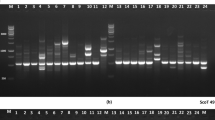
Amplified fragments were visualized using ethidium bromide staining and photographed using a Gel Doc 2000 (Bio-Rad).
Data analysis
The banding patterns were scored as present (1) or absent (0) for each primer pair. Only polymorphic bands were considered in the genetic analysis even though monomorphic markers were scored. The distance matrix and dendrogram were constructed using the Numerical Taxonomy Multivariate Analysis System (NTSYSpc) version 2.1 (Exeter Software, Setauket, N.Y.) software package (Rohlf 2000). Genetic similarities between cultivars were measured by the Dice similarity coefficient based on the proportion of shared alleles (Dice 1945; Nei and Li 1979), with SIMQUAL module. The Dice similarity coefficient was calculated as follows:
where N AB is the number of bands shared by samples, N A represents amplified fragments in sample A and N B represents fragments in sample B. The FIND module was used to identify all trees and the trees were compiled by CONSEN module to test the robustness of tree topology. Observed heterozygosities, test for Hardy-Weinberg Equilibrium (HWE), F ST estimates and analysis of molecular variance (AMOVA) (Excoffier et al. 1992) were performed based on 1,000 random permutations. For population identity assignment, zero frequencies were adjusted to 1/2 N (N = the number of individual sampled). Spearman’s rank correlation coefficients were calculated in order to compare indices on genetic relatedness obtained from molecular-marker types. Cophenetic values were calculated from all four dendrograms and were used to compare dendrograms by the Mantel test of significance (Mantel 1967). Coefficient of variation (CV) of the mean genetic similarity calculated from the bootstraps resamples were computed from each sub sample outlined in Tivang et al. (1994). Confusion probabilities of each marker system were calculated as described by Tessier et al. (1999).
Results and discussion
PCR-based marker analysis used in this study revealed significant genetic divergence between seeded and vegetative biotypes. These results were consistent with the morphological and ecological divergence. Compared to the vegetative biotypes, genetic variation within and among seeded biotypes was higher. Genotypes generally deviated from HWE with a bias of homozygosity. This was not unexpected because selection in a small breeding population will result in excess homozygosity. However, a big proportion of locus-by-cultivar comparison deviated from HWE toward heterozygosity. Detecting heterozygosity in many plants is not unexpected (Eguiarte et al. 1993). The cross-pollinating nature of buffalograss promotes excess heterozygosity.
AMOVA was performed to analyze the portion of each marker-system variation in vegetative and seeded biotypes. Although most of the genetic diversity was attributable to differences among genotypes, significant variation between seeded and vegetative biotypes for all markers used in this study demonstrated the existence of the biotype differences. Variation among population was significant for all markers used. All loci tested in this study were significant between variations of genotype components. All pairwise F ST, which are not expected to be affected by marker type used (albeit Nei’s genetic distance is expected to give different results using different marker system), and RST values were significantly greater than 0 (Mohammadi et al. 2003). This result indicates that pairs of cultivars were distinguished based on allelic frequencies.
ISSR analysis
Thirty out of 46 ISSR markers (Blair et al. 1999) were used for estimation of genetic relationships among and within buffalograss biotypes. A total of 256 bands were observed; 207 of these (81%) were polymorphic among seeded and vegetative biotypes and were shared between at least two individuals.
Genetic similarities among all individuals ranged from 0.03 to 0.99, with a mean similarity of 0.51. For the ISSR dendrogram, cophenetic correlation was estimated at r=0.90, corresponding to a very good fit. ISSR markers revealed a high level of genetic distances between most cultivars, indicating a diverse genetic background. ‘Cody’ (seeded biotype) and ‘NE91-118’ (vegetative biotype) were found to span the extremes of the dendrogram (Fig. 1a). The UPGMA clustering algorithm grouped the genotypes into four clusters. Grouping of genotypes revealed by the present analysis generally agreed with the biotypes with few discrepancies. ‘Cody’ and ‘Frontier Turfallo’ (seeded biotypes) grouped at a high level of similarity (99%). ISSR markers could not distinguish ‘Legacy’ and ‘NE86-120’ (both vegetative biotypes), which might be expected since they were selected from the same location. ‘Tatanka’ and ‘Texoka’ (seeded biotypes) grouped at a high level of similarity. ‘Bison’ and ‘Bowie’ (seeded biotypes) also grouped at an 86% level of similarity. This was expected because the male and female parents of ‘Mesa’ buffalograss and a superior plant of each sex selected from ‘Texoka’ constitute the parents of ‘Bison’ (Taliaferro et al. 1994). ‘Bowie’ has two male parents, which are selections of ‘Texoka’ (Stock Seed Farms).





UPGMA dendrograms of the unweighted pair-group method with arithmetic averaging cluster analysis revealed by a inter-simple sequence repeats, b simple sequence repeats, c random amplified polymorphic DNA, d sequence-related amplified polymorphisms, and e combined data sets of the four marker techniques
SSR transferability and analysis
The evolutionary implications of genome synteny have indicated that all grasses in the four subfamilies contain genes mostly in the same order, despite huge differences in their DNA content and chromosome number (Gale and Devos 1998). By utilizing colinearity among the Paniceae, Maydeae, and Andropogoneae tribes, the ability of SSR markers from maize, sorghum, pearl millet, and sugarcane to amplify SSRs loci in buffalograss (Chlorideae) was tested. One hundred and eighty maize (Senior and Heun 1993), 180 sorghum (Bhattramakki et al. 2000), 66 pearl millet (Allouis et al. 2001; Qi et al. 2001; Budak et al. 2003), and 26 sugarcane (Cordeiro et al. 2000) SSR markers were tested using four buffalograss genotypes for marker transferability. Of the 180, 112 maize (63%) and 143 (79%) sorghum SSR markers were amplified in buffalograss. Of the 66 pearl millet, 55 (82%) and 20 out of 26 (77%) sugarcane SSR markers could be amplified in buffalograss (Table 2). As expected, some of the markers could not be amplified. One reason for lack of amplification could be rapid genome modification. Even though changing annealing temperature may increase transferability of SSR, it is most likely to have fragments that are not anticipated. This could have led to a miscalculation of genetic diversity within and among buffalograss biotypes. A total of 180 SSR markers derived from maize, sorghum, pearl millet, and sugarcane were integrated to buffalograss and could be used for marker-assisted selection (MAS), conservation of genetic resources, and assessment of genetic relationships. Here we have demonstrated that a set of transferable SSR markers from related species could be used for determination of genetic diversity in buffalograss. This approach can be used to exploit these different species’ resources for map-based cloning of major genes in buffalograss. This method is cost effective and saves time. To the best of our knowledge, this approach has not been used for buffalograss germplasm improvement.
Of the 66 pearl millet SSR markers, 30 (Allouis et al. 2001; Qi et al. 2001; Budak et al. 2003) were used for estimation of genetic relationships among and within buffalograss biotypes. A total of 194 bands were observed; 168 of these (87%) were polymorphic among the seeded and vegetative biotypes and were shared between at least two individuals. The number of alleles revealed in buffalograss ranged from one to five per locus, with an average of three.
Genetic similarities among all individuals ranged from 0.04 to 0.99, with a mean similarity of 0.52. Cophenetic correlation was estimated at r=0.88, corresponding to a good fit. The SSR markers used revealed high genetic distances between most cultivars, indicating the cultivars developed from a number of germplasms that had diverse genetic backgrounds. ‘Cody’ (hexaploid) and ‘378’ (pentaploid) were found to span the extremes of the dendrogram (Fig. 1b). The UPGMA clustering algorithm grouped the genotypes into six clusters. The SSR markers identified the uniqueness of ‘NE 91-118’ among the remaining ones. ‘Cody’, ‘Bison’, ‘Frontier Turfallo’, and ‘Bowie’ (seeded biotypes) clustered together at a 61% level of similarity. Like ISSR, genetic variation within and among seeded biotypes was higher than vegetative biotypes. Although there was no distinct pattern, cultivars from the same geographic region tended to cluster together. Buffalograss is a relatively new emerging species and might not have an extensive evolutionary history. Hence, ecological distribution might be due to geographic origins or breeding origins.
RAPD analysis
Thirty RAPD primers were used for estimation of genetic relationships among and within buffalograss cultivars. A total of 210 bands were generated; 165 of these (79%) were polymorphic among the seeded and vegetative biotypes and were shared between at least two individuals. The number of alleles ranged from two to nine, with an average number of five per primer.
Genetic similarities among all individuals ranged from 0.15 to 0.99, with a mean similarity of 0.57. For the RAPD dendrogram, cophenetic correlation was estimated at r=0.86, corresponding to a very good fit. The UPGMA clustering algorithm grouped the genotypes into five clusters. Grouping of genotypes revealed by the present analysis generally agreed with the biotypes with few discrepancies. ‘Cody’ (hexaploid, seeded biotype) and ‘Density’ (diploid, vegetative biotype) were found to span the extremes of the dendrogram (Fig. 1c). However, RAPD could not separate ‘Topgun’ and ‘SWI 2000’, ‘Legacy’ and ‘NE86-120’, and ‘Bison’ and ‘Bowie’. Unlike ISSR analysis, RAPD separated ‘Cody’ and ‘Frontier Turfallo’ from each other. However, the similarity between ‘Cody’ and ‘Frontier Turfallo’ was high (92%).
SRAP analysis
Thirty out of 34 SRAP primer combinations (Budak et al. 2004) were tested. A total of 263 bands were observed; 249 of these (95%) were polymorphic within the collection and were shared between at least two individuals. SRAP markers used revealed a relatively high level of genetic distance among most genotypes. The number of alleles ranged from one to nine, with an average number of five.
Genetic similarities among all individuals ranged from 0.25 to 0.95, with a mean similarity of 0.62. ‘Cody’ and ‘Density’ were found to span the extremes of the dendrogram (Fig. 1d). For this dendrogram, cophenetic correlation was estimated at r=0.92, corresponding to a very good fit. The UPGMA clustering algorithm grouped the genotypes into three clusters. SRAP markers clearly separated vegetative biotypes from seeded biotypes with the exception of ‘609’. ‘Tatanka’, a seeded biotype, grouped with ‘609’, a vegetative biotype. ‘Density’ was singled out from the rest, probably because either it is a diploid vegetative biotype or from a different location and background. However, buffalograss with different ploidy levels grouped together in a previous study (Budak et al. 2004). Genotypes from different geographical regions grouped together, although there was a less-clear pattern in this regard. This response might be due to ecotype selection and interchange of the germplasm. Breeding origin could explain the tendency that genotypes developed from the same region group together. SRAP markers were the only marker system that differentiated all genotypes studied including those with a similar pedigree.
Combined analysis
Few studies have compared the results obtained from individual versus combined molecular-markers data sets for the purpose of genetic diversity analysis (Franco et al. 1997; Russell et al. 1997; Ajmone-Marsan et al. 1998). In this study, individual data sets were combined to examine congruence and incongruence among the results. For this dendrogram (Fig. 1e), cophenetic correlation was estimated at r=0.94, corresponding to a very good fit. In a combined analysis, genetic similarities among all individuals ranged from 0.15 to 0.93, with a mean similarity of 0.59. As in SRAP and RAPD analysis, ‘Cody’ and ‘Density’ were found to span the extremes of the dendrogram (Fig. 1e). ‘Topgun’ and ‘SWI 2000’ grouped at a high level of similarity (91%). This result was similar to the result obtained from RAPD analysis (Fig. 1c), with the exception of the level of similarity, which is 99%. ‘Density’ was grouped alone, which might be due to either its ploidy level or background and location where it is selected. Combined analysis demonstrated that the seeded biotypes are separated from the vegetative biotypes. ‘Bison’ and ‘Bowie’, both seeded biotypes, grouped together as in the RAPD, SSR, and ISSR. These two genotypes closely related in the SRAP analysis but were not in the same cluster. ‘Legacy’ and ‘NE86-120’ were clustered together with a high level of similarity (91%). Although there are divergent opinions about the utility of combining data sets (Mohammadi et al. 2003), our results demonstrated that combined data were concurrent with individual data when correlation among procedures were high.
Comparison of ISSR, SSR, RAPD, and SRAP dendrograms
All four markers proved to be highly effective in discriminating the 15 cultivars used in this study. All genotypes having more than 70% similarity by SSR data shared 100% of their SRAP bands. Some differences in the association of SSRs and RAPDs groupings and SRAPs and RAPDs groupings were apparent for similarity values less than 30%. In order to obtain a more robust comparison, the Mantel matrix correspondence test was used in matrices of cophenetic values generated from ISSRs, SSRs, RAPDs, and SRAP dendrograms. The correlation coefficients were statistically significant for all four marker system. Correlations between SRAPs and RAPDs and between SSRs and ISSRs dendrograms were 0.73 and 0.66 (P<0.05), respectively. This indicates that there is a concordance between SRAP and RAPDs and between SSRs and ISSRs. On the other hand, the correlation between SSR and RAPD and between ISSR and SRAP were low at 0.24 and 0.09, respectively (Table 3). Buffalograss is a cross-pollinated, highly heterogeneous species with a diverse genetic background (Budak et al. 2004). With this diversity, a high level of similarity among the different marker techniques might not be seen. Degani et al. (2001) reported absence of correlation between markers in strawberry, an allogamous crop.
All four marker types showed a high degree of similarity in dendrogram topologies (Fig. 1a–d), though with some differences in the positioning of some genotypes. The values of average discriminating power followed the pattern SRAP>SSR>ISSR>RAPD, as a direct consequence of their confusion probabilities.
The 15 buffalograss cultivars analyzed were unequivocally fingerprinted using each marker technique. The range and genetic similarity were similar for SRAP and RAPD, but a much wider range was found for SSRs and ISSRs, leading to a lower mean genetic similarity. Similar results have been found by Russell et al. (1997) for barley. In this study, estimated mean genetic similarities ranged from 0.51 to 0.57, based on SSRs and RAPD data, respectively, and to 0.52 for SSRs and 0.62 for SRAPs. A comparison of genetic similarities using different genetic materials is difficult because most estimates are based on randomly detected ploymorphic bands instead of all randomly detected bands. Despite the great and similar discriminating power of each marker systems used, there were some differences detected among them. For instance, the number of total polymorphic and discriminant fragments was higher for ISSRs than RAPDs. One of the possible explanations could be that ISSRs have a higher capacity of revealing polymorphism and demonstrate a great potential to determine the intra- and intergenomic diversity as opposed to the other arbitrary primers, such as RAPD (Zietkicwicz et al. 1994). In this study, SRAPs had the highest polymorphic and discriminating fragments compared to all marker systems tested. In addition, since RAPDs and ISSRs are dominant marker systems; ancestral dissection of the polyploids might be difficult compared to SRAPs and SSRs. Hence, the codominant nature of SRAPs and SSRs would make them the marker of choice for segregation studies and genome mapping in buffalograss.
The effects of probability of population identity and sampling
Sampling strategy is one the most important criteria for accurate and unbiased estimates of genetic diversity. Sampling distribution is largely unknown in most measures of genetic diversity. The effect of sampling on an accurate genotype assignment depends on the number of loci used for genotyping, the number of individuals sampled per genotype, mating system, and effective population size (Weir 1990). Information on the identity of individuals is important for the management of population. In population assignment tests, whether a buffalograss individual had a genotype that better represents the genetic characteristics of a different cultivar or its own cultivar was tested. RAPD analyses using all loci demonstrated relatively poor assignment compared to SSR or SRAP with correct assignment. Using a subset of SSR, ISSR, SRAP, and RAPD data randomly taken from the full data set, the effects of the fewer loci and fewer individuals were evaluated. This analytical approach may be useful for cultivar identification and property rights protection. For instance, when 20 individuals per genotype were sampled at five loci, the test indicated 71.6% correctly assigning individuals for SSRs. On the other hand, the test indicated 99.1% correctly assigning individuals when 18 loci were used. Basically, there is a tradeoff between the number of loci and the number of individuals. From the SSR, SRAP, and ISSR data sets, we used 29 loci per individual and 20 individuals per genotype and obtained 100% accurate assignments. RAPD data were clearly inferior for population assignment tests. The best probability for correct assignment tests using RAPD was 86%, whereas the worst probabilities of for ISSR and SSRs were 94% and 96%, respectively. A precise estimate of the genetic relationships, ideal population sample size, number of samples, or number of loci should be taken into account. Although there are no simple recommendations, it should be noted that adding or reducing the number of loci analyzed does not significantly influence the standard deviation of estimate for a reliable estimate of genetic diversity.
In the present study, coefficient of variation (CV) of the mean genetic similarity was also estimated on bootstrap resamples to evaluate the effect of the sample size. Threshold of CV was held to 10% for accurate estimates of genetic similarities (Tivang et al. 1994). When the number of bands increased, CV decreased (CV=5.75% for SRAP, CV=9.84% for RAPD, CV=11.23% for SSR, CV=10.87% for ISSR); using all of the bands indicated that all bands are necessary for accurate estimate of genetic similarity for all markers tested. SSR and ISSR bootstrap probabilities were low compared to SRAP.
In summary, the four molecular-marker types used can be successfully employed in assaying genetic differentiation for cultivar identification and property rights protection with a tremendous help of genotype assignment analytical method. However, using different generations of biotypes for property rights protection would be helpful to determine the sensitivity of the test. Our results have shown that all cultivars can be identified using SRAP technology, which will prove useful for varietal identification. It can provide a better approximation to true variation within and among buffalograss cultivars. Further, we emphasize that combined data sets are concurrent with individual data set and have given us greater confidence. In the presence of linkage disequilibrium, equally spaced markers will give a better estimate compared to randomly spaced ones. However, in buffalograss, this situation may not be the case, since buffalograss is highly heterogeneous. Much could be gained from a convergence between buffalograss genetic mapping and diversity studies (Budak et al. 2004), where markers could be chosen according to their distribution to ensure that marker sampling errors are very minimum or not committed. It is now our intention to (1) build a buffalograss genetic linkage map to test utility and distribution of available markers and (2) classify available markers into different categories, for instance, based on mode of transmission, or evolution with respect to different selective pressures.
References
Ajmone-Marsan P, Castiglino P, Fusari F, Kuipper M, Motto M (1998) Genetic diversity and its relationship to hybrid performance in maize as revealed by RFLP and AFLP markers. Theor Appl Genet 96:219–227
Allouis S, Qi X, Lindup S, Gale MD, Devos K M (2001) Construction of BAC library of pearl millet, Pennisetum glaucum. Theor Appl Genet 102:1200–1205
Bhattramakki D, Dong J, Chhabra A, Hart G (2000) An integrated SSR and RFLP linkage map of Sorghum bicolor (L.) Moench. Genome 43:988–1002
Blair MW, Panaud O, McCouch SR (1999) Inter-simple sequence repeat (ISSR) amplification for analysis of microsatellite motif frequency and fingerprinting in rice (Oryza sativa L.). Theor Appl Genet 98:780–792
Budak H, Pedraza F, Baenziger PS, Cregan PB, Dweikat I (2003) Development and utilization of SSR to estimate genetic diversity in a collection of pearl millet germplasm. Crop Sci 43:2284–2290
Budak H, Shearman RC, Parmaksiz I, Gaussoin RE, Riordan TP, Dweikat I (2004) Molecular characterization of buffalograss germplasm using sequence related amplified polymorphism markers. Theor Appl Genet 108:328–334
Corderio GM, Taylor GO, Henry RJ (2000) Characterization of microsatellite markers from sugarcane (Saccharum sp.), a highly polyploidy species. Plant Sci 155:161–168
Degani C, Rowland LJ, Saunders JA, Hokanson SC, Ogden E, Golan-Goldrish A, Galetta GL (2001) A comparison of genetic relationship measures in strawberry (Frageria ananassa Duch.) based on AFLPs, RAPDs and pedigree data. Euphytica 117:1–12
Dice LR (1945) Measures of the amount of ecological association between species. Ecology 26:297–302
Excoffier L, Smouse P, Qattro J (1992) Analysis of molecular variance inferred for metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Eguirta LE, Perez-Naser N, Pinero D (1993) Genetic structure, outcrossing rate and heterosis in Astrocaryum mexicanum (tropical palm): implications for evolution and conservation. Heredity 47:75–87
Franco J, Crossa J, Villasenor J, Taba S, Eberhart SA (1997) Classifying Mexican maize accessions using hierarchical and density search methods. Crop Sci 37:972–980
Gale MD, Devos KM (1998). Plant comparative genetics after 10 years. Science 282:656–659
Gupta PK, Varshney RK (2000) The development and use of microsatellite markers for genetic analysis and plant breeding with emphasis on bread wheat. Euphytica 113:163–185
Huff DR, Peakall R, Smouse PE (1993) RAPD variation within and among natural populations of outcrossing buffalograss [Buchloe dactyloides (Nutt.) Engelm.]. Theor Appl Genet 86:927–934
Johnson PG, Kenworthy KE, Auld DL, Riordan TP (2001) Distribution of buffalograss polyploid variation in the southern Great Plains. Crop Sci 41:909–913
Li G, Quiros CF (2001) Sequence-related amplified polymorphism (SRAP) a new marker system based on a simple PCR reaction: its application to mapping and gene tagging in Brassica. Theor Appl Genet 103:455–461
Mantel M (1967) The detection of disease clustering and generalized regression approach. Cancer Res 27:209–220
Mohammadi SA, Prasanna BM (2003) Analysis of genetic diversity in crop plants-salient stataistical tools and considerations. Crop Sci 43:1235–1248
Nei M, Li WH (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci USA 76:5269–5273
Peakall R, Smouse PE, Huff DR (1995) Evolutionary implications of allozyme and RAPD variation in diploid populations of dioecious buffalograss Buchloe dactyloides. Mol Ecol 4:135–147
Qi X, Lindup S, Pittaway TS, Allouis S, Gale MD, Devos KM (2001) Development of simple sequence repeat markers from bacterial artificial chromosomes without subcloning. BioTechniques 31:355–362
Riordan TP, de Shazer SA, Johnson-Cicalese JM, Shearman RC (1993) An overview of breeding and development of buffalograss. Int Turfgrass Soc Res J 7:816–822
Rohlf JF (2000) NTSYSpc: numerical taxonomy and multivariate analysis system. Exeter Software, Setauket, New York, USA
Rossetto M (2001) Sourcing of SSR markers from related plant species. In: Henry R (ed) Plant genotyping the DNA fingerprinting of plants. CAB International, Wallingford, UK, pp 211–224
Russell JR, Fuller JD, Macaulay M, Hatz BG, Jahoor A, Powell W, Waugh R (1997) Direct comparison levels of genetic variation among barley accessions detected by RFLPs, AFLPs, SSRs and RAPDs. Theor Appl Genet 95:714–722
Senior ML, Heun M (1993) Mapping maize microsatellites and polymerase chain reaction confirmation of the targeted repeats using CT primer. Genome 36:884–889
Taliaferro CM, Springer TL, Ahring RM (1994) Registration of bison buffalograss. Crop Sci 34:304
Tessier C, David J, This P, Boursiquot JM, Charrier A (1999) Optimization of the choice of molecular markers for varietal identification in Vitis vinifera L. Theor Appl Genet 98:171–177
Tivang JG, Nienhuis J, Smith OS (1994) Estimation of sampling variance of molecular marker data using the bootstrap procedure. Theor Appl Genet 89:259–264
Van Deynze AE, Sorrels ME, Park WD, Ayres NM, Fu H, Cartinhour SW, Paul E, McCouch SR (1998) Anchor probes for comparative mapping of grass genera. Theor Appl Genet 97:356–369
Wagner DB, Furnier MA, Saghai-Maroof SA, Williams SM, Dancik BP, Allard RW (1987) Chloroplast DNA polymorphisms in lodgepole and jack pines and their hybrids. Proc Natl Acad Sci USA 84:2097–2100
Weir BS (1990) Genetic data analysis. Sinauer, Sunderland, Mass.
Wu L, Lin H (1994) Identifying buffalograss [Buchloe dactyloides (Nutt.) Engelm.] cultivar breeding lines using random amplified polymorphic DNA (RAPD) markers. J Am Soc Hortic Sci 119:126–130
Zietkievicz E, Rafalski A, Labuda D (1994) Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification. Genome 20:176–183
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by B. Friebe
A contribution of the University of Nebraska Agricultural Research Division, Lincoln, Nebraska 68583. Journal Series No. 14398.
Rights and permissions
About this article
Cite this article
Budak, H., Shearman, R.C., Parmaksiz, I. et al. Comparative analysis of seeded and vegetative biotype buffalograsses based on phylogenetic relationship using ISSRs, SSRs, RAPDs, and SRAPs. Theor Appl Genet 109, 280–288 (2004). https://doi.org/10.1007/s00122-004-1630-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-004-1630-z