
Abstract
The genetic linkage map of European beech (Fagus sylvatica L.) that we report here is the first to our knowledge. Based on a total of 312 markers (28 RAPDs, 274 AFLPs, 10 SSRs) scored in 143 individuals from a F1 full-sib family. Two maps (one for each parent) were constructed according to a “two-way pseudo-testcross” mapping strategy. In the male map 119 markers could be clustered in 11 major groups (971 cM), while in the female map 132 markers were distributed in 12 major linkage groups (844 cM). In addition, four and one minor linkage groups (doublets and triplets) were obtained for the male and female map respectively. The two maps cover about 82% and 78% of the genome. Based on the position of 15 AFLP and 2 SSR loci segregating in both parents, seven homologous linkage groups could be identified. In the same pedigree we investigated the association with genetic markers of several quantitative traits: leaf area, leaf number and shape in 2 different years, specific leaf area, leaf carbon-isotope discrimination and tree height. A composite interval-mapping approach was used to estimate the number of QTLs, the amount of variation explained by each of them, and their position on the genetic linkage maps. Eight QTLs associated with leaf traits were found that explained between 15% and 35% of the trait variation, five on the female map and three on the male map.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
European beech (Fagus sylvatica L.) is one of the most important broad-leave trees in Europe. Pure and mixed beech stands cover more than 12 million hectares, from Southern Scandinavia to the Iberian, Italian and Balkan Peninsulas, and from Southern England to Ukraine. Beech prefers oceanic climates (no extreme winter temperatures, cool and foggy springs and mild summers) with mean annual rainfall higher than 1,500 mm. Beech does not tolerate extreme water stress and grows better in deep and well-drained soils (Teissieur du Cros 1981). In central Europe, beech is a component of lowland forests, while in the Mediterranean area it is a typical mountain species.
In spite of the importance of the species as timber, the relationship between genetic markers and quantitative traits has received relatively little attention (Bucci et al. 1999). This is probably due to the lack of breeding programs for the species. Natural regeneration is extensively used in managed forests and, if planting occurs, material from natural seed sources is used. However, selection by foresters does occur, for instance when choosing seed-producing plants. Information on the genetic control of adaptive traits will provide new tools for foresters to enhance wood production and to preserve adaptive capacity stands. QTLs could be thought of as surrogates for adaptive traits. One of the most commonly used strategy for identifying the genetic base of quantitative trait loci (QTLs) is the study of their association with anonymous markers.
Genetic mapping represents a new tool to help traditional tree breeding methods through the identification of quantitative trait loci (Bradshaw and Stettler 1995) and their integration into marker assisted selection programs (Plomion et al. 1996).
Molecular markers such as AFLPs, RAPDs, SSRs and ESTPs have been used extensively for the preparation of linkage maps of a number of tree species. A comprehensive list is available at http://dendrome.ucdavis.edu and http://www.pierroton.inra.fr/genetics/labo/mapreview.html. Within the Fagaceae family linkage maps have been published for the genus Castanea (Kubisiak et al. 1997; Casasoli et al. 2001) and for the genus Quercus (Barreneche et al. 1998).
For forest trees, given the high genetic load and long generation time, segregating populations derived from crosses between inbred lines are not available. To circumvent this limitation a “two-way pseudo-test-cross” approach is generally used to construct linkage maps and for QTL analysis. QTLs have been mapped for growth and yield, wood quality, water use efficiency (δ 13C) and bud phenology traits in forest trees (Bradshaw and Stettler 1995; Plomion et al. 1996; Frewen et al. 2000; Sewell et al. 2000, 2002; Jermstad et al. 2001; Brendel et al. 2002).
Here we report the first linkage map for the genus Fagus and a preliminary investigation of the relationship between genetic markers (AFLPs, RAPDs and Microsatellites) and the variation of some important QTLs such as growth, leaf size and shape, and carbon isotope discrimination (δ 13C).
Materials and methods
Plant material and DNA extraction
Our mapping population consisted of 143 full-sibs from a controlled cross (Ceroni et al. 1997), part of a complete diallel cross 4×4 carried out in 1996, using parent trees from a population in northern Italy at 1,650 m a.s.l., just below the tree-line. Individuals were grown for 6 years in well-watered soil and optimal nutritional conditions in a nursery.
Genomic DNA was isolated from fresh buds and leaves using the NucleoSpin Plant kit from Macherey-Nagel. DNA concentration was estimated by comparing the fluorescence intensities of ethidium bromide-stained samples to those of λ DNA standards, on a 0.8% agarose gel.
RAPD analysis
We used 19 highly reproducible RAPD markers initially screened for amplification and polymorphisms in the two parents of the mapping population (Di Masso 1999). Procedures for detecting RAPDs followed Williams et al. (1990). Fragments were separated on 1.5% agarose gels, stained with ethidium bromide and observed using ultraviolet light.
AFLP analysis
AFLP analysis was performed according to Vos et al. (1995) with some modifications. Briefly, 300 ng of genomic DNA were incubated for 2 h 30′ at 37°C with 5 U of the endonuclease EcoRI and 5 U of the endonuclease Tru9I in the universal One-Phor-All-Buffer (Pharmacia). Next a 10 µl of a solution containing 5 pmol of EcoRI-adapters, 50 pmol of Tru9I -adapters, 1 U of T4 DNA ligase, 1 mM of ATP in the universal One-Phor-All buffer were added, and the incubation was continued overnight at room temperature. Digested-ligated DNA fragments were diluted 1:4 in water and used as the template for the first amplification reaction (pre-amplification). Pre-amplification was conducted using standard AFLP EcoRI and Tru9I primers with one additional selective nucleotide at the 3′end (EcoRI+A, Tru9I+C). The adaptor and primer sequences used were based on the core primer designed as described by Vos et al. (1995). Pre-amplification was performed in 20 µl of 1×Buffer, 1.5 mM of MgCl2, 0.2 mM of each dNTP, 5 pmol of each primer, 0.4 U of Taq DNA polymerase and 2 µl of diluted restriction-ligation mixture. PCR amplification consisted of 20 cycles of a 30 s DNA denaturation at 94°C, 30 s annealing at 56°C, a 2 min primer extension at 72°C and a final-step of 30 min at 60°C.
The pre-amplification products were diluted 1:10 in de-ionized water and 5 µl were used in the selective amplification step performed using various combinations of primer pairs having three selective additional nucleotides at both EcoRI and Tru9I adaptor sides. EcoRI primers (forward primer) were fluorescent labeled with Cy-5 while Tru9I primers were unlabeled. Reaction mixture was as described above for pre-amplification, with the difference that only 0.8 pmol of the labeled EcoRI primer and 0.8 U of Taq DNA polymerase were used. PCR amplification was performed for 36 cycles with a touchdown profile following Vos et al. (1995). One-microliter of the AFLP products was added to 40 µl of de-ionized formamide in the CEQ sample plate followed by 0.5 µl of the CEQ DNA Size Standard-400 (P/N 608098) and run on a CEQ 2000 8-capillary Analysis System (Beckman Coulter). Samples were injected onto a 33-cm capillary at 2.0 kV for 60 s and electrophoresed at 6.0 kV for 35 min at 50°C. For loading samples to a 96-well CEQ plate we used a liquid handling Laboratory Automation Workstation (Biomek 2000).
PCR fragment sizes were obtained by comparison with internal size standards by using the local Southern algorithm (Southern 1979). Data were then exported to GENOGRAPHER software for scoring (version 1.6, abailable from http://hordeum.oscs.montana.edu/genographer/). Lanes were normalized considering the total signal intensity of the lane.
A total of 43 primer combinations were used to screen the mapping population based on a pre-screening of 80 combinations. The density, complexity and number of polymorphic AFLP fragments were used as selection criteria. The AFLP marker name refers to the primers used: the first two letters refer to the last two selective nucleotides of the EcoRI primer, the following two letters refer to the last two selective nucleotides of the Tru9I primer, and the last numbers refer to the molecular weight of the polymorphic band scored.
For one AFLP combination (E-AAC/T-CTT) 18 individuals previously run on the CEQ 2000 were also run on a LI-COR 4000 automated sequencer to compare AFLP reproducibility between different detection systems (fluorescent samples resolved by capillary electrophoresis and by slab gel electrophoresis).
SSR analysis
Seven nuclear microsatellites developed for Fagus by Pastorelli et al. (2003) were used in this study. PCR and separation conditions are reported in Pastorelli et al. (2003). Two SSRs developed in Quercus, QrZAG31 and Quru0A01 (Kampfer et al. 1998; Aldrich et al. 2002) and one SSR developed in Castanea sativa, CsCAT14 (Marinoni et al. 2003), were also used. These SSRs were amplified using fluorescent IRD labeled primers (IRD800). Amplification reactions were performed in a total volume of 10 µl containing 1×Buffer, 2.5 mM of MgCl2, 0.2 mM of dNTPs, 3 pmol of IRD-labeled primer, 3 pmol of unlabeled primer, 0.5 U of Taq polymerase and 20 ng of DNA. Amplification conditions were as previously described (Kampfer et al. 1998; Aldrich et al. 2002; Marinoni et al. 2003). Gel electrophoresis was performed on a LI-COR 4000 automated sequencer. Samples were electrophoresed on 25-cm-long 8% polyacrylamide gels containing 6 M urea.
Segregation analysis and map construction
Mendelian segregation was tested using χ2 goodness-of-fit to a 1:1 ratio at a 5% and 1% significance level.
Linkage analysis was performed using MAPMAKER/EXP 3.0 (Lander et al. 1987) and JOINMAP 3.0 (Van Ooijen and Voorrips 2001) software. Maps were constructed according to a two-way pseudo-testcross strategy (Grattapaglia and Sederoff 1994). RAPD, AFLP and SSR markers, segregating 1:1 and 1:1:1:1 in the offspring, were used for the construction of parental maps. Preliminary grouping of marker loci (two-point analysis) was performed using MAPMAKER (LOD threshold 3.0, maximum recombination fraction 0.40). Markers were ordered within linkage groups using a multipoint analysis (minimum LOD 2.0). Markers that could not be ordered were reported as accessory markers linked to a specific marker on the map and were not included in the estimation of the overall genetic length of the maps. Distances between adjacent marker loci were calculated from recombination fractions using Kosambi’s mapping function. Markers that showed a departure from the 1:1 ratio (0.01<P<0.05) were also used for map construction but their position was defined after establishing gene order for the other markers. MAPMAKER does not accept mixed segregation data. Intercross dominant markers (i.e. AFLP and RAPD markers segregating 3:1) were given an approximate location on both maps using JOINMAP. Intercross markers segregating in both parents were used to align the two parental maps but were not used to integrate the two maps.
Genome length (L) was estimated from partial linkage data using the equation:
where n is the number of framework markers, n(n−1) the number of pair-wise comparisons, d the maximum distance between two adjacent framework markers in centimorgans at a certain minimum LOD score, and k the number of markers pairs with a LOD value at the same minimum LOD score (Hulbert et al. 1988; Chakravarti et al. 1991). A LOD score of 3.0 was chosen to estimate genome length. Only pair-wise comparisons between framework markers were considered to avoid an overestimation of genome size.
Quantitative traits and QTL mapping
Quantitative traits were measured on the same progeny for the construction of the linkage map.
As a consequence of ongoing mortality, the number of individuals was not the same for all traits. Leaf area was estimated from individual leaf length and width. The relationship between leaf area, and length and width, was calibrated for each year using 50 leaves with known area (measured in scanned leaf images). Leaf counts were available for 1999 and for 2002. Leaf shape was estimated using the leaf length-width ratio. Dry weight was determined drying leaves at 70°C for 24 h. Specific leaf area (SLA) was calculated from the ratio of total leaf area and total leaf dry mass. Height was measured for the 118 surviving seedlings at the end of the growing season in year 2002. Carbon isotope ratios were estimated on leaves collected in the fall of 2001. Leaves were dried for 48 h at 70°C and ground using mortar and pestle. For each individual a sub-sample of 0.1 mg was analyzed for 13C composition using an elemental analyzer “on line” with a gas-isotope-ratio mass spectrometer (ThermoFinnigan Delta plus XP). Carbon isotope composition was calculated (δ 13C) following Craig (1957). Quantitative trait measurements were transformed to obtain normal distributions.
QTL analysis was performed using QTL Cartographer 1.12 (Basten et al. 1994, 2002). Composite Interval Mapping (CIM) analysis (Zeng 1994) was performed separately on each map using background markers (from both maps) associated with the trait found using simple and stepwise regression. CIM includes neighboring markers and uses the remaining background markers as co-factors in order to remove the effects of multiple QTLs. The number of background markers employed varied from 1 to 5. Significant thresholds (with overall P=0.05) were established using a permutation test (1,000 runs) (Churchill and Doerge 1994) including a permutation of the background markers (Basten et al. 2002).
Results
Molecular markers
For map construction we used a total of 33 polymorphic RAPD markers obtained from 19 primers [1.7 (±0.9 SD) polymorphic bands per primer]. Of the 33 RAPD loci scored 27 were in a testcross configuration (14 in one parent and 13 in the other), while six were segregating in both parents (Table 1).
A total of 80 primer combinations compatible with the E+A/T+C pre-amplification were screened by selective amplification of DNA from the two parents and six progenies. The 43 for which we observed reproducible polymorphisms were used for the construction of the map. Of the 319 AFLP loci scored, 272 were heterozygous in one parent and null in the other (139 and 133 respectively: testcross configuration segregating 1:1), while 47 were segregating in both (intercross configuration segregating 3:1) (Table 1).
The two detection methods (capillary electrophoresis by CEQ 2000 and slab gel electrophoresis by the LI-COR 4000 automated sequencer) gave consistent fingerprint patterns for all samples.
All tested microsatellites (ten) were polymorphic in the two parental trees. In the mapping population, six were in testcross configuration (three in one parent and three in the other) and four were fully informative. No significant deviation from the expected Mendelian ratio was observed.
Segregation distortion and map construction
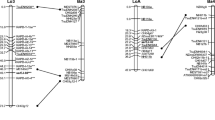
Twenty-nine markers showed limited segregation distortion (0.01<P<0.05) and were used for mapping. Fifty markers (45 AFLPs and 5 RAPDs) with high segregation distortion (P≤0.01) were discarded (Table 1). We used a total of 312 markers (28 RAPDs+274 AFLPs+10 SSRs) for linkage analysis, of which 265 were in 1:1 and 1:1:1:1 configuration. In the female map 112 test-cross markers (71 framework and 41 accessory) were grouped in 12 linkage groups, with more than three markers) (nine markers per group, with a length size of 844 cM on average and marker distance of 12 cM). One triplet was also obtained while 26 markers were unlinked. In the male map 98 test-cross markers (66 framework and 32 accessory) were grouped in 11 linkage groups, with more than three markers (nine markers per group, length size of 971 cM on average, and marker distance of 14 cM). Two doublets and two triplets were obtained while 20 markers were unlinked. The distance between adjacent markers varied from 2.2 to 36.8 cM in the female map and from 3.4 to 35.1 cM in the male map. Using JOINMAP an additional 47 (43 AFLPs+4 RAPDs) intercross dominant markers (3:1 ratio) were added to the two parental maps but their relative positions were used as additional anchor loci only. In both maps no linkage with framework loci was found for 21 intercross loci. Accessory markers were located next to their closest framework markers (Fig. 1). Twenty markers with limited distortion (0.01<P<0.05) were positioned on the linkage maps: ten in the female and ten in the male one.

Genetic linkage map of F. sylvatica. Female and male linkage groups are indicated with “F” and “M”, respectively. Re-coded markers are marked by “r”. Loci showing segregation distortion at 0.01<P<0.05 are indicated by a star (*). Accessory markers (testcross and intercross) are listed beside their closest framework marker with the distance in cM. Intercross dominant markers are underlined; bridge markers are interconnected with bold lines. Map units (cM) shown on the left of each linkage group were calculated by Kosambi mapping function
Considering only framework markers, the female map spanned 844 cM and the male map 971 cM. Since our estimate of beech genome size is 1,081 (female) and 1,185 (male) cM, the 71 framework loci of the female map covered about 78% while the 66 framework loci of the male map covered 82% of the genome.
Based on the position of 15 AFLP and two SSR loci which segregated in both parents, seven homologous linkage groups could be identified. (Fig. 1).
QTL analysis
For each phenotypic trait, mean, standard deviation and sample size are reported in Table 2. Beside the expected significant correlations between height, leaf area and leaf number within each year, significant correlations were found between leaf number measured in 1999 and 2002 (r=0.29, P=0.03) and leaf area measured in 1999 and 2002 (r=0.38, P<0.01). Specific leaf area (SLA) was correlated with leaf area in 2002 as expected from the formula (r=0.35, P<0.001). Correlation between leaf shape in the 2 years was not significant. Leaf number and area measured in 1999 were significantly correlated with δ 13C measured in 2001 (leaf number r=0.45, P<0.01; leaf area r=0.50, P<0.001).
By Composite Interval Mapping [(P<0.05) set by the permutation test] we found eight QTLs: five on the female map and three on the male map. Results are reported in Table 3. Three QTLs (leaf area in 1999 and 2002, and SLA) were located on a single region on LG1-F between AFLP markers acgg229 and atcc197. Additive QTL effects for leaf area in 1999 and 2002 have opposite signs but their location seems to be more than 7 cM apart. In the genomic window between 115 and 130 cM on LG1-F, estimated additive effects for leaf area change sign from negative to positive in both years (although not always significant). Similar results were also found in the single marker analysis (data not reported). A further QTL for leaf area for 2002 is located in a homologous linkage group (LG1-M) on the male map. Two other QTLs for specific leaf area (SLA) are located in different linkage groups. No QTL was found for height, and δ 13C.
Discussion
Marker variation and the linkage map
To our knowledge no previous genetic linkage map for the genus Fagus has been reported. It is known that AFLP markers reveal a larger number of polymorphic bands than other techniques like RAPDs. In tree species the average number of fragments per primer combination ranged from six (Salix, Tsarouhas et al. 2002) to 26 (Larix, Arcade et al. 2000), while for RAPD polymorphic fragments per assay ranged from 1.3 (Pinus thunbergii, Kondo et al. 2001) to 4.8 (Pinus pinaster, Costa et al. 2000). In our study the average number of fragments per primer combination was 7.4 and 1.7 for AFLPs and RAPDs, respectively. RAPD reproducibility is a weak aspect of this technique (Isabel et al. 1999; Troggio et al. 2001). We only used highly reproducible RAPD markers (Di Masso 1999). SSRs markers produced well-scorable bands [no or weak stutter bands, quality 1 or 2 (Smulders et al. 1997)] with a high level of polymorphism typical of these markers. We found null alleles at three loci (FS1-25, FS4-46, FS1-03). The presence of null alleles in SSRs is a weakness in a marker otherwise considered optimal from most other standpoints. In beech we found a frequency of null alleles higher than that reported for other species. (Scotti et al. 1999).
We only used markers with relatively minor distortion (between 1% and 5%). High levels of segregation distortions have frequently been reported in tree species. In two different interspecific crosses in Populus (Cervera et al. 2001) the percentage of AFLP markers showing segregation distortion varied from 18.8% to 7.8%. In Salix (Tsarouhas et al. 2002) and in Quercus (Barreneche et al. 1998) 18% and in Eucalyptus (Marques et al. 1998) approximately 15% of the segregating loci showed significantly skewed segregation.
We found 12 linkage groups for the female map and 11 for the male map. The number of groups is close to the number of chromosomes (2n=24; Ohri and Akuja 1991). This is in line with what was reported for other broad-leaf trees: in C. sativa (2n=24) 12 linkage groups have been identified for both the female and male framework maps, with a total length of 720 and 721 cM (76% and 68% of the genome) (Casasoli et al. 2001); in Quercus robur (2n=24) 12 linkage groups have also been identified for a total length of 893 and 921 cM in the female and male genomes (a 85–90% genome coverage) (Barreneche et al. 1998); in Salix (2n=38) two maps have been constructed from an interspecific cross and 26 and 18 major linkage groups found with a 70–80% genome coverage (Tsarouhas et al. 2002). Intercross dominant markers (which segregate in both parents in a 3:1 ratio) were only used to identify homology between parental maps, and were mapped as accessory markers in the two parental maps. Intercross dominant markers provide limited information for estimating recombination frequency (Maliepaard et al. 1998). Of the 47 intercross dominant AFLP and RAPD loci, 15 (32%) were found associated with framework markers in both maps (LOD>3.0). Twenty one did not show any strong and unambiguous linkage with framework markers segregating 1:1; probably because of the low information content of the linkage between marker pairs segregating 1:1 and 3:1.
QTL mapping
Using a pseudo-testcross approach we carried out a preliminary investigation of the linkage relationship between molecular markers and loci that control quantitative traits in F. sylvatica.
QTL mapping is problematic in forest trees. F2 would be the ideal mapping population for outcrossing species. In forest trees, since the F2 population is in general not available, two-generation pedigrees are used. For this type of mapping population a pseudo-testcross configuration has been shown to be effective (Bradshaw and Stettler 1995). It is common practice to produce a separate QTL analysis for each parent.
In our study we focused on the analysis of quantitative traits related to biomass production (height, leaf traits) and to water use efficiency (δ 13C measurements that provide time-integrated estimates of water use efficiency). The investigated traits have obvious adaptive value (Parkhurst and Loucks 1972; Sun et al. 1996).
The genetic control of height and leaf traits is hard to evaluate because of the low heritability of these traits. Our data do not allow meaningful heritability estimates. Narrow-sense heritability in four full-sib families of Eucalyptus was estimated as 0.19 for height and 0.36 for leaf area (Byrne et al. 1997). For δ 13C, narrow-sense heritability estimates in Picea mariana (Johnsen et al. 1999) and in P. pinaster and (Brendel et al. 2002) were 0.54 and 0.17 respectively.
We found eight QTLs for leaf number, leaf area and shape, and SLA, each explaining about 30% of the total phenotypic variance while no QTLs have been identified for height and δ 13C. Considering both the female and male map, it appears that there is a cluster of QTLs related to leaf characteristics on linkage group 1. Clustering of QTLs has been described for hybrid poplar (Bradshaw and Stettler 1995; Wu et al. 1997). In particular, in the latter study a cluster for various leaf traits was observed. Clusters of QTLs were observed also for different species of the genus Eucalyptus for wood traits, growth traits (Grattapaglia et al. 1996; Verhaegen et al. 1997) and leaf area traits (Byrne et al. 1997), for blight resistance in the genus Castanea (Kubisiak et al 1997) and for wood traits in Pinus taeda (Sewell et al. 2000). Bradshaw and Stettler (1995) suggest for their QTLs that the combination of position and mode of action could indicate pleiotropy of a single QTL as the origin for the QTL clustering, and is more likely than simple linkage to explain the correlations between some of the traits. In our study, the clustered QTLs for leaf traits (mean leaf dimension, leaf area and SLA for year 2002) could suggest the possible presence of only one QTL having pleiotropic effect on different traits. QTLs for leaf area for 1999 and for 2002 have positive and negative effects in the 2 years. Although in the same linkage group they map about 7-cM apart, indicating that we probably located two different QTLs.
The percentage of total variance explained by our leaf trait QTLs is in the range of what was observed in other tree species. In Eucalyptus (Byrne et al. 1997) two QTLs for leaf area (each explaining between 8.9 and 11.6% of the phenotypic variance) were identified. In an inter-specific cross in Populus, QTLs that underlie various leaf traits (i.e. leaf area, shape, orientation, color) and growth (accounting for a relatively large part of trait variation) have been found (Bradshaw and Stettler 1995; Wu et al. 1997). In hybrid poplar, the percentage of explained variance for leaf traits was between 18 and 62.6% (Wu et al. 1997). In Salix (Tsarouhas et al. 2002) ten QTLs for growth-related traits were identified (each explaining between 14 and 22% of the phenotypic variance).
Our results could be useful for the identification of genotypes linked to faster growth rate in the first developmental stages, a very important trait for seedling establishment in naturally regenerated forests, as often European beech forests are.
QTLs for δ 13C in forest tree species were first reported by Brendel et al. (2002). The four significant QTLs explained nearly one-third of the phenotypic variation observed for this trait. Two significant QTLs were found for ring width. No co-location of QTLs for δ 13C and growth was found, whereas a strong genetic correlation between δ 13C and tree growth (height and diameter) was found for P. mariana (Johnsen et al. 1999), but not when water availability was high. In agreement with the results reported by Johnsen et al. (1999), we found significant phenotypic correlations for leaf traits measured in 1999 and δ 13C. For δ 13C, which was demonstrated to be highly correlated in plants with water use efficiency (WUE) (Farquhar and Richards 1984), the absence of QTL location could be related to the lack of variability for the trait in parent trees. Alternatively this can be due to the high plasticity of the trait, that could show variation only in water depletion condition, a condition not reached in our experiment (Silim et al. 2001).
We did not find QTLs for height. In poplar and Eucalyptus (Bradshaw and Stettler 1995; Byrne et al. 1997), leaf area traits clustered in the same linkage groups with height, suggesting the presence of closely linked genes or one gene having a pleiotropic effect on both height and leaf area. In beech we cannot reach firm conclusions: the absence of QTLs can be due to a lack of variability for this trait in the parents of the controlled cross progeny or simply to the small size of our sample (Beavis 1995), and not necessarily to lack of genomic clustering for these genes.
The small size of our sample could also lead to an underestimation of the number of QTLs per trait and to an overestimation of the phenotypic effect associated with each QTL. From simulation studies, Beavis (1995) demonstrated that the power of QTL detection in experiments with small sample sizes decreases as the number of QTLs increases. The estimation of the size of the phenotypic effect is similarly biased, with an estimated effect much greater than the actual simulated effect.
QTL stability over years was evaluated by comparing the locations of putative QTLs for traits measured in different years. The genomic region associated with leaf traits detected in 1999 was close to the one detected in 2002, but the sign of the effect is opposite. This has been previously reported in forest trees. Bradshaw and Stettler (1995) and Kaya et al. (1999) did not find the same QTLs in consecutive growing seasons for height growth or basal area in hybrid Populus, or for height or diameter increment in P. taeda. Similarly, QTLs for total height in P. pinaster for different developmental stages were located on distinct linkage groups (Plomion et al. 1996). To explain this phenomenon, Plomion et al. (1996) hypothesized that maturation in forest trees may induce a progressive shift of the genetic control of growth traits, which may provide an explanation for the low year-to-year correlation. Verhaegen et al. (1997) proposed that a set of regulatory genes may differentially control the temporal expression of the genes controlling a trait; alternatively, a different set of regulatory factors may be involved at different stages of development.
References
Aldrich PR, Michler CH, Sun WL, Romero-Severson J (2002) Microsatellite markers for northern red oak (Fagaceae: Quercus rubra). Mol Ecol Notes 2:472–474
Arcade A, Anselin F, Faivre Rampant P, Lesage MC, Pâques LE, Prat D (2000) Application of AFLP, RAPD and ISSR markers to genetic mapping of European and Japanese larch. Theor Appl Genet 100:299–307
Barreneche T, Bodenes C, Lexer C, Trontin JF, Fluch S, Streiff R, Plomion C, Roussel G, Steinkellner H, Burg K, Favre JM, Glössl J, Kremer A (1998) A genetic linkage map of Quercus robur L. (pedunculate oak) based on RAPD, SCAR, microsatellite, minisatellite, isozyme and 5 S rRNA markers. Theor Appl Genet 97:1090–1103
Basten CJ,Weir BS, Zeng ZB (1994) Zmap-a QTL cartographer. In: Smith C, Gavora JS, Benkel B, Chesnais J, Fairfull W, Gibson JP, Kennedy BW, Burnside EB (eds) Proc 5th World Congress on Genetics Applied to Livestock Production: Computing Strategies and Software. Volume 22. 5th World Congress on Genetics Applied to Livestock Production, Guelph, Ontario, Canada, pp 65–66
Basten CJ,Weir BS, Zeng ZB (2002) QTL cartographer, version 1.16. Department of Statistics, North Carolina State University, Raleigh, North Carolina
Beavis WD (1995) The power and deceit of QTL experiments: lessons from comparative QTL studies. Proc 49th Annu Corn and Sorghum Industry Res Conf 1994:304–312
Bradshaw HD, Stettler RF (1995) Molecular genetics of growth and development in Popolus. IV. Mapping QTLs with large effects on growth, form and phenology traits in a forest tree. Genetics 139:963–973
Brendel O, Pot D, Plomion C, Rozenber P, Guehl JM (2002) Genetic parameters and QTL analysis of δ 13 C and ring width in maritime pine. Plant Cell Environ 25:945–953
Bucci G, Raddi S, Vendramin GG, Leonardi S, Giannini R Menozzi P (1999) Aspetti genetici del faggio in Italia. In “Funzionalità dell’ecosistema faggeta” (a cura di G. Scarascia Mugnozza), Edagricole, Bologna pp 35–69
Byrne M, Murrell JC, Owen V, Kriedemann P, Williams ER, Moran GF (1997) Identification and mode of action of quantitative trait loci affecting seedling height and leaf area in Eucalyptus nitens. Theor Appl Genet 93:674–681
Casasoli M, Mattioni C, Cherubini M, Villani F (2001) A genetic linkage map of European chestnut (Castanea sativa Mill.) based on RAPD, ISSR and isozyme markers. Theor Appl Genet 102:1190–1199
Ceroni M, Leonardi S, Piovani P, Menozzi P (1997) Incrocio diallelico in faggio: obiettivi, metodologie, primi risultati. Monti e Boschi 48:46–51
Cervera MT, Storme V, Ivens B, Gusmao J, Liu BH, Hostyn V, Van Slycken J, Van Mantagu M, Boerjan W (2001) Dense genetic linkage maps of three populus species (Populus deltoids, P. nigra and P. trichocarpa) based on AFLP and microsatellite markers. Genetics 158:787–809
Chakravarti A, Lasher LK, Reefer JE (1991) A maximum likelihood method for estimating genome length using genetic linkage data. Genetics 128:175–182
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Costa P, Pot D, Bubos C, Frigerio JM, Pionneau C, Bodenes C, Bertocchi E, Cervera MT, Remington DL, Plomion C (2000) A genetic map of Maritime pine based on AFLP, RAPD and protein markers. Theor Appl Genet 100:39–48
Craig H (1957) Isotopic standards for carbon and oxygen, and corrections factors for mass spectrometric analysis of carbone dioxide. Geochim Cosmochim Ac 12:133–149
Di Masso E (1999) Fenologia e marcatori genetici in Fagus sylvatica in una popolazione dell’Appennino settentrionale. Dipartimento di Scienze Ambientali, Università di Parma, PhD thesis
Farquhar GD, Richards RA (1984) Isotopic composition of plant carbon correlates with water-use efficiency of wheat genotypes. Aust J Plant Physiol 11:539–552
Frewen BE, Chen THH, Howe GT, Davis J, Rohde A, Boerjan W, Bradshaw Jr HD (2000) Quantitative trait loci and candidate gene mapping of bud set and bud flush in Populus. Genetics 154:837–845
Grattapaglia D, Sederoff R (1994) Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: mapping strategy and RAPD markers. Genetics 137:1121–37
Grattapaglia D, Bertolucci FLG, Penchel R, Sederoff R (1996) Genetic mapping of quantitative trait loci controlling growth and wood quality traits in Eucalyptus grandis using a maternal half-sib family and RAPD markers. Genetics 144:1205–1214
Hulbert SH, Ilott TW, Legg EJ, Lincoln SE, Lander ES, Michelmore RW (1988) Genetic analysis of the fungus, Bremia lactucae, using restriction fragment length polymorphisms. Genetics 120:947–958
Isabel N, Beaulieu J, Theriault P, Bousquet J (1999) Direct evidence for biased gene diversity estimates from dominant amplified polymorphic DNA (RAPD) fingerprints. Mol Ecol 8:477–483
Jermstad KD, Bassoni DL, Jech KS, Wheeler NC, Neale DB (2001) Mapping of quantitative trait loci controlling adaptive traits in coastal Douglas-fir. I. Timing of vegetative bud flush. Theor Appl Genet 102:1142–1151
Johnsen KH, Flanagan LB, Huber DA, Major JE (1999) Genetic variation in growth, carbon isotope discrimination, and foliar N concentration in Picea mariana: analyses from half-diallel mating-system design using field-grown trees. Can J For Res 29:1727–1735
Kampfer S, Lexer C, Glössi J, Steinkellner H (1998) Characterization of (GA)n microsatellite loci from Quercus robur. Hereditas 129:183–186
Kaya Z, Sewell MM, Neale D (1999) Identification of quantitative trait loci influencing annual height- and diameter-increment growth in loblolly pine (Pinus taeda L.). Theor Appl Genet 98:586–592
Kondo T, Terada K,·Hayashi E, Kuramoto N, Okamura M, Kawasaki H (2001) RAPD markers linked to a gene for resistance to pine needle gall midge in Japanese black pine (Pinus thunbergii). Theor Appl Genet 102:871–875
Kubisiak TL, Hebard FV, Nelson CD, Zhang J, Bernatzky R, Huang H, Anagnostakis SL, Doudrick RL (1997) Molecular mapping of resistance to blight in an interspecific cross in the genus Castanea. Phytopathology 87:751–759
Lander E, Green P, Abrahamson J, Barlow A, Daley M, Lincoln S, Newburg L (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Maliepaard C, Alston FH, van Arkel G, Brown LM, Chevreau E, Dunemann F, Evans KM, Gardiner S, Guilford P et al. (1998) Aligning male and female linkage maps of apple (Malus pumila Mill.) using multi-allelic markers. Theor Appl Genet 97:60–73
Marinoni D, Akkak A, Bounous G, Edwards KJ, Botta R (2003) Development and characterization of microsatellite markers in Castanea sativa (Mill). Mol Breed 11:127–136
Marques CM, Araújo, Ferreira JG, Whetten R, O’Malley DM, Liu BH, Sederoff R (1998) AFLP genetic maps of Eucalyptus globulus and E. tereticornis. Theor Appl Genet 96:727–737
Ohri D, Ahuja MR (1991) Giemsa C-banding in Fagus sylvatica L., Betula pendula Roth and Populus tremula L. Silvae Genet 40:72–75
Parkhurst DF, Loucks DL (1972) Optimal leaf size in relation to environment. J Ecol 60:5505–5537
Pastorelli R, Smulders MJM, Van’t Westende WPC, Vosman B, Giannini R, Vettori C, Vendramin GG (2003) Characterisation of microsatellite markers in Fagus sylvatica L. and Fagus orientalis Lipsky. Mol Ecol Notes 3:76–78
Plomion C, Durel CE, O’Malley D (1996) Genetic dissection of height in maritime pine seedlings raised under accelerated growth conditions. Theor Appl Genet 93:849–858
Scotti I, Paglia G, Magni F, Morgante M (1999) Microsatellite markers as a tool for the detection of intra- and interpopulational genetic structure. In: Gillet EM (ed) Which marker for which purpose? http://webdoc.sub.gwdg.de/ebook/y/1999/whichmarker/index.htm
Sewell MM, Bassoni DL, Megraw RA, Wheeler NC (2000) Identification of QTLs influencing wood property traits in loblolly pine (Pinus taeda L.). I. Physical wood properties. Theor Appl Genet 101:1273–1281
Sewell MM, Davis MF, Tuskin GA, Wheeler NC, Elam CC, Bassoni DL, Neale DB (2002) Identification of QTLs influencing wood property traits in loblolly pine (Pinus taeda L.). II. Chemical wood properties. Theor Appl Genet 104:214–222
Silim SN, Guy RD, Patterson TB, Livingston NJ (2001) Plasticity in water-use efficiency of Picea sitchensis, P. glauca and their natural hybrids. Oecologia 128:317–325
Smulders MJM, Bredemeijer G, Rus-Kortekaas W, Arens P, Vosman B (1997) Use of short microsatellites from database sequences to generate polymorphisms among Lycopersicon esculentum cultivars and accessions of other Lycopersicon species. Theor Appl Genet 94:264–272
Southern EM (1979) Measurement of DNA length by electrophoresis. Anal Biochem 100:319–323
Sun ZJ, Livingston NJ, Guy RD, Ethier GJ (1996) Stable carbon isotopes as indicators of increased water use efficiency and productivity in white spruce [Picea glauca (Moench) Voss] seedlings. Plant Cell Environ 19:887–894
Teissier du Cros E, Le Tacon F, Nepveu G, Pardè J, Perrin R, Timbal J (1981) Le hetre, INRA Department des Reserches Forestieres, Paris
Troggio M, Kubisiak TL, Bucci G, Menozzi P (2001) Randomly amplified polymorphic DNA linkage relationships in different Norway spruce populations. Can J For Res 31:1456–1461
Tsarouhas V, Gullberg U, Lagercrantz U (2002) An AFLP and RFLP linkage map and quantitative trait locus (QTL) analysis of growth traits in Salix. Theor Appl Genet 105:277–288
Van Ooijen JW, Voorrips RE (2001) JoinMap 3.0, software for the calculation of genetic linkage maps. Plant Research International, Wageningen, The Netherlands
Verhaegen D, Plomion C, Gion JM, Poitel M, Costa P, Kremer A (1997) Quantitative trait dissection analysis in Eucalyptus using RAPD markers. 1. Detection of QTL in interspecific hybrid progeny, stability of QTL expression across different ages. Theor Appl Genet 95:597–608
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–14
Williams JGK, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV (1990) DNA plymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res 18:6531–6535
Wu R, Bradshaw JR HD, Stettler RF (1997) Molecular genetics of growth and development in Populus (Salicaceae). V. Mapping quantitative trait loci affecting leaf variation. Am J Bot 84:143–153
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Acknowledgements
The first two authors contributed equally to the work. This research was supported by the European Union (QLRT-1999-01210, DYNABEECH) and the project M.U.R.S.T. COFIN2000 (Coordinator: Prof. Marco Borghetti). We declare that the experiments comply with the current laws of Italy where the experiments were carried out.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by D. B. Neale
An erratum to this article is available at http://dx.doi.org/10.1007/s00122-003-1583-7.
Rights and permissions
About this article
Cite this article
Scalfi, M., Troggio, M., Piovani, P. et al. A RAPD, AFLP and SSR linkage map, and QTL analysis in European beech (Fagus sylvatica L.). Theor Appl Genet 108, 433–441 (2004). https://doi.org/10.1007/s00122-003-1461-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-003-1461-3