
Abstract
Gene tagging is the basis of marker-assisted selection and map-based cloning. To develop PCR-based markers for Pm4a, a dominant powdery mildew resistance gene of wheat, we surveyed 46 group 2 microsatellite markers between Pm4a near-isogenic line (NIL) CI 14124 and the recurrent parent Chancellor (Cc). One of the markers, gwm356, detected polymorphism and was used for genotyping an F2 population of 85 plants derived from CI 14124 × Cc. Linkage mapping indicated that Xgwm356 was linked to Pm4a at a distance of 4.8 cM. To identify more PCR-based markers for Pm4a, we also converted the restriction fragment length polymorphism marker BCD1231 linked to it into a sequence-tagged site (STS) marker. The STS primer designed based on the end sequences of BCD1231 amplified an approximately 1.6-kb monomorphic band in both parents. Following digestion of the products with the four-cutter enzymes HaeIII and MspI, it was discovered that the band from CI 14124 consisted of at least two products, one of which had a digestion pattern different from the band from Cc. In the F2 population, the cleaved polymorphism revealed by the STS marker between the parents co-segregated with powdery mildew resistance. To design Pm4a-specific PCR markers, the 1.6-kb band from Cc and the fragment associated with Pm4a in CI 14124 were sequenced and compared. Based on these sequences a new PCR marker was identified, which detected a 470-bp product only in the Pm4a-containing lines. These PCR-based markers provide a cost-saving option for marker-assisted selection of Pm4a.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Phenotypic selection based on morphological characteristics has been a common practice in plant breeding. Because the phenotype of a plant is determined by both its genotype and the environment, the efficiency of phenotypic selection is compromised by many factors such as, for example, the number of genes for the specific traits, the effect of individual genes, the degree to which the environments affect their expression, and the ease with which the traits can be evaluated. The indirect selection of target traits using genetic markers, commonly known as marker-assisted selection (MAS), has been an attractive strategy for breeders ever since it was proposed by Sax (1923) with the hope of speeding up and simplifying the breeding process. However, because of the lack of markers available to breeders, MAS was not very widely applied for breeding purposes until the 1980s when the field of molecular genetic markers began to rapidly expand, and molecular markers such as restriction fragment length polymorphism (RFLPs) and PCR-based marker systems such as randomly amplified polymorphic DNA (RAPDs), amplified fragment length polymorphism (AFLPs), microsatellite DNA or simple sequence repeats (SSRs), and single nucleotide polymorphism (SNPs) (Kwok et al. 1996) became widely available. A very comprehensive review on molecular markers and their applications in wheat breeding can be found in Gupta et al. (1999). Using these markers, breeders have been able to construct genetic maps, some of them of a very high density, for almost every crop species, and a large number of traits important for crop production have been mapped or tagged. This progress has stimulated research on MAS in breeding programs (Bernacchi et al. 1998; Cregan et al 1999; Chen et al. 2000).
Powdery mildew, a worldwide wheat disease caused by Blumeria graminis (DC.) E.O. Speer f. sp. tritici, poses a serious threat to wheat production due to the widespread use of high-yielding, semi-dwarf wheat (Triticum aestivum L. em. Thell) and the application of high levels of fertilizer. More than 30 loci conferring resistance have been described against this disease. Hartl et al (1993) and Ma et al (1994) were the first to use RFLP markers in mapping powdery mildew resistance genes. Since then, tightly linked molecular markers have been identified for Pm1–Pm4, Pm6, Pm8, Pm12, Pm13, Pm17, Pm21, Pm24–Pm27, Pm29 and Pm30 (McIntosh et al. 2003). Even though PCR-based markers are favored for MAS because of their simplicity and relatively low cost, many of the markers available for the powdery mildew resistance genes are RFLP probes. The development of PCR-based gene-specific markers has not been easy for wheat because of its hexaploid nature.
To identify PCR-based markers for Pm4a, a dominant resistance gene mapped to the long arm of chromosome 2A of wheat, we surveyed SSR markers and subsequently isolated a marker tightly linked to this gene. In addition, the RFLP marker xbcd1231, which co-segregates with Pm4a (Ma et al. 1994), was converted into a Pm4a-specific PCR marker.
Materials and methods
Plant materials and disease screening
Near-isogenic line (NIL) CI 14124 possessing Pm4a, Chancellor (Cc), their F2 and F3 progenies, nine pure lines with or without Pm4a, and a Triticum monococcum L. accession were used in this study (Table 1). CI 14124 was developed by Briggle (Briggle 1969) through backcrossing durum wheat cv. Yuma for eight generations using Cc as the recurrent parent. All of the plants were inoculated at the one- to two-leaf stage with a local Blumeria graminis mixture in a greenhouse of the Nanjing Agricultural University, Nanjing, China. Cc was used as the susceptible control and CI 14124 as the resistant control. 15–20 plants from each F2 individual were used for progeny test. Data were collected 7–14 days after inoculation, when the susceptible control showed distinct disease symptoms and the resistant control was still clear. Rating of the seedling reactions was simplified to two classes—resistant and susceptible—as there was a clear distinction between these two categories (Ma et al. 1994).
DNA extraction, sequencing, and primer design
Tissues from young seedlings of the plants were harvested for DNA extraction following the procedures described in Ma and Sorrells (1995). Sequencing was carried out by the CASarray Co., Shanghai (MegaBACE1000 DNA analyzers; Molecular Dynamics, UK). The sequences were analyzed using macvector ver. 7.0 (Accelrys, UK), which was also used in designing primers for the PCR analyses.
STS analysis
STS primers were designed based on the end sequences of BCD1231 (Ma and Sorrells, unpublished data), which is now deposited in GenBank (BE438906). The forward primer is 5′-TCCAGTGACCCCATCTGCTCATAC-3′, and the reverse primer is 5′-TCTCAGCGTTCCTTGTGATTCC-3′. The PCR analyses were carried out in a PE 9600 thermal cycler (Perkin Elmer, Norwalk, Conn.) in a volume of 25 µl containing 15–20 ng of template, 5 pmol of each of the primers, 5 nmol of each of the dNTPs, 37.5 nmol MgCl2, 0.13 U Taq DNA polymerase (Promega, Madison, Wis.) and 1× PCR buffer supplied together with the enzyme. The PCR conditions was as follows: one cycle of 2.5 min at 94°C, followed by 35 cycles of 50 s at 94°C, 1 min at 55°C, 1.5 min at 72°C, and a final extension for 5 min at 72°C. PCR products were resolved in 1% agarose gels run in 0.5× NEB buffer (50 mM Tris, 0.5 mM EDTA, 6.3 mM NaAc, pH 8.1). Two 4-bp cutter enzymes, HaeIII and MspI, were used to digest the PCR products according to the enzyme supplier’s manual. The digested products were separated in 1.2% agarose gels or 8% non-denaturing polyacrylamide gels (0.8 mm thick) in a M16 vertical gel electrophoresis system (Gibco-BRL, Gaithersburg, Md.) run at room temperature in a standard 1× TBE buffer, then stained with ethidium bromide.
Cloning of PCR products
PCR products were excised from the gel and purified through phenol extraction. The products were then cloned using the pGEM-T Easy cloning vector (Promega) following the manufacturer’s recommendation. Positive clones based on blue-white selection were picked out for a size check. Those plasmids containing inserts of the right size or bacteria cells harboring the positive inserts were chosen for sequencing.
SSR analysis
Forty-six pairs of wheat microsatellite primers mapped to the long arm of chromosome group 2 (Röder et al. 1998) were surveyed. PCR was conducted under conditions similar to those for the STS analysis except that the 35-cycle amplification profile consisted of 35 cycles of 40 s at 94°C, 50 s at 50–60°C (depending on the specific primer), and 1 min at 72°C. The PCR products were separated in 8% non-denaturing polyacrylamide gels.
Linkage analysis
Marker linkage analysis was done with mapmaker Macintosh V2.0 (Lander et al. 1987). The Kosambi function was used for calculating the map distance, and a LOD score of 3.0 was used as the threshold for declaration of linkage.
Results
Evaluation for powdery mildew resistance
A total of 85 F2 individuals were obtained from the cross of CI 14124 with Cc. Of these, 61 plants showed complete resistance, which, on the basis of the χ2 test, indicated a 3:1 segregation for resistance. No resistance segregation was observed for 21 F3 progenies derived from the resistant F2 plants. Combining both the F2 and F3 data, we concluded that the F2 population consisted of 21 Pm4aPm4a plants, 24 pm4apm4a plants, and 40 Pm4apm4a plants, which fit a 1:2:1 segregation.
The T. monococcum accession was susceptible, and the other plant lines evaluated showed the resistance reactions expected based on their genotypes at the Pm4a locus. Since T. monococcum contains the A genome in which Pm4a is located, the PCR product resulting from the amplification of this accession’s DNA was used as the reference for judging the allelic relationship between the DNA fragment linked to Pm4a from CI 14124 and fragments from the susceptible parent Chancellor. This is described below in more detail.
Identification of a SSR marker linked to Pm4a
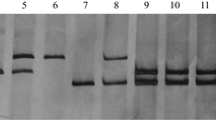
In the 46 pairs of wheat microsatellite primers tested, gwm356 amplified three and two products in CI 14124 and Cc, respectively. One of the products, a fragment of about 130 bp, is common between the two parents. The remaining two products in CI 14124 co-segregated with each other and were allelic to the second band in Cc (Fig. 1). When the 85 F2 plants were genotyped with gwm356, the genotypes of most of the plants at the Pm4a locus were in agreement with the marker genotypes of gwm356, but four plants heterozygous and three homozygous at the Pm4a locus were homozygous and heterozygous for the Xgwm356-CI 14124 allele, respectively. Thus, we determined the genetic distance between Xgwm356 and Pm4a to be 4.8 cM. When gwm356 was tested on the nine pure lines, all but CI14118, CI14119 and OG66, which do not have the Pm4a gene, produced the CI 14124 banding pattern (data not shown).

Genotyping the F2 population using the gwm356 marker. Arrow 1 indicates the Cc allele, arrows 2 and 3 indicate the CI 14124 allele
Conversion of BCD1231 to a STS marker
The products amplified by the original STS primer pair derived from BCD1231 were about 1.6 kb long and not polymorphic between CI 14124 and Cc (Fig. 2). Therefore, BCD1231 could not be directly used as a genetic marker. The digestion of CI 14124 using MspI resulted in four distinct bands of approximately 1,400 bp, 770 bp, 650 bp and 600 bp, respectively, and a less distinct one of approximately 500 bp (Fig. 2). In Cc, only bands of approximately 650 bp and 600 bp were observed. The polymorphic pattern between the two parents implied that this cleaved polymorphism was dominant with respect to inheritance. A similar polymorphic pattern was revealed by HaeIII digestion but was less readable under the separation conditions we used.

The PCR products amplified by the STS primers derived from BCD1231 before and after MspI digestion. Analysis was conducted using a 1.2% agarose gel
Segregation of the cleaved polymorphism was examined in 69 plants of the F2 population. All of the plants shared the 600-bp and 650-bp bands. The 770-bp band was observed only in resistant plants, irrespective of whether the resistance was segregating or not in their F3 progenies, and it was also detected in all of the pure lines possessing Pm4a and not in lines without it. When separated in a polyacrylamide gel, the 500-bp band could be easily seen and appeared consistently together with the 770-bp band. Thus, these two bands co-segregated with Pm4a, indicating that the PCR fragment that they derived from locates at the same position as Xbcd1231. Segregation of the 1,400-bp band (see Fig. 2) was observed among the F2 plants but showed no relation to powdery mildew resistance.
Sequence analysis of the amplified fragments
The variation in restriction sites between the resistant and susceptible plants suggested that it could be used for designing primers specific for Pm4a. To achieve this goal, the 1.6-kb bands from both parents were cloned. Positive clones were checked for insert size and MspI fingerprinting pattern. The Pm4a-linked insert, hereafter called Pm4a-A, was identified by the existence of the 770-bp band following digestion.
The Pm4a-A insert, five inserts (C2, C4, C5, C18, C36) from Cc and one insert (Mono) from the T. monococcum accession were sequenced. All of these sequences are homologous to BCD1231, thereby supporting their relationship to Xbcd1231. Complete sequences were obtained for the Pm4a-A, C2, C4 and Mono inserts. The Pm4a-A insert is actually 1,594 bp in length. Except for C18, which resulted from a single-primer amplification event, all of the remaining four inserts from Cc are different from one another, indicating that at least four homologous copies of the Pm4a-A loci may exist in Cc. Both C2 and C4 are 1,515 bp in length and are different from each other in only seven bases. The Mono sequence, which is 132 bp longer than C2 and C4, is more homologous to Pm4a-A than the C2 and C4 fragments based on full sequence comparison (1,270 conserved bases for Mono versus 1,187 or 1,186 for C2 or C4, respectively). By taking a closer look at the initial 480-bp sequences of C2 and C4, we noticed that the fragment immediately after 435 bp, which falls into an intron region, displays no noticeable homology in nucleotide composition to either Pm4a-A or Mono, even though only one nucleotide difference could be found between Pm4a-A and Mono in this region (Fig. 3). The C5 and C36 sequences were even further from Pm4a-A in terms of clustw alignment results. These results indicate that none of the Cc inserts sequenced was allelic to the Pm4a-A insert.

Comparison of the 480-bp sequence at the 5′ end of Pm4a-A, C4, C2 and Mono and the positions of the primers designed. The shaded portion indicates the conserved bases when compared with the Pm4a-A or Mono sequences, numbers on top indicate the nucleotide position of Pm4a-A. The common primer is the same as the reverse primer for STS amplification. R1, R3 Based on Pm4a-A, R2, R4 based on C4 and Mono, respectively
PCR markers based on sequence variation
Four different reverse primers based on sequence variation shown in Fig. 3 were combined with the forward STS primer as a common one to perform the PCR reactions. The R1 and R2 primers were derived from the intron region and the R3 and R4 primers were derived from the exon sequence.
All of these primers, except for R3, produced a single band of the expected size. The R2 primer based on the C4 sequence and R4 primer based on the Mono sequence amplified a monomorphic fragment in CI 14124 and Cc. The R3 primer, again based on the Pm4a-A sequence, produced a monomorphic fragment in both parents and an extra fragment in CI 14124, which is slightly larger in size and could be visualized by using a higher percentage of agarose gel in the electrophoresis. The R1 primer based on the Pm4a-A sequence amplified a 470-bp fragment in CI 14124 but not in Cc and thus could be used as a Pm4a-specific marker. PCR primers based on the C2 and Mono sequences corresponding to the R1 primer region produced no polymorphism (data not shown). These results support our premise that C2 and C4 are not allelic to Pm4a-A.
The F2 individuals from CI 14124 × Cc were tested with the R1 primer pair. The results show that the polymorphic band segregated in agreement with the segregation of Pm4a (Fig. 4).

Segregation of the fragment amplified in CI 14124 in a sub-sample of the F2 population. φX174 Marker, R resistant, S susceptible
Discussion
PCR-based markers are ideal tools for breeders. Since the advent of PCR-based marker technologies, various marker methods—for example RAPD, STS, and SSR—have been employed for developing PCR-based markers for traits of interest. Sixteen powdery mildew resistance genes of wheat have been mapped or tagged using molecular markers, but PCR-based markers are available for only eight of them, among which five were tagged with either RAPDs or AFLPs, or both.
Here, we report the identification of a SSR marker tightly linked to Pm4a and the successful conversion of the RFLP marker BCD1231 of Pm4a to a STS marker and an allele-specific marker. The SSR marker is co-dominant and can be used for separating homozygous and heterozygous types. The allele-specific marker is dominant and produces a single band only in the Pm4a-carrying lines, which makes it possible to detect the gene using only staining without the need for electrophoresis (Gu et al. 1995). BCD1231 has been successfully applied in a wheat breeding program (Liu et al. 2000). We showed through analysis of the breeding lines and the segregation population that the STS markers identified were genetically consistent with the RFLP markers. Therefore, they can replace BCD1231 in breeding programs. The use of an allele-specific marker will make the selection process more simple and cost-effective.
Many wheat traits have been mapped using RFLP or RAPD markers. To facilitate the use of molecular markers in large-scale screening, RFLP markers need to be converted into PCR-based markers. Talbert et al (1994) were the first to convert wheat RFLP probes to STS markers. Since then, quite a few laboratories have generated STS primers from either wheat clones (Feuillet et al, 1995; Talbert et al. 1996), RAPD markers (Talbert et al. 1996; Hu et al. 1997; Dweikat et al. 2002), or AFLP bands (Prins et al. 2001; Parker and Langridge 2002; Smith et al. 2002). All of these conversions were based on wheat DNA sequences. Seyfarth et al. (1999) reported the development of a STS marker of BCD260 for adult plant leaf rust resistance gene Lr35 of wheat. The RFLP marker BCD1434 for Pm3a (Ma et al. 1994) was also converted to a STS marker (Ma and Sorrells, unpublished data). Generally speaking, the conversion of heterologous probes, such as barley clones (for example, the BCD probes) or oat clones (for example, the CDO probes), is more difficult because sequence polymorphism may exist between the homoeologous loci of wheat and barley or oat. In addition, the amplified products may not originate from the same loci the RFLP probes mapped (Erpelding et al. 1996). Byran et al. (1999) pointed out that the PCR primers based on wheat cDNA have a tendency to produce several PCR products in similar size. This is also true for the original STS marker of BCD1231 developed in this study. By using it, we obtained more than four products with a high level of homology, and these may include both the orthologous and the paralogous sequences. Thus, enzyme digestion of the PCR products had to be used to exploit the sequence variation among them. To alleviate this problem, other strategies, such as single-strand conformation polymorphism, could also be exploited (Forsstrom et al. 2003).
The allele-specific design for BCD1231 was complicated by its intra-chromosome repetition. As reported in Ma et al (1994), two loci detected by BCD1231, including xbcd1231-Pm4a, were tightly linked to each other in 2AL, which explained why more than three kinds of homologous sequences were amplified by the first STS marker. Since the approximately 1.6-kb sequences cover three full introns, to test the feasibility for developing allele-specific markers using the sequence information on hand, we designed two SNP (R3 and R4) primers based on an exon sequence and two (R1 and R2) primers based on the intron sequence (starting at +410 in Fig. 3). The R1 primer produced the expected results, which only amplified a 470-bp product in Pm4a-carrying lines.
The low level of DNA sequence variation in wheat (Bryan et al 1999), especially when the coding regions are targeted, is another limitation in designing allele-specific primers. Our study implies that the large variation in the intron regions could make it easier to develop such primers and that it should be taken advantage of. Since introns often evolve faster and contain more sequence variations, the rich polymorphism of the intron regions in homoeologous loci is particularly useful in design locus-specific primers.
References
Bernacchi D, Beck-Bunn T, Emmatty D, Eshed Y, Inai, Lopez J, Petiard V, Sayama H, Uhlig J, Zamir D, Tanksley S (1998) Advanced backcross QTL analysis of tomato. II. Evaluation of near-isogenic lines carrying single-donor introgressions for desirable wild QTL-alleles derived from Lycopersicon hirsutum and L. pimpinellifolium. Theor Appl Genet 97:170–180
Briggle LW (1969) Near-isogenic lines of wheat with genes for resistance to Erysiphe graminis f. sp. tritici. Crop Sci 9:70–72
Bryan GJ, Stephenson P, Collins A, Kirby J, Smith JB, Gale MD (1999) Low levels of DNA sequence variation among adapted genotypes of hexaploid wheat. Theor Appl Genet 99:192–198
Chen S, Lin XH, Xu CG, Zhang QF (2000) Improvement of bacterial blight resistance of ‘Minghui 63’, an elite restorer line of hybrid rice by molecular marker-assisted selection. Crop Sci 40:239–244
Cregan PB, Mudge J, Fickus EW, Danesh D, Denny R, Young ND (1999) Two simple sequence repeat markers to select for soybean cyst nematode resistance conditioned by the rhg1 locus. Theor Appl Genet 99:811–818
Dweikat I, Zhang W, Ohm H (2002) Development of STS markers linked to Hessian fly resistance gene H6 in wheat. Theor Appl Genet 105:766–770
Erpelding JE, Blake NK, Blake TK, Talbert LE (1996) Transfer of sequence tagged site PCR markers between wheat barley. Genome 39:802–810
Feuillet C, Messmer M, Schacehermayr G, Keller B (1995) Genetic and physical characterization of the LR1 leaf rust resistance locus in wheat (Triticum aestivum L.). Mol Gen Genet 248:553–562
Forsstrom PO, Koebner R, Merker A (2003) The conversion of wheat RFLP probes into STS markers via the single-stranded conformation polymorphism technique. Genome 46:19–27
Gu WK, Weeden NF, Yu J, Wallace DH (1995) Large-scale, cost-effective screening of PCR products in marker-assisted selection applications. Theor Appl Genet 91:465–470
Gupta PK, Varshney RK, Sharma PC, Rameshi B (1999) Molecular markers and their applications in wheat breeding. Plant Breed Z Pflanzenzuecht 118:369–390
Hartl L, Weiss H, Zeller FJ, Jahoor A (1993) Use of RFLP markers for the identification of alleles at the Pm3 locus conferring powdery mildew resistance in wheat (Triticum aestivum L.). Theor Appl Genet 86:959–963
Hu XY, Ohm, HW, Dweikat I (1997) Identification of RAPD markers linked to the gene Pm1 for resistance to powdery mildew in wheat. Theor Appl Genet 94:832–840
Kwok PY, Deng Q, Zakeri H, Taylor SL, Nickerson DA (1996) Increasing the information content of STS-based genome maps: identifying polymorphisms in mapped STSs. Genomics 31:123–6
Lander E, Green P, Abrahamson J, Barlow A, Daley M, Lincoln S and Newburg L (1987) mapmaker: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Liu J, Liu D, Tao W, Li W, Wang S, Chen P, Cheng S, Gao D (2000) Molecular marker-facilitated pyramiding of different genes for powdery mildew resistance in wheat. Plant Breed Z Pflanzenzuecht 119:21–24
Ma Z-Q, Sorrells ME (1995) Chromosomal regions linked to restoring genes in two restorers for Triticum timopheevii cytoplasm sterility of wheat. Crop Sci 35:1137–1143
Ma Z-Q, Sorrells ME, Tanksley SD (1994) RFLP marker linked to powdery mildew resistance genes Pm1, Pm2, Pm3 and Pm4 in wheat. Genome 37:871–875
McIntosh RA, Yamazaki Y, Devos KM, Dubcovsky J, Roders WJ, Appels R (2003) In: Pogna NE, Romanò, Pogna EA, Galterio G (eds) Catalogue of gene symbols for wheat. In: 10th Int Wheat Genet Symp, vol 4. Istituto Sperimentale per la Cerealicoltura, Rome, p34
Parker GD, Langridge P (2002) Development of a STS marker linked to a major locus controlling flour color in wheat (Triticum aestivum L.). Mol Breed 6:169–174
Prins R, Groenewald JZ, Marais GF, Snape JW, Koebner RMD (2001) AFLP and STS tagging of Lr19, a gene conferring resistance to leaf rust in wheat. Theor Appl Genet 103:618–624
Röder MS, Korzun V, Wendehake K, Plaschke J, Tixier MH, Leroy P, Ganal MW (1998) A microsatellite map of wheat. Genetics 149:2007–2023
Sax K (1923) The association of size differences with seed-coat pattern and pigmentation in Phaseolus vulgaris. Genetics 8:552–560
Seyfarth R, Feuillet C, Schachermayr G, Winzeler M, Keller B (1999) Development of a molecular marker for the adult plant leaf rust resistance gene Lr35 in wheat. Theor Appl Genet 99:554–560
Smith H, Koebner D, Boyd A (2002) The development of a STS marker linked to a yellow rust resistance derived from the wheat cultivar Moro. Theor Appl Genet 104:1278–1282
Talbert LE, Blake NK, Chee PW, Blake TK, Magyar GM (1994) Evaluation of “sequence-tagged-site” PCR products as molecular markers in wheat. Theor Appl Genet 87:789–794
Talbert LE, Bruckner PL, Smith LY, Sears R, Martin TJ (1996) Development of PCR markers linked to resistance to wheat streak mosaic virus in wheat. Theor Appl Genet 93:463–467
Acknowledgements
This project was partially supported by the Outstanding Youth Fund (no. 30025030) of NSFC, the ‘863’ project 2003AA207100, and ‘2000’ Trans-century Talent Development Program of MOE, China. We thank X.W. Gu and L. Xiong for their help in resistance screening.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by F. Salamini
Rights and permissions
About this article
Cite this article
Ma, ZQ., Wei, JB. & Cheng, SH. PCR-based markers for the powdery mildew resistance gene Pm4a in wheat. Theor Appl Genet 109, 140–145 (2004). https://doi.org/10.1007/s00122-004-1605-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-004-1605-0